

Abstract
The aim of this project was to gain an improved understanding of how the efficiency of hole and electron transfer from the solvation layer to DNA decreases as a function of distance from DNA. The packing of DNA in crystals of known structure makes it possible to calculate the degree of DNA hydration with a precision that is significantly greater than that achievable for amorphous samples. Previous work on oligodeoxynucleotide crystals has demonstrated that the efficiency of free radical trapping by DNA exposed to ionizing radiation at 4 K is relatively insensitive to base sequence, conformation, counterion, or base stacking continuity. Having eliminated these confounding variables, it is now possible to ascertain the degree of radical transfer that occurs from ionized water as a function of DNA hydration (Γ, in mol water/mol nucleotide). EPR is used to measure the hydroxyl radical concentration in crystals irradiated at 4 K. From a lack of hydroxyl radicals trapped in the inner hydration mantle, we determine that hole transfer to DNA is complete for water molecules located within 8 Å. This corresponds to Γ = 9–11 and indicates that hole transfer is 100% (as efficient as direct ionization of DNA) for water molecules adjacent to DNA. Beyond ~8 Å (Γ > 10), hydroxyl radicals are observed; thus deprotonation of the water radical cation is seen to compete with hole transfer to DNA as soon as one water intervenes between the ionized water and DNA. The boundary for 0% hole transfer is projected to occur somewhere between 15 and 20 waters per nucleotide. Electron transfer, on the other hand, is 100% efficient across the entire range studied, 4.2 ≤ Γ ≤ 15.6.
INTRODUCTION
There are two categories of damage to hydrated DNA resulting from interaction with ionizing radiation. The first is direct-type damage, resulting from direct ionization of the DNA or its inner hydration layer (1, 2). The holes and electrons produced in the inner hydration layer transfer to the DNA, and once trapped by DNA are indistinguishable from holes and electrons produced by direct DNA ionization. The second category is indirect-type damage. The holes formed in the more distant bulk water lead to radical intermediates, such as the hydroxyl radical (HO·), which may then react with the DNA (3). In cells, DNA is tightly wrapped around core proteins, and the hydration state is low (4, 5). It has been estimated that up to half the damage to DNA in this environment is due to direct-type interactions (6).
Identifying the primary factor(s) that influences the free radical yield of DNA, G(free radicalDNA), has been a focus of our research (1, 7-10). A number of potentially confounding variables have been investigated, including conformation, base sequence, base stack continuity, packing, and degree of hydration (given as Γ in mol water/mol nucleotide). In studying free radical yields in DNA films as a function of Γ, it was postulated that the large scatter in the data was due to variability in the way the DNA is packed (7). Subsequent work on NaDNA films led to reduced scatter and showed that free radical yields increase as Γ increases from 0 to 15 (8, 9); this increase had also been observed in lyophilized powders (11). There is little question, therefore, that hydration of DNA influences radical yields not only by radical transfer but also by affecting other variables. Previous work in our laboratory demonstrated that G(free radicalDNA) is not very sensitive to conformation, base sequence, or base stacking of the DNA (10), and we have proposed that the packing of DNA is a critical variable (8, 9). In this paper, knowledge of the crystal structure in DNA oligodeoxynucleotide crystals provides relatively precise information on the DNA packing and is used to better define how the hydration layer influences radical yields.
Although a substantial fraction of holes initially formed in the hydration layer migrate to the DNA, recent work has shown that for Γ > 9 hydroxyl radicals (HO·) are trapped by the hydration shell in lyophilized powders (12, 13). That is, for the more distant waters of the hydration shell, formation of HO· competes with hole transfer. But there are difficulties in interpreting results obtained using powder DNA: The packing is not well characterized, and the extent to which local values of Γ deviate from the average Γ is unknown.
This paper revisits the question of the distance dependence of hole transfer through the hydration shell using oligodeoxynucleotide crystals. The crystals again offer some advantages. For the first time, hole transfer through the hydration layer is studied in samples for which a more precise knowledge of the local distribution of water, as well as the global average, is available. We are able to use the known spacing within the crystal lattice to correlate hole transfer with distance between the water and DNA.
MATERIALS AND METHODS
Crystal growth and mounting have been described previously (10). Mother liquor was removed from the crystal surfaces using fine paper wicks. The weights of these relatively pure crystalline samples were measured with a Cahn C-30 microbalance to an accuracy of ±1 μg. Typical samples weighed 150–300 μg.
Some samples were placed under dehydrating conditions in chambers containing NaOH or P2O5 for several weeks to remove lattice water. From the change in total weight, typically 7–12%, the amount of water lost was calculated. The dehydrated samples retained a crystalline appearance, although they often became darker or even opaque. Estimated water loss for the dehydrated samples is shown in Table 1.
TABLE 1.
Crystal Characteristics
| Crystal sequence | Γ (calculated) | Test sphere radius (Å) | Free radical yield (μmol/J)a | Dehydration water loss (%)b | Dehydrated free radical yield (μmol/J)c | Reference |
|---|---|---|---|---|---|---|
| CGCG | 7.1 | 5.5 | 0.65 ± 0.15 | ~45 | 0.57 ± 0.02 (2) | (24) |
| CACGCG:GTGCGC | 4.2 | 6.0 | 0.61 ± 0.03 | NA | (25) | |
| CGCGCG (I) | 6.2 | 6.0 | 0.73 ± 0.10 | NA | (26) | |
| CGCGCG (II) | 7.2 | 6.0 | 0.70 ± 0.06 | NA | (27) | |
| CGCGCG (III) | 5.2 | 6.0 | 0.57 ± 0.05 | NA | (20) | |
| CTCTCGAGAG | 6.7 | 6.0 | 0.70 ± 0.06 | NA | (28) | |
| GCGGGCCCGC (I) | 9.6 | 7.0 | 0.55 ± 0.10 | NA | (29) | |
| CCAACGTTGG | 6.8 | 7.5 | 0.78 ± 0.05 | NA | (30) | |
| CGCGAATTCGCG | 9.0 | 7.5 | 0.66 ± 0.06 | 22 | 0.63 ± 0.05 (2) | (31) |
| CCCTAGGG | 9.3 | 8.0 | 0.70 ± 0.04 | ~50 | 0.74 ± 0.07 (2) | (32) |
| GTGCGCAC | 10.5 | 8.5 | 0.72 ± 0.07 | 21 | 0.58 ± 0.03 (1) | (33) |
| GCACGCGTGC | 14.0 | 12.5 | 0.57 ± 0.04 | 23 | 0.62 ± 0.12 (6) | (34) |
| GCGGGCCCGC (II) | 15.6 | 13.0 | 0.58 ± 0.08 | 29 | 0.56 ± 0.06 (1) | (29) |
| CTCGAG | 15.2 | 14.0 | 0.75 ± 0.14 | NA | (35) |
The degree of DNA hydration (Γ) was not measured directly due to inherent difficulties in determining densities. Instead, the value of Γ was calculated using structural information obtained by X-ray crystallography. The coordinates of all ordered non-hydrogen atoms were downloaded from the Nucleic Acids Database (Rutgers, NJ) into the programs Quanta 97 and InsightII 97.0 (Molecular Simulations Inc., San Diego) on a Silicon Graphics workstation. The symmetry elements of the crystallographic models were generated and the empty regions of the unit cell were filled with water molecules following routines provided with the InsightII software package. An example of such a fit is shown in Fig. 1. The water content of each crystal cell was determined by adding the number of water molecules generated by the InsightII routine to the number of water molecules assigned by X-ray crystallography. The hydration state (Γ) of the DNA crystal was determined by dividing the total number of water molecules in the cell by the number of base moieties in the cell. Water molecules placed by InsightII that made van der Waals contact with other atoms were removed from the calculation, so the hydration states reported here should be considered as slightly underestimating the true hydration. The values of Γ of the crystals are given in Table 1. Note that counterions located in the crystal structure by X-ray crystallography have been included in the calculations of DNA hydration, but no additional counterions were simulated in the disordered regions of the structure. Calculations have shown that the addition of two or three counterions per molecule did not significantly alter the hydration state of the crystals. In any event, few counterions are expected farther than 10 Å from the DNA (14).
FIG. 1.

X-ray crystallographic structure of d(CTCGAG). The atoms located by X-ray crystallography are shown in green and the water molecules fitted by InsightII in blue.
Irradiations were carried out using an OEG-76 tungsten filament X-ray tube operated at 20 mA and 70 keV with a dose rate of 26 kGy/h at a temperature of 4 K. EPR (electron paramagnetic resonance) measurements were made using a Q-band Varian E-12 spectrometer equipped with a Janis cryostat. Free radical yields were determined at 4 K by comparing the intensity of the EPR spectra of the crystalline oligodeoxynucleotides with a ruby standard mounted on the side of the detection cavity [for more information on the experimental setup, see refs. (10, 15)].
Hydroxyl radical yields were measured by comparing the EPR spectral intensity of pure ice with the spectral intensity of an oligodeoxynucleotide crystal irradiated with the same dose, 22 kGy. The downfield features of the hydroxyl radical spectrum, which are well outside any features found from the spectra of DNA radicals, are shown in Fig. 2. Of these features, one was used as the reference feature for comparing the two spectra (7). Alignment of the spectra was made using the spectral lines of H· trapped in the quartz capillaries. The intensity of the central hydroxyl radical peak increases at the same rate as the downfield reference line in the dose range employed. The yields of hydroxyl radicals were measured for 11 different oligodeoxynucleotides. Given the need for relatively large samples and significant limitations on amounts of material, 1–4 measurements were made for each oligodeoxynucleotide. The measurements are subject to relatively large errors primarily because the spectral components of interest are broad and weak. To estimate the error, the comparison of the pure ice signal to that of the DNA crystal was pushed to the upper and lower limit, yielding a maximum and minimum hydroxyl radical concentration. In pure ice, we measure G(HO·) = 0.032 μmol/J, similar to the yields obtained by others (13).
FIG. 2.
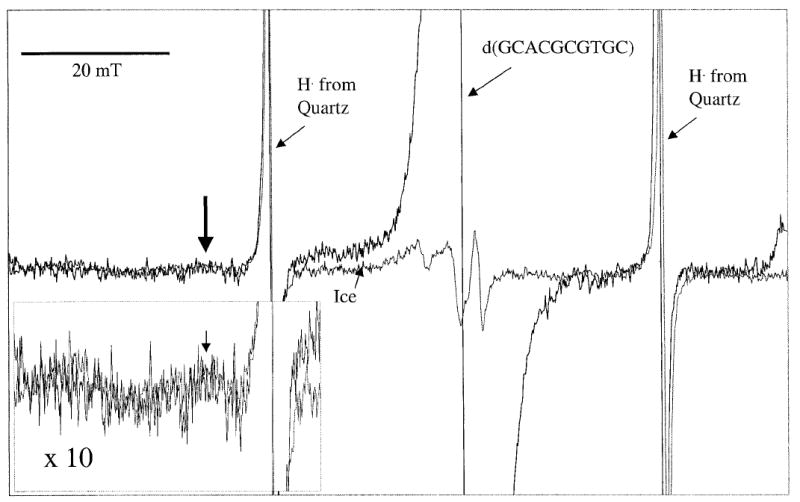
EPR spectrum of d(GCACGCGTGC) crystal irradiated with 22 kGy at 4 K overlaid with a spectrum of ice irradiated with the same dose. Scan width is 100 mT, and the spectra were recorded at 4 K. The inset depicts the downfield portion of the spectra magnified by a factor of 10. The arrow points to the feature of the hydroxyl radical spectrum used in determining the hydroxyl radical yields. The hydroxyl radical spectrum is quite broad and contains multiple features. It is responsible for all but the sharp singlet in the spectrum of ice.
RESULTS AND DISCUSSION
The completeness of hole transfer from the hydration layer to the DNA was measured by monitoring the production of hydroxyl radicals in the oligodeoxynucleotide crystals. Previous work has suggested that complete hole transfer occurs up to a Γ of 9 (13). Consistent with that finding, we observe very little, if any, hydroxyl radical production up to Γ = 9.3. In crystals with hydration beyond Γ = 9.3, there is an increase of hydroxyl radical production, indicating that deprotonation of H2O·+ competes with hole transfer and results in trapped HO·.
To better define the distance dependence of this competition, the crystal structures were used to determine the maximal distance a radical generated in the hydration waters must travel to reach the DNA. A test sphere created by the program Quanta 97 was inserted into the hydrated region of the DNA structures, and its radius was allowed to expand until the surface of the test sphere could no longer be moved without touching an atom of a base or sugar. The fitted test sphere radii are given in Table 1 and are accurate to ±0.5 Å. An example of this fit is shown in Fig. 3. A plot of hydroxyl radical concentration as a function of the test sphere radius (maximum distance a hole must travel to reach DNA) is given in Fig. 4.
FIG. 3.

Example of the determination of the test sphere size using Quanta 97. The size of this sphere gives the maximal distance at which a radical could be created from a base or sugar of the DNA. The atoms of the d(GCGGGCCCGC) (II) crystal structure are indicated in blue. The test sphere is shown in orange. The radius of this sphere is increased until the surface contacts a base or sugar atom.
FIG. 4.
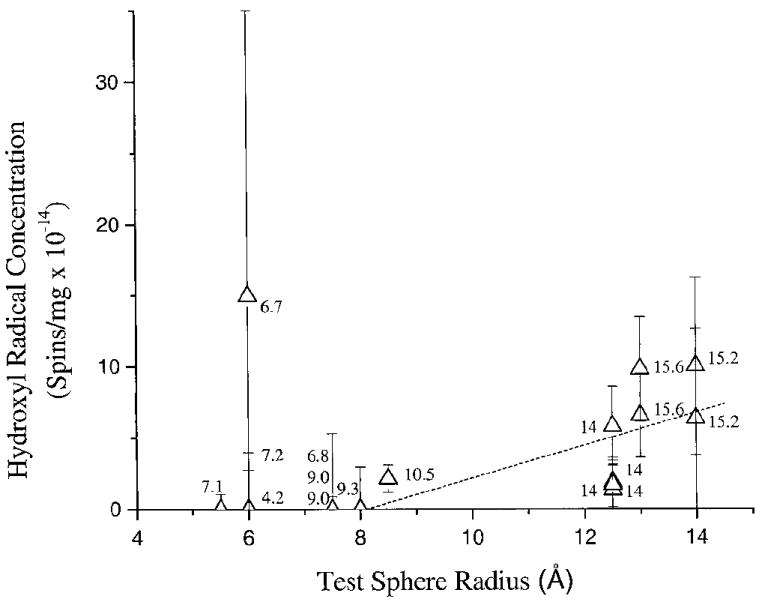
Plot of the hydroxyl radical concentration as a function of the test sphere size. The error in the absolute number of spins is greater than the relative errors between the samples. Errors are estimated as described in the Materials and Methods. The numbers alongside the data refer to the degree of hydration (Γ) of the individual crystalline samples; the sequences are given in Table 1 and can be identified by the respective values of Γ. The dotted line is a linear least-squares fit to the data for the crystals of radius 9 Å or more. Note that there is an apparent increase in the hydroxyl radical concentration as the solvent space increases in size.
For values of Γ ≤ 9.3, the hole traverses at least 8 Å, which correlates with water molecules that are in direct contact with the DNA. There appears to be a general trend of increasing hydroxyl radical concentration as the distance increases from 12.5 to 14 Å, with an onset of hole trapping as close as 8.5 Å. In Fig. 4, a least-squares line is fitted to the data >8 Å. Over this span, Γ increases from 9.3 to 15.6. While the actual fit to the data may not be linear, it appears that the boundary for complete hole transfer to DNA appears to be, at minimum, between 8 and 9 Å, with a relatively rapid decrease in hole transfer between 12.5 and 14 Å.
In all of the crystals displaying the ability to trap HO·, there are waters not in direct contact with the DNA. The sole exception appears to be the d(CTCTCGAGAG) crystal, but there is reason to believe that the measurement of the yield in this case was influenced by the presence of mother liquor. Most samples used for the measurement of hydroxyl radical yields were single crystals, or a small number of crystals gathered together. Owing to their extremely small size, over a dozen of the d(CTCTCGAGAG) crystals had to be used. Such a collection makes removal of all mother liquor much less certain. For this reason, we treat the HO· yield for d(CTCTCGAGAG) as an aberrant point and conclude that hole transfer is complete for water in direct contact with the DNA.
The average hydroxyl radical yield for the crystals with Γ > 10 was estimated from the data shown in Fig. 4 and the number of water molecules beyond Γ = 9 in each sample. The result averaged over all eight data points is G(HO·) ~ 0.27 ± 0.15 μmol/J. This value for the hydroxyl radical yield in the hydration layer is somewhat higher than that determined by La Vere et al. in lyophilized powders (0.09 μmol/J) (13), but the agreement is fairly good considering the differences in sample type. In particular, lyophilized powders are likely to have a larger disparity in the local values of Γ.
The average yield of hydroxyl radicals in the crystals calculated above may be compared to the initial yield of radicals (prior to any combination events). The expected initial yield is G(H2Ototal) = 1.2 μmol/J (16) and, since half are holes, the initial value of G(hole) would be 0.6 μmol/J. Dividing the range of G(HO·) measured for the DNA crystals with Γ > 10 (0.12–0.42 μmol/J) by 0.6 μmol/J indicates that 20–70% of the initial holes produce hydroxyl radicals in the outer hydration waters of the DNA crystals. This points out that deprotonation of the water radical cation competes effectively with both recombination and hole transfer to DNA. The fact that G(HO·) is an order of magnitude greater in DNA crystals than in ice is almost certainly a consequence of electron scavenging by the DNA inhibiting the combination reaction between electrons and HO·.
The total free radical yields, G(free radicaltotal), for 14 different types of oligodeoxynucleotide crystals [determined previously: see refs. (10, 17)] are plotted as a function of Γ in Fig. 5 and are depicted as solid squares. The total free radical yield includes the radicals trapped on the DNA as well as the hydroxyl radicals trapped in the hydration layer, since the bulk of the hydroxyl radical EPR signal is coincident with the DNA EPR spectrum (see Fig. 2). The data have been fitted by least-squares to a straight line. G(free radicaltotal) remains relatively constant for crystals hydrated from 4.2 to 15.6 waters/nucleotide. A portion of the hydroxyl radical signal is not included in the measurement of DNA-trapped radical intensity because it is far from the center of the DNA spectrum (the DNA spectrum is integrated over ~16 mT while the HO· spectra extend over >90 mT). In effect, the more holes that are trapped and incapable of reaching the DNA, the more signal in the extreme wings of the EPR spectra that is not counted in the total free radical yield. The values of G(free radicalDNA), when the contribution of the hydroxyl radical signal is subtracted out, are displayed as open circles in Fig. 5.
FIG. 5.
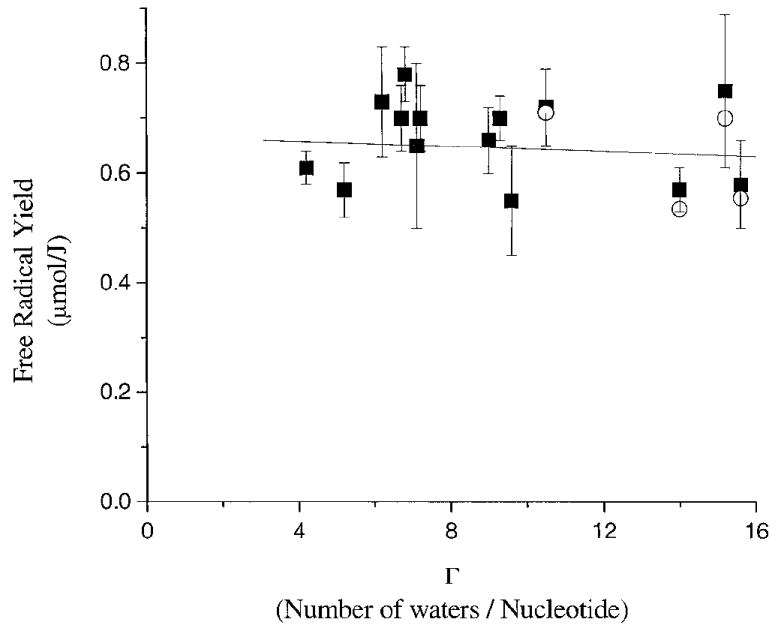
Plot of the free radical yields of 14 oligodeoxynucleotide crystals as a function of hydration. The crystal sequences are found in Table 1. The yields G(free radicaltotal) as determined in ref. (10) are shown as solid squares. A linear least-squares has been fitted to G(free radicaltotal). The open circles are the yields G(free radicalDNA) after subtraction of the hydroxyl radical contribution.
The flat dose response seen in Fig. 5 contrasts with that found for DNA films (8, 9) and lyophilized powders (18); in the latter, G(free radicaltotal) increased as Γ increased from 0 to 15. Films of NaDNA are known to increase in density over this same range, changing from 1.2 g/cm3 at Γ = 4 to 1.3 g/cm3 at Γ = 15 (19). Although the densities of the crystals we employed are not generally known and are difficult to measure, they can be estimated using the unit cell volume and assuming that the disordered space is occupied by water (see the Materials and Methods). The calculated densities of the crystals fall in the range of 1.1–1.4 g/cm3 and are similar to estimates made by others (20). Our calculated densities are lower than the actual densities because some counterions, polycations (spermine), and waters are neglected (see the Materials and Methods). Regardless, the films and crystals are close in density.
The difference between the crystals and films is the uniformity and constancy of the DNA packing. Packing in the films must be more variable, with clusters of strands separated by distances comparable to those for crystals in some regions, and other regions of much larger separation. It has been proposed that this variability is the reason that yield measurements in films fluctuate more than those in crystals. The increase in the yield of the films from Γ = 0 to Γ = 15 can be ascribed to increasing the amount of DNA that is in tightly packed arrays (8). The crystals have a regular pattern of strand separation that is constant for any given crystal type and relatively constant between different crystal types. The standard deviations on yield measurements are thereby smaller than deviations obtained for films. Since crystals lack the regions of wide separation that occur in films and powders, the yields are higher (9, 10).
In the interval 4.2 < Γ < 15.6, the fractional mass of water increases from 20% to 48% in the crystals, yet the variation of G(free radicaltotal) appears to be random and remains between 0.55 and 0.77 μmol/J. Because all the salient variables are known, particularly packing, this result provides a clearer definition of the role of water. That is, water up to at least Γ = 15.6 transfers radiation-generated electrons and holes to the DNA. The radicals formed from water-derived holes and electrons cannot be distinguished from radicals produced by direct ionization of DNA. The yield of DNA trapped radicals originating from the ionization of water is, to the first approximation, the same as those originating from direct ionization of DNA. This is why the slope of the line in Fig. 5 is approximately zero. These results, by virtue of eliminating problems having to do with covariance of packing and Γ, prove that the proposal made by Gregoli (21) and Hüttermann (22) is correct; direct ionization of the hydration layer is transferred to DNA. More precisely, the efficiency for hole and electron trapping by DNA via hole and electron transfer from the solvation layer is the same as that for trapping via direct ionization of DNA up to at least Γ = 9.3. At higher hydration, the efficiency of hole transfer decreases.
As described in the Materials and Methods, several DNA crystals were dehydrated. The free radical yields of the dehydrated crystals were determined and are shown in Table 1. There is no obvious change in G(free radicaltotal) for the dehydrated crystals when compared to the fully hydrated crystals. These results are further evidence that transfer from the hydration layers is comparable in efficiency to direct ionization of the DNA with respect to excess electron and hole trapping.
The yield of DNA trapped electrons, G(eDNA), produced by water ionization is effectively equal to that produced by direct DNA ionization. There are no competing reactions for Γ < 16. Evidence for this is the lack of formation of hydrogen atoms in the crystalline DNA samples (data not shown). This is consistent with our previous observation that the threshold for formation of hydrogen atoms in DNA films occurs at Γ > 20 (8). While electron transfer is effectively complete, i.e., all excess electrons that are trapped are trapped by DNA, this is not the case for hole transfer.
To estimate the fraction of holes transfer from distant waters to DNA, the following calculation was performed. The average hydroxyl radical yield was calculated from the data (Fig. 4) from four d(GCACGCGTGC) crystals (Γ = 14). The water above and beyond Γ = 9 (14−9 = 5 waters/nucleotide) was used as the radiation-absorbing mass. Using this mass and the measured concentration of HO·, the average value of G(HO·) in this crystal is 0.16 ± 0.11 μmol/J. The G(free radicaltotal) of d(GCACGCGTGC) is 0.57 μmol/J (see Table 1), where free radicaltotal includes DNA trapped radicals and HO·. Because [free radicalDNA] ≫ [HO·], G(free radicalDNA) ≈ G(free radicltotal) = 0.57 μmol/J. Based on the observation that crystals hydrated between 4 and 9 waters/nucleotide yield no HO·, we assume that the inner 9 waters in this crystal contribute equally [G(free radicalDNA) of 0.57 μmol/J] to the yield of DNA-trapped holes and electrons, i.e., G(hDNA) = G(eDNA) = 0.28 μmol/J for Γ ≤ 9.
The outer waters (Γ = 10–14) do not trap the electron and therefore trapping of water generated electrons is as efficient as trapping DNA-generated electrons, i.e., G(efromH2O) = G(eDNA) = 0.28 μmol/J. This is not the case for holes produced in the outer water layer. The difference between G(hDNA) and G(HO·) provides an estimate of the yield of DNA trapped holes originating from the outer waters [G(hfromH2O)]. That is, G(hfromH2O) = G(hDNA) − G(HO·) = [0.28 − (0.16 ± 0.11)] μmol/J = 0.12 ± 0.11 μmol/J. Thus 43 ± 40% of the holes transfer to the DNA from the waters in the Γ = 10–14 shell [based on DNA having about the same stopping power as water (23): This calculation assumes that the initial radical yield in DNA is the same as water]. While the distance dependence for hole transfer is probably not a step function, it appears to drop quickly and is most likely zero for Γ beyond 15–20 waters per nucleotide.
CONCLUSIONS
This work supports the hypothesis that both electrons and holes, formed by ionization of the first hydration layer of DNA, transfer to the DNA, where they are trapped with an efficiency equal to the trapping efficiency for electrons and holes produced by direct ionization of the DNA itself. The efficiency of electron transfer remains constant out to Γ > 15. In contrast, the efficiency of hole transfer begins to drop somewhere between 8 and 12.5 Å from the DNA, corresponding to a Γ of 9 to 14 waters per nucleotide. Beyond Γ of ~ 10, hole transfer is in competition with deprotonation of the H2O·+ and formation of HO·. The boundary for 0% hole transfer is projected to occur somewhere between 15 and 20 waters per nucleotide.
Acknowledgments
The authors would like to thank Kermit R. Mercer for his invaluable technical assistance. This study was supported by PHS Grant 2-R01-CA32546, awarded by the National Cancer Institute. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute.
References
- 1.Mroczka NE, Mercer KR, Bernhard WA. The effects of lattice water on free radical yields in X-irradiated crystalline pyrimidines and purines: A low-temperature electron paramagnetic resonance investigation. Radiat Res. 1997;147:560–568. [PubMed] [Google Scholar]
- 2.Mroczka NE, Bernhard WA. Electron paramagnetic resonance investigation of X-irradiated poly(U), poly(A) and poly(A):poly(U): Influence of hydration, packing and conformation on radical yield at 4 K. Radiat Res. 1995;144:251–257. [PubMed] [Google Scholar]
- 3.von Sonntag C. The Chemical Basis of Radiation Biology. Taylor and Francis; New York: 1987. [Google Scholar]
- 4.Texter J. Nucleic acid–water interactions. Prog Biophys Mol Biol. 1978;33:83–97. doi: 10.1016/0079-6107(79)90026-9. [DOI] [PubMed] [Google Scholar]
- 5.Cullis PM, Langman S, Podmore ID, Symons MC. Effects of ionising radiation on deoxyribonucleic acid. Part VI.—Effects of hydroxyl radical scavengers on radiation damage to DNA. J Chem Soc Faraday Trans. 1990;86:3267–3271. [Google Scholar]
- 6.Krisch RE, Flick MB, Trumbore CN. Radiation chemical mechanisms of single- and double-strand break formation in irradiated SV40 DNA. Radiat Res. 1991;126:251–259. [PubMed] [Google Scholar]
- 7.Mroczka N, Bernhard WA. Hydration effects on free radical yields in DNA X-irradiated at 4 K. Radiat Res. 1993;135:155–159. [PubMed] [Google Scholar]
- 8.Milano MT, Bernhard WA. The effect of packing and conformation on free radical yields in films of variably hydrated DNA. Radiat Res. 1999;151:39–49. [PMC free article] [PubMed] [Google Scholar]
- 9.Milano MT, Bernhard WA. The influence of packing on free radical yields in solid-state DNA: Film compared to lyophilized frozen solution. Radiat Res. 1999;152:196–201. [PMC free article] [PubMed] [Google Scholar]
- 10.Debije MG, Bernhard WA. Free radical yields in crystalline DNA X-irradiated at 4 K. Radiat Res. 1999;152:583–589. [PMC free article] [PubMed] [Google Scholar]
- 11.Wang W, Yan M, Becker D, Sevilla MD. The influence of hydration on the absolute yields of primary free radicals in gamma-irradiated DNA at 77 K. II. Individual radical yields. Radiat Res. 1994;137:2–10. [PubMed] [Google Scholar]
- 12.Becker D, La Vere T, Sevilla MD. ESR detection at 77 K of the hydroxyl radical in the hydration layer of gamma-irradiated DNA. Radiat Res. 1994;140:123–129. [PubMed] [Google Scholar]
- 13.La Vere T, Becker D, Sevilla MD. Yield of ·OH in gamma-irradiated DNA as a function of DNA hydration: Hole transfer in competition with ·OH formation. Radiat Res. 1996;145:673–680. [PubMed] [Google Scholar]
- 14.Young MA, Jayaram B, Beveridge DL. Intrusion of counterions into the spine of hydration in the minor groove of DNA: fractional occupancy of electronegative pockets. J Am Chem Soc. 1997;119:59–69. [Google Scholar]
- 15.Mercer KR, Bernhard WA. Design and operation of a variable temperature accessory for Q-band ESR. J Magn Reson. 1987;74:66–71. [Google Scholar]
- 16.Chatterjee A, Koehl P, Magee JL. Theoretical consideration of the chemical pathways for radiation-induced strand breaks. Adv Space Res. 1986;6:97–105. doi: 10.1016/0273-1177(86)90282-6. [DOI] [PubMed] [Google Scholar]
- 17.Debije MG, Milano MT, Bernhard WA. DNA responds to ionizing radiation as an insulator not as a ‘molecular wire’. Angew Chem Int Ed Engl. 1999;38:2752–2756. [PMC free article] [PubMed] [Google Scholar]
- 18.Wang W, Becker D, Sevilla MD. The influence of hydration on the absolute yields of primary ionic free radicals in γ-irradiated DNA at 77 K. I. Total radical yields. Radiat Res. 1993;135:146–154. [PubMed] [Google Scholar]
- 19.Lee SA, Lindsay SM, Powell JW, Weidlich T, Tao NJ, Lewen GD. A Brillouin scattering study of the hydration of Li- and Na-DNA films. Biopolymers. 1987;26:1637–1665. doi: 10.1002/bip.360261002. [DOI] [PubMed] [Google Scholar]
- 20.Egli M, Williams LD, Gao Q, Rich A. Structure of pure-spermine form of Z-DNA (magnesium free) at 1Å resolution. Biochemistry. 1991;30:11388–11402. doi: 10.1021/bi00112a005. [DOI] [PubMed] [Google Scholar]
- 21.Gregoli S, Olast M, Bertinchamps A. Radiolytic pathways in γ-irradiated DNA: Influence of chemical and conformational factors. Radiat Res. 1982;89:238–254. [PubMed] [Google Scholar]
- 22.Hüttermann J, Roehrig M, Köhnlein W. Free radicals from irradiated lyophilized DNA: influence of water of hydration. Int J Radiat Biol. 1992;61:299–313. doi: 10.1080/09553009214550981. [DOI] [PubMed] [Google Scholar]
- 23.La Verne JA, Pimblott SM. Electron energy-loss distributions in solid, dry DNA. Radiat Res. 1995;141:208–215. [PubMed] [Google Scholar]
- 24.Crawford JL, Kolpak FJ, Wang A, Quigley G, van Boom J, van der Marel G, Rich A. The tetramer d(CpGpCpG) crystallizes as a left-handed double helix. Proc Natl Acad Sci USA. 1980;77:4016–4020. doi: 10.1073/pnas.77.7.4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sadasivan C, Gautham N. Sequence-dependent microheterogeneity of Z-DNA: The crystal and molecular structures of d(CACGCG)d(CGCGTG) and d(CGCACG)d(CGTGCG) J Mol Biol. 1995;248:918–930. doi: 10.1006/jmbi.1995.9894. [DOI] [PubMed] [Google Scholar]
- 26.Wang AH, Quigley GJ, Kolpak FJ, Crawford JL, van Boom JH, van der Marel G, Rich A. Molecular structure of a left-handed double helical DNA fragment at atomic resolution. Nature. 1979;282:680–686. doi: 10.1038/282680a0. [DOI] [PubMed] [Google Scholar]
- 27.Gessner RV, Frederick CA, Quigley GJ, Rich A, Wang AH-J. The molecular structure of the left-handed Z-DNA double helix at 1.0Å atomic resolution. J Biol Chem. 1989;264:7921–7935. doi: 10.2210/pdb1dcg/pdb. [DOI] [PubMed] [Google Scholar]
- 28.Goodsell DG, Grzeskowiak K, Dickerson RE. Crystal structure of CTCTCGAGAG. Implications for the structure of the Halliday junction. Biochemistry. 1995;34:1022–1029. doi: 10.1021/bi00003a037. [DOI] [PubMed] [Google Scholar]
- 29.Ramakrishnan B, Sundaralingam M. Evidence for crystal environment dominating base sequence effects on DNA conformation: Crystal structures of the orthorhombic and hexagonal polymorphs of the A-DNA decamer d(GCGGGCCCGC) and comparison with their isomorphous crystal structures. Biochemistry. 1993;32:11458–11468. doi: 10.1021/bi00093a025. [DOI] [PubMed] [Google Scholar]
- 30.Privé GG, Yanagi K, Dickerson RE. Structure of the B-DNA decamer CCAACGTTGG and comparison with isomorphous decamers CCAAGATTGG and CCAGGCCTGG. J Mol Biol. 1991;217:177–199. doi: 10.1016/0022-2836(91)90619-h. [DOI] [PubMed] [Google Scholar]
- 31.Shui X, McFail-Isom L, Hu GG, Williams LD. The B-DNA dodecamer at high resolution reveals a spine of water on sodium. Biochemistry. 1998;37:8341–8355. doi: 10.1021/bi973073c. [DOI] [PubMed] [Google Scholar]
- 32.Tippin DB, Sundaralingam M. Structure of d(CCCTAGGG): Comparison with nine isomorphous octamer sequences reveals four distinct patterns of sequence-dependent intermolecular interactions. Acta Crystallogr. 1996;D52:997–1003. doi: 10.1107/S0907444996005033. [DOI] [PubMed] [Google Scholar]
- 33.Bingman C, Li X, Zon G, Sundaralingam M. Crystal and molecular structure of d(GTGCGCAC): Investigation of the effect of base sequence on the conformation of octamer duplexes. Biochemistry. 1992;31:12803–12812. doi: 10.1021/bi00166a014. [DOI] [PubMed] [Google Scholar]
- 34.Ban C, Sundaralingam M. Crystal structure of the self-complementary 5(-purine start decamer d(GCACGCGTGC) in the A-DNA conformation. Biophys J. 1996;71:1222–1227. doi: 10.1016/S0006-3495(96)79351-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wahl M, Rao S, Sundaralingam M. Crystal structure of the B-DNA hexamer d(CTCGAGG): Model for an A-to-B transition. Biophys J. 1996;70:2857–2866. doi: 10.1016/S0006-3495(96)79855-7. [DOI] [PMC free article] [PubMed] [Google Scholar]