

Abstract
Gap junction redistribution and reduced expression, a phenomenon termed gap junction remodeling (GJR), is often seen in diseased hearts and may predispose toward arrhythmias. We have recently shown that short-term pacing in the mouse is associated with changes in connexin43 (Cx43) expression and localization but not with increased inducibility into sustained arrhythmias. We hypothesized that short-term pacing, if imposed on murine hearts with decreased Cx43 abundance, could serve as a model for evaluating the electrophysiological effects of GJR. We paced wild-type (normal Cx43 abundance) and heterozygous Cx43 knockout (Cx43+/−; 66% mean reduction in Cx43) mice for 6 h at 10–15% above their average sinus rate. We investigated the electrophysiological effects of pacing on the whole animal using programmed electrical stimulation and in isolated ventricular myocytes with patch-clamp studies. Cx43+/− myocytes had significantly shorter action potential durations (APD) and increased steady-state (Iss) and inward rectifier (IK1) potassium currents compared with those of wild-type littermate cells. In Cx43+/− hearts, pacing resulted in a significant prolongation of ventricular effective refractory period and APD and significant diminution of Iss compared with unpaced Cx43+/− hearts. However, these changes were not seen in paced wild-type mice. These data suggest that Cx43 abundance plays a critical role in regulating currents involved in myocardial repolarization and their response to pacing. Our study may aid in understanding how dyssynchronous activation of diseased, Cx43-deficient myocardial tissue can lead to electrophysiological changes, which may contribute to the worsened prognosis often associated with pacing in the failing heart.
Keywords: ventricular myocytes, gap junction
downregulation of connexin43 (Cx43), the major cardiac gap junction protein subtype, has been described in cardiac disease including ischemia, infarction, hibernating myocardium, and dilated cardiomyopathy (20, 21, 24, 30). Altered expression of Cx43 in cardiac disease is a potentially significant contributor to arrhythmogenicity (19, 33). Experimental models have demonstrated the relationship between decreased cardiac Cx43 expression and increased susceptibility to ventricular arrhythmias. In a heart-specific knockout of Cx43, we observed that ventricular expression levels of Cx43 are inversely related to inducibility into malignant ventricular arrhythmias (8). Additionally, gap junction remodeling (GJR), a process marked by decreased expression and intracellular redistribution of Cx43, corresponds spatially to the central common pathway of reentrant circuits in the canine peri-infarct zone (30).
We recently demonstrated that alterations in Cx43 expression in the wild-type mouse heart could be induced by ventricular pacing over a short period of time. However, pacing-induced changes in Cx43 expression were not accompanied by decreased cardiac function, prolonged refractoriness, or increased inducibility into sustained arrhythmias (22). In the present study, we hypothesized that alterations in Cx43 expression induced by pacing might have more profound effects on ventricular function and electrophysiology in the setting of reduced basal levels of Cx43. To test our hypothesis, we paced heterozygous Cx43 knockout mice (Cx43+/−), with Cx43 expression at 34.3 ± 4.4% of wild-type Cx43 levels, and compared them with paced wild-type mice.
Pacing in Cx43+/− hearts was not associated with significant changes in global ventricular function. However, in Cx43+/− hearts, pacing resulted in a significant prolongation of ventricular effective refractory period (VERP) compared with unpaced and paced wild-types and unpaced Cx43+/− hearts. To investigate the mechanisms whereby pacing in Cx43+/− hearts influences refractoriness, we studied action potentials from isolated ventricular myocytes and the potassium currents that represent the primary determinants of action potential duration (APD) (28). As our laboratory has previously demonstrated (9), APD was significantly shorter and steady-state and inward rectifier potassium currents (Iss and IK1, respectively) were increased in Cx43+/− compared with wild-type myocytes. However, we found that myocytes isolated from paced Cx43+/− hearts had significant prolongation of APD and decreased Iss compared with unpaced Cx43+/− myocytes. These changes were not seen in paced versus unpaced wild-type cardiomyocytes. Thus short-term pacing in Cx43+/− mice, but not in wild-types, was associated with significant changes in cardiac refractoriness and repolarization. These data suggest that Cx43 plays a critical role in the regulation of repolarizing currents and their response to pacing. In the clinical setting, dyssynchronous activation of diseased myocardium with reduced Cx43 expression may lead to electrophysiological changes, which underlie the worsened prognosis often associated with pacing in the failing heart.
MATERIALS AND METHODS
Procedure for short-term pacing in Cx43-deficient mice.
We investigated the effect of reduced Cx43 expression on pacing-induced remodeling of repolarizing currents using Cx43+/− mice (23) and their wild-type littermates, all of which were maintained in a mixed background consisting of C57BL/6J, SV129, and FVB strains. Mice aged 3–6 mo were used for these experiments. All studies were approved by the Institutional Animal Care and Use Committee of the New York University School of Medicine (New York, NY) and performed in accordance with their regulations.
Cx43+/− mice and wild-type littermates were paced for 6 h using the subdiaphragmatic approach as described in detail elsewhere (13). In brief, a stimulating electrode (UE-GM1; Frederick Haer, Bowdoinham, ME) mounted on a micromanipulator (Fine Science Tools, North Vancouver, BC, Canada) was inserted transdiaphragmatically through a 1-cm midline abdominal incision directly into contact with the surface of the right ventricle. Pacing was performed with a Model 2352 Programmable Stimulator (Medtronic, Minneapolis, MN).
Pacing was performed with the output set at twice the stimulating threshold and with a pulse width of 1.0 ms. Unpaced Cx43+/− and wild-type controls were prepared identically to their paced littermates, but the stimulator was left off for the 6-h experimental period. Electrocardiographic recordings were performed at baseline and during and after the pacing protocol as previously described (8). Electrocardiographic signals were monitored to ensure 1:1 capture, and pacing cycle lengths were maintained at 10–15% above the sinus rate for the duration of the experiment.
In vivo electrophysiology.
Programmed electrical stimulation (PES) for the determination of VERP and to assess for inducible arrhythmias was performed as described previously (13).
Echocardiography.
Echocardiography was performed according to a previously described protocol (12) on paced and sham-paced C57BL/6J WT mice (n = 6 in each group) before and immediately following 6-h pacing and sham protocols. Follow-up studies were also performed in Cx43+/− mice and wild-type littermates 2 h after cessation of pacing. Mice were imaged using a Philips HDI 5000 echocardiography machine equipped with a 15-MHz linear probe.
Cardiac myocyte isolation.
Ventricular myocytes were isolated from Cx43+/− and wild-type littermate hearts immediately following the pacing or sham-pacing procedures using standard enzymatic techniques. Mice were first anticoagulated with 1,000 U/kg heparin administered intraperitoneally 20 min before removal of the heart. Under isoflurane anesthesia, hearts were rapidly removed and perfused in a constant-pressure Langendorff system with Minimum Essential Medium Eagle (Joklik Modification; Sigma) containing (in mM) 0.44 EGTA, 34.5 NaHCO3, 1 MgSO4, and 10 2,3-butanedione monoxime at 37°C for 5 min. The hearts were then perfused with the same solution containing 0.05% (wt/vol) collagenase (Worthington Type 2), 0.1% bovine serum albumin, and 12.5 μM CaCl2, but without EGTA, at 37°C for 15 min. The heart was removed from the Langendorff system and dispersed mechanically. The dissociated heart tissue was then incubated in medium containing 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) and 12.5 μM CaCl2 for 10 min at RT, followed by an additional 10 min incubation in medium containing 5% fetal bovine serum and 12.5 μM CaCl2. Isolated myocytes were then incubated for 30 min at RT in Kraft-Bruhe (KB) solution, which contained (in mM) 20 taurine, 50 glutamine, 10 glucose, 10 HEPES, 0.5 EGTA, 3 MgSO4, 30 KH2PO4, 30 KCl, and 85 KOH (pH 7.2 adjusted with KOH). The KB solution was gradually replaced with modified Tyrode's solution containing (in mM) 140 NaCl, 4 KCl, 10 HEPES, 1.1 MgCl2, 1.8 CaCl2, and 10 glucose (pH 7.4 adjusted with NaOH). For action potential measurements, ventricular myocytes were isolated from both ventricles. For potassium currents, cells were isolated exclusively from the right ventricle. Cells were used for patch-clamp experiments within 6 h of isolation.
Cellular electrophysiology.
Isolated myocytes were placed in a bath (∼500 μl volume) on the stage of an inverted microscope (Nikon Diaphot, Tokyo, Japan), and whole-cell patch-clamp recordings (15) were obtained at RT. Experiments were performed using an EPC 9/2 amplifier and Pulse 8.79 software (HEKA Elektrotonik, Lambrecht, Germany). For recording action potential, modified Tyrode's solution was used as the bath solution (components as in Cardiac myocyte isolation). Recording pipettes were fabricated from borosilicate capillary glass (Warner Instruments, Novato, CA) using a Sutter Model P-97 micropipette puller (Novato, CA) and polished using a MF-830 Micro Forge (Narishige, Japan) to resistances of 2–5 MΩ when filled with pipette solution. The pipette solution for recording APs consisted of (in mM) 135 KCl, 4 MgCl2, 5 EGTA, 10 glucose, 10 HEPES, 5 Na2-ATP, and 3 Na2-creatine phosphate (pH 7.2 with LiOH). For recording potassium currents, the bath solution contained (in mM) 135 choline chloride, 5.4 KCl, 1.1 MgCl2, 1.8 CaCl2, 0.001 ryanodine, 0.01 atropine sulfate, 0.5 CdCl2, 10 glucose, and 10 HEPES (pH 7.4 with NaOH) and the pipette solution contained (in mM) 115 potassium aspartate, 5.0 KCl, 7 MgCl2, 5 EGTA, 10 HEPES, and 4 Na2-ATP (pH 7.2 with KOH).
After establishing the whole-cell configuration, membrane capacitances (Cm) were determined by integrating capacitance transients recorded during a brief 5-mV step from a holding potential (HP) of −70 mV. The cell capacitance was calculated as the ratio of the integral of the capacitance transient divided by the voltage step. Series resistance (Rs) was estimated from the decay of the capacitative transients. In each cell, Rs was compensated electronically by 80–90%. Tip potentials were zeroed before membrane-pipette seals were formed. The liquid junction potential was calculated to be −18 mV (Axoscope; Axon Instruments). Membrane potentials were not corrected for the liquid junction potential. Only data obtained from cells with seal resistances >1 GΩ were analyzed.
Action potential and potassium current recordings were performed in the whole-cell configuration. Action potentials were recorded at RT in the current-clamp mode by stimulation with a 3-ms pulse at a frequency of 1 Hz. Peak outward potassium current (IKpeak) was recorded using 1-s depolarizing voltage steps at 10-mV increments between −50 and +50 mV from a HP of −60 mV at 0.01 Hz. The interval between voltage steps was set at 10 s to allow complete recovery of outward potassium current. Steady-state inactivation of the outward potassium current was examined with a standard two-pulse protocol consisting of 1-s prepulses from −80 to +5 mV in 5-mV increments from a HP of −60 mV followed by a 500-ms test pulse to +40 mV. Recovery of transient outward potassium current (Ito) from inactivation was measured by using two depolarizing pulses to +40 mV from a HP of −60 mV separated by sequentially increasing intervals of 5 to 185 ms in 10-ms increments (26). Outward potassium current was recorded before and after the application of 2 mM 4-aminopyridine (4-AP) to separate the 4-AP-insensitive current (Iss) from the 4-AP-sensitive current (Ito) (36). IK1 was recorded in response to voltage steps to potentials between −20 and −140 mV from a HP of −60 mV. Currents were low-pass filtered at 2.9 kHz, digitized at 10 kHz, and stored for subsequent offline analysis.
Electrophysiology data analysis.
Data were analyzed using PulseFit 8.79 (HEKA). Iss was measured 100 ms from the end of each voltage step after the application of 4-AP. The peak Ito was calculated as the maximal current amplitude after the subtraction of the 4-AP-insensitive current from IKpeak at each depolarizing voltage step. IK1 was measured 100 ms from the end of each voltage step. Potassium current densities were obtained by dividing the measured currents by Cm.
Inactivation of Ito was fitted using the following equation to current traces after the subtraction of the 4-AP-insensitive current from IKpeak: y(t) = a0 + a1 exp (−t/τf) + a2 exp (−t/τs), where t is time; τf and τs are the time constants of inactivation of Ito; a1 and a2 are the amplitudes of the inactivating current components; and a0 is the residual amplitude of the steady-state, noninactivating component of the total outward potassium current after the subtraction of the 4-AP-insensitive component.
Steady-state inactivation curves were obtained by using a two-pulse protocol as described above. Curve fitting after the normalization of the current was performed using the Boltzmann equation, 1/[1 + exp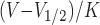 ], where V is the prepulse potential, V1/2 is half-maximal inactivation potential, and K is the slope factor for steady-state inactivation curves.
], where V is the prepulse potential, V1/2 is half-maximal inactivation potential, and K is the slope factor for steady-state inactivation curves.
The time course of recovery of Ito was described by a first-order exponential function as follows (26): y = 1 − exp−x/τ, where y is the normalized current, x is time, and τ is the time constant of recovery from Ito inactivation.
Immunoblot analysis.
Immunoblot analysis of ventricular lysates was performed using a custom-manufactured rabbit polyclonal antibody directed against an epitope on the carboxy-terminus of Cx43 (14) and a monoclonal antibody directed against GAPDH (Chemicon/Millipore, Billerica, MA). Horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA) was then applied to immunoblots, followed by HyGlo chemiluminescent processing (Denville Scientific, Metuchen, NJ) and autoradiography. Cx43 band intensities from three separate experiments were quantified by densitometry and normalized to the relative intensity of the corresponding GAPDH band for each sample.
Statistics.
Data are expressed as means ± SE. Electrocardiographic indexes and echocardiographic measurements obtained after the pacing protocol were compared with baseline measurements with paired two-tailed t-tests (Microsoft Excel). Electrocardiography, echocardiography, PES, and cellular electrophysiology results in the paced and unpaced Cx43+/− mice and their wild-type littermates were compared between groups with ANOVA followed by Fisher's protected paired least significant difference posttest using StatView 5.0 (SAS Institute, Cary, NC). A post hoc analysis was performed to compare IK1 in wild-type versus Cx43+/− myocytes. IK1 in unpaced versus paced subgroups was first compared using ANOVA, followed by pooling and comparison of all data from wild-type versus Cx43+/− myocytes at each voltage using ANOVA. P < 0.05 was considered statistically significant.
RESULTS
Short-term pacing in Cx43+/− mice results in prolonged ventricular refractoriness.
To determine whether short-term pacing in Cx43-deficient mice is associated with altered cardiac electrophysiology in vivo, we first examined surface electrocardiograms and then PES in Cx43+/− mice and matched wild-type littermates. Cx43+/− mice (both paced and unpaced) had a slightly but significantly reduced QRS duration compared with that of paced but not unpaced wild-types (Table 1). This finding contrasts with those of one of the coinvestigators on this study (G. E. Morley), who found no difference in QRS duration between wild-type and Cx43+/− mice (25), and others who observed a prolonged QRS interval in Cx43+/− mice compared with wild-types (11). However, given the poor correlation of the QRS duration with direct epicardial measurements of cardiac conduction (10), the significance of the changes in QRS interval in this model is unclear.
Table 1.
Electrocardiographic indexes and electrophysiological data in WT and Cx43+/− unpaced and paced mice
| WT Unpaced | WT Paced | Cx43+/− Unpaced | Cx43+/− Paced | |
|---|---|---|---|---|
| QRS duration, ms | 12.0±0.3 (0.3±0.6) | 13.2±1.3 (0.5±1.4) | 9.8±0.8*(−0.6±0.7) | 10.1±0.7*(0.2±1.4) |
| RR interval, ms | 136±4.3 (5.7±6.3) | 138±6.9 (4.2±7.3) | 134±9.8 (−2.9±5.9) | 142±13.2 (2.9±14.7) |
| QTc, ms | 116±3.7 (1.2±6.0) | 127±5.3 (1.8±9.2) | 130±11.1 (11.4±13.0) | 167±23.3 (48.3±34.6) |
| VERP100, ms | 36.6±3.2 | 34.3±1.9 | 30.3±1.8 | 49.4±5.1† |
| VERP80, ms | 37.2±3.0 | 35.7±2.2 | 29.7±1.0 | 49.8±5.4† |
Postpacing data (and changes from prepacing baseline) are presented as group means ± SE. For electrocardiographic indexes, n = 6 wild-type (WT) unpaced, 6 WT paced, 7 connexin43 (Cx43)+/− unpaced, and 9 Cx43+/− paced; for electrophysiological data [ventricular effective refractory period (VERP)], n = 9 in each WT group and 6 in each Cx43+/− group. Comparisons between groups were performed with ANOVA.
P < 0.05 compared with WT paced;
P < 0.01 compared with all other groups. QTc, rate-corrected QT interval; VERP80 and VERP100, VERP at a pacing cycle lengths of 80 and 100 ms, respectively.
There were no significant differences among the groups in RR interval or rate-corrected QT interval (QTc; Table 1). Interestingly, there was a nonsignificant trend toward increased QTc in the paced Cx43+/− mice compared with baseline recordings and to controls, suggesting increased ventricular refractoriness in the paced Cx43 mutants.
To directly test the effect of pacing in the Cx43+/− mice on refractoriness, we employed PES. After pacing, the VERP at a pacing cycle length of 100 ms (VERP100) in Cx43+/− mice was significantly prolonged (P < 0.01) at 49.4 ± 5.1 ms compared with that of unpaced Cx43+/− mice (30.3 ± 1.8 ms), unpaced wild-types (36.6 ± 3.2 ms), and paced wild-types (34.3 ± 1.9 ms; Figure 1 and Table 1). Similarly, the VERP at a pacing cycle length of 80 ms (VERP80) was significantly prolonged (P < 0.01) in the paced Cx43+/− mice (49.8 ± 5.4 ms) compared with unpaced Cx43+/− mice (29.7 ± 1.0 ms), unpaced wild-types (37.2 ± 3.0 ms), and paced wild-types (35.7 ± 2.2 ms; Table 1). Two hours after the cessation of pacing, VERP remained elevated in the Cx43+/− mice (VERP100 = 50.0 ± 9.0 ms and VERP80 = 51.3 ± 7.4 ms; n = 3). No sustained arrhythmias were induced by either single or double extrastimuli in the wild-type or Cx43+/− groups.
Fig. 1.

Prolonged refractoriness in paced connexin43 (Cx43)+/− mice. A: surface electrocardiograms demonstrated a slight but statistically significant shortening of the QRS duration in unpaced and paced Cx43+/− mice compared with the paced wild-type (WT) group and a nonsignificant trend compared with unpaced WT. Other electrocardiographic indexes were unchanged. B: programmed electrical stimulation revealed significantly prolonged refractoriness in paced Cx43+/− mice compared with each of the other groups.
Since short-term pacing in Cx43+/− mice results in significant prolongation of refractoriness, we investigated whether pacing of Cx43+/− mice might also affect systolic performance. To do so, we imaged Cx43+/− mice and matched wild-type littermates with transthoracic echocardiography before and immediately following the pacing and sham-pacing protocol. We found no significant differences in baseline echocardiographic characteristics including ventricular dimensions, fractional shortening, or wall thicknesses in any of the wild-type or Cx43+/− subgroups (supplemental table; all supplementary material can be found with the online version of this article). Similarly, after pacing or sham-pacing in the wild-type and Cx43+/− mice, indexes of ventricular dimensions, fractional shortening, and wall thicknesses were unchanged when compared between groups and compared with baseline values (Table 2).
Table 2.
Postpacing echocardiographic measurements in WT and Cx43+/− mice
| WT Unpaced | WT Paced | Cx43+/− Unpaced | Cx43+/− Paced | |
|---|---|---|---|---|
| IDD, mm | 3.4±0.10 (−0.05±0.10) | 3.2±0.1 (−0.12±0.21) | 3.3±0.12 (−0.16±0.21) | 3.5±0.15 (0.28 ±0.14) |
| IDS, mm | 1.9±0.09 (0.08±0.06) | 1.8±0.14 (0.05±0.18) | 1.8±0.11 (−0.04±0.18) | 2.2±0.21 (0.41±0.13) |
| Fractional shortening, % | 43.9±1.4 (−3.2±1.7) | 43.6±1.2 (−1.5±2.7) | 45.8±1.7 (−1.5±2.7) | 38.4±3.5 (−7.0±2.9) |
| AWTs, mm | 1.0±0.02 (−0.02±0.07) | 0.98±0.06 (−0.20±0.12) | 1.1±0.08 (−0.03±0.08) | 1.1±0.08 (−0.02±0.11) |
| AWTd, mm | 0.81±0.08 (−0.07±0.08) | 0.80±0.08 (0.0±0.12) | 0.79±0.06 (−0.03±0.05) | 0.88±0.06 (−0.01±0.08) |
| PWTs, mm | 1.3±0.09 (−0.23±0.13) | 1.6±0.10 (0.03±0.08) | 1.4±0.10 (−0.14±0.15) | 1.4±0.08 (−0.17±0.10) |
| PWTd, mm | 0.83±0.09 (−0.25±0.17) | 1.2±0.14 (0.23±0.18) | 0.97±0.08 (−0.19±0.12) | 1.1±0.08 (−0.14±0.12) |
Postpacing data (and changes from prepacing baseline) are presented as group means ± SE; n = 6 WT unpaced, 6 WT paced, 7 Cx43+/− unpaced, and 9 Cx43+/− paced. Comparisons between groups were performed with ANOVA; no significant differences among the groups were detected. IDD and IDS, intraventricular dimensions at end diastole and end systole, respectively; AWTs and AWTd, anterior wall thicknesses at end systole and end diastole, respectively; PWTs and PWTd, posterior wall thicknesses at end systole and end diastole, respectively.
As expected, Cx43+/− hearts had significantly reduced Cx43 protein levels compared with matched samples from littermate wild-types (34.3 ± 4.4% of wild-type Cx43 levels; P < 0.01; Fig. 2A). Pacing in Cx43+/− hearts did not significantly affect overall Cx43 protein levels (Fig. 2B).
Fig. 2.

Cx43 expression in Cx43+/− hearts. A: Cx43 abundance as determined by Western blotting followed by densitometry and normalization to GAPDH is significantly lower in Cx43+/− compared with WT hearts. B: no significant differences between unpaced and paced Cx43+/− hearts in expression of Cx43 protein levels were detected by Western blotting.
Taken together, these data suggest that short-term pacing in the setting of reduced Cx43 expression, but not wild-type levels of Cx43, results in the prolongation of refractoriness without significantly affecting overall ventricular function.
APD is reduced in unpaced Cx43+/− myocytes and lengthened by pacing.
To investigate how pacing in the setting of reduced Cx43 expression influences electrophysiological properties, we used whole-cell patch-clamp techniques to record action potentials in ventricular myocytes obtained from unpaced and paced wild-type and Cx43+/− mice (Fig. 3 and Table 3). Myocytes isolated from unpaced Cx43+/− hearts had significantly shorter action potentials than unpaced wild-type cells. In wild-type myocytes, pacing had no effect on APD. Myocytes isolated from unpaced wild-type hearts demonstrated APD measurements that were no different from those of paced wild-type myocytes [APD at 50% duration (APD50) = 9.7 ± 1.0 ms in unpaced wild-type vs. 9.9 ± 0.8 ms in paced wild-type and APD at 90% duration (APD90) = 54.1 ± 4.8 ms in unpaced wild-type vs. 55.3 ± 3.0 ms in paced wild-type; P = not significant (NS) for both comparisons]. By contrast, Cx43+/− myocytes demonstrated significant prolongation of APD after pacing compared with unpaced Cx43+/− myocytes (APD50 = 5.8 ± 0.9 ms in unpaced Cx43+/− vs. 8.5 ± 0.6 ms in paced Cx43+/−; P < 0.05; and APD90 = 35.0 ± 3.2 ms in unpaced Cx43+/− vs. 52.7 ± 4.1 ms in paced Cx43+/−; P < 0.01). Action potential amplitude and resting membrane potential were no different among the unpaced and paced wild-type and Cx43+/− subgroups. Thus, although pacing resulted in no significant change in APD in wild-type cardiac myocytes, the imposition of pacing on Cx43-deficient cells resulted in a significant lengthening of APD compared with its unpaced baseline.
Fig. 3.

Action potentials in unpaced and paced WT and Cx43+/− cardiac myocytes. A: representative action potentials from unpaced and paced WT and Cx43+/− cardiac myocytes. Action potentials were recorded from adult ventricular myocytes in current-clamp mode, with cells stimulated by 3-ms, 3–5-mV pulses at a frequency of 1 Hz. B: summary data of action potential duration at 50 percent (APD50) and at 90 percent (APD90) of repolarization. Unpaced Cx43+/− cells had shorter APD50 and APD90 values compared with the unpaced WT cells. APD increased significantly in the Cx43+/− cells with pacing, whereas there was no significant change with pacing in the WT cells.
Table 3.
Comparison of action potential parameters in WT and Cx43+/− unpaced and paced mice
| WT Unpaced | WT Paced | Cx43+/− Unpaced | Cx43+/− Paced | |
|---|---|---|---|---|
| Amplitude, mV | 101.8±3.8 | 97.2±1.7 | 97.6±4.0 | 97.8±2.0 |
| RMP, mV | −72.0±0.5 | −72.5±1.0 | −71.5±1.0 | −72.0±1.0 |
| APD20, ms | 2.7±0.3 | 2.6±0.2 | 2.0±0.2 | 2.3±0.2 |
| APD50, ms | 9.7±1.0 | 9.9±0.8 | 5.8±0.9* | 8.5±0.6 |
| APD90, ms | 54.1±4.8 | 55.3±3.0 | 35.0±3.2† | 52.3±4.1 |
Data are presented as group means ± SE; n = 6 WT unpaced, 11 WT paced, 7 Cx43+/− unpaced, and 10 Cx43+/− paced. Comparisons between groups were performed with ANOVA.
P < 0.05 compared to all other groups;
P < 0.01 compared to all other groups. RMP, resting membrane potential; APD20, APD50, and APD90, action potential duration at 20%, 50%, and 90% duration, respectively.
IK1 is increased in Cx43+/− myocytes.
Since APD in cardiac cells is largely determined by voltage-dependent potassium channels (28), we examined whether changes in repolarization in unpaced and paced Cx43+/− myocytes might result from altered potassium currents. We measured IK1 in the whole-cell configuration, in which we calculated cell capacitances as 144.6 ± 7.6 pF in wild-type right ventricular myocytes (n = 28) and 145.9 ± 5.5 pF in Cx43+/− right ventricular myocytes (n = 29; P = NS).
We first evaluated the background IK1, which is responsible for maintaining the resting membrane potential and influences the late phase of repolarization (16). Initially, we did not observe significant differences in a four-way comparison of IK1 in unpaced wild-type (peak current = −14.3 ± 1.4 pA/pF), paced wild-type (−13.7 ± 1.3 pA/pF), unpaced Cx43+/− (−17.7 ± 1.2 pA/pF), and paced Cx43+/− (−16.3 ± 1.3 pA/pF) myocytes (Fig. 4, A–E). Because IK1 was not statistically different in unpaced versus paced wild-type myocytes across all of the tested voltages, the data from these two groups were pooled at each voltage to increase statistical power. IK1 values in unpaced and paced Cx43+/− myocytes were also pooled at each voltage, since they were not significantly different (except at −30 mV; P = 0.028). A post hoc analysis of the pooled values from wild-type versus Cx43+/− myocytes revealed significantly increased IK1 in the Cx43+/− myocytes compared with wild-types (Fig. 4F). Thus increased IK1 in Cx43+/− myocytes may contribute to APD shortening. However, there was no significant effect of pacing on IK1 in either wild-type or Cx43+/− myocytes.
Fig. 4.

Effect of pacing on the inward rectifier potassium current (IK1) in WT and Cx43+/− right ventricular cardiac myocytes. A and B: representative recordings of IK1 from WT (A) and Cx43+/− (B) unpaced and paced right ventricular myocytes at a potential of −140 mV. C: voltage-clamp protocol consisted of steps of 10-mV increments between −140 and −20 mV (1-s duration; 0.01 Hz) from a holding potential of −60 mV. D and E: IK1 amplitude, measured at the end of a 1-s pulse, was plotted as a function of applied voltage for WT unpaced (n = 5) and paced (n = 5) right ventricular myocytes (D) and Cx43+/− unpaced (n = 5) and paced (n = 6) myocytes (E). The results indicated that there were no significant differences between any of the 4 subgroups. F: post hoc analysis of pooled IK1 values comparing results from all WT vs. Cx43+/− cells at each voltage showed significantly increased IK1 in the Cx43+/− myocytes compared with WT at voltages of ≤−70 mV. *P < 0.05.
Iss is increased in unpaced Cx43+/− myocytes but diminished after pacing.
Other currents that are critical in determining APD include two components of the total voltage-dependent outward potassium current (Fig. 5A), which are distinguished by sensitivity to 4-AP. Outward potassium current in cardiac myocytes comprises a 4-AP-insensitive component (Iss; Fig. 5B) and a 4-AP-sensitive component (Ito; Fig. 5C) (2, 26, 36). Representative traces in Fig. 5 were recorded from an adult murine right ventricular myocyte. The 4-AP-sensitive current, Ito, was no different in amplitude among the unpaced wild-type, paced wild-type, unpaced Cx43+/−, or paced Cx43+/− subgroups (Fig. 6). Furthermore, inactivation kinetics of Ito and indexes of the recovery of Ito from inactivation were no different in any of the groups tested (Fig. 7 and Table 4).
Fig. 5.

Outward potassium current in adult murine right ventricular cardiac myocytes. A: total outward potassium current, as shown, is termed IKpeak. B: a 4-AP-insensitive steady-state current (Iss) was obtained after 2-mM 4-AP was applied. C: the transient outward potassium current, Ito, was obtained offline by subtracting Iss from IKpeak. D: voltage-clamp protocol consisted of steps of 10-mV increments between −50 and +50 mV (1-s duration; 0.01 Hz) from a holding potential of −60 mV.
Fig. 6.

Effect of pacing on Ito in WT and Cx43+/− right ventricular cardiac myocytes. A and B: representative Ito traces from WT (A) and Cx43+/− (B) unpaced and paced right ventricular myocytes at +52 mV. C and D: Ito peak amplitude was measured and plotted as a function of applied voltage for WT unpaced and paced (n = 6 and 5, respectively; C) and Cx43+/− unpaced and paced (n = 6 and 7, respectively; D) myocytes. There were no significant differences between any of the subgroups.
Fig. 7.

Effect of pacing on kinetics of Ito inactivation properties and recovery of Ito from inactivation in WT and Cx43+/− myocytes. A: representative Ito recording made during a 2-pulse voltage-clamp protocol to assess voltage dependence of steady-state inactivation. B: the 2-pulse protocol consisted of a 1-s prepulse delivered from a holding potential of −60 mV in 5-mV increments from −80 to +5 mV, followed by a 500-ms test pulse to +40 mV. C and D: steady-state inactivation curves obtained from peak current obtained during second pulse in WT unpaced and paced (n = 6 each; C) and Cx43+/− unpaced and paced (n = 6 and 5, respectively; D) myocytes. Data were normalized to peak current amplitude at a prepulse of −80 mV, plotted as a function of prepulse potentials, and fitted with the best nonlinear least-squares fit of a Boltzmann function. E: representative tracings of recovery of Ito from inactivation. F: recovery of Ito from inactivation was measured by using 2 depolarizing pulses to +40 mV, from a holding potential of −60 mV, separated by intervals of increasing duration from 5 to 185 ms in 10-ms increments. G and H: recovery of Ito current was obtained from WT unpaced and paced (n = 8 and 7, respectively; G) and Cx43+/− unpaced and paced (n = 8 each; H) cells. Data were normalized to maximum current value, plotted as a function of recovery time, and fitted with a first-order exponential function. No significant differences among the subgroups were identified in inactivation properties or recovery kinetics of Ito.
Table 4.
Comparison of Ito inactivation, steady-state inactivation, and recovery from inactivation in WT and Cx43+/− unpaced and paced mice
| WT Unpaced | WT Paced | Cx43+/− Unpaced | Cx43+/− Paced | ||||
|---|---|---|---|---|---|---|---|
| Ito Inactivation Kinetics | |||||||
| τf, ms | 47.4±6.7 | 49.2±5.5 | 45.9±3.8 | 43.6±1.3 | |||
| τs, ms | 1,138±188 | 1,161±182 | 1,104±126 | 1,190±144 | |||
| a0, nA | 0.4±0.06 | 0.4±0.05 | 0.4±0.04 | 0.4±0.04 | |||
| a1, nA | 1,226±304 | 1,176±305 | 1,295±275 | 1,450±309 | |||
| a2, nA | 1.7±0.4 | 1.6±0.3 | 1.5±0.4 | 1.6±0.4 | |||
| Steady-State Inactivation | |||||||
| Vh, mV | −26.3±2.0 | −27.9±2.4 | −30.5±3.5 | −26.8±2.3 | |||
| K, mV | 11.3±1.2 | 11.0±1.3 | 9.5±1.0 | 8.3±1.2 | |||
| Recovery from Inactivation | |||||||
| τrecovery, ms | 31.5±4.6 | 30.3±4.9 | 44.3±5.5 | 36.6±3.7 | |||
Data are presented as group means ± SE; n = 8 per group. Ito, transient outward K+ current; τf and τs, time constants of the fast and slow component of inactivation, respectively; a0, residual amplitude of the steady-state, noninactivating component of the total outward K+ current after subtraction of the 4-aminopyridine-insensitive component; a1 and a2, amplitudes of the inactivating current components; Vh, half-maximal inactivation potential; K, slope factor for steady-state inactivation curves at Vh; τrecovery, time constant of recovery from inactivation. Comparisons between groups were performed with ANOVA. P = not significant for all comparisons.
Significant elevations in the 4-AP-insensitive current, Iss, have been described in right ventricular myocytes from mice with conditional loss of Cx43 expression in the heart (9). Similarly, we found that Iss was significantly increased in unpaced Cx43+/− myocytes (peak current = 4.2 ± 0.5 pA/pF) compared with unpaced wild-type myocytes (2.4 ± 0.2 pA/pF; P < 0.01). Pacing did not affect a significant change in Iss current density in wild-type myocytes (2.7 ± 0.1 pA/pF after pacing; Fig. 8, A and C). However, pacing was associated with a significant diminution of Iss current density in the Cx43+/− myocytes (2.9 ± 0.2 pA/pF; P < 0.01 compared with unpaced Cx43+/− myocytes; Fig. 8, B and D) to levels that were not statistically different from those of the wild-type myocytes. Thus changes in the Iss density in unpaced and paced Cx43+/− myocytes most likely represent a major mechanism underlying corresponding alterations in APD.
Fig. 8.

Effect of pacing on Iss. A and B: representative recordings of Iss from WT unpaced and paced (n = 6 each; A) and Cx43+/− unpaced and paced (n = 5 and 6, respectively; B) myocytes at +50 mV. C and D: Iss amplitude was measured at the end of the depolarization pulse and plotted as a function of applied voltage for WT (C) and Cx43+/− (D) cells. The results indicated that Iss was not altered by pacing in the WT cells. However, Iss was elevated in the Cx43+/− unpaced cells compared with all other groups and was diminished significantly by pacing in the Cx43+/− myocytes. *P < 0.05; †P < 0.01.
DISCUSSION
In this paper, we describe an interaction between reduced basal levels of Cx43 and pacing that influences ventricular electrophysiology. Pacing in the setting of heterozygous loss of Cx43, but not in wild-type mice, is associated with prolonged refractoriness. To investigate the prolongation of refractoriness associated with pacing in the setting of reduced Cx43 expression, we studied action potentials and potassium currents from myocytes isolated from paced and unpaced Cx43 mutant and wild-type hearts. Isolated myocytes from Cx43-deficient murine hearts have shortened APDs compared with those measured in wild-type cells, most likely due to increased Iss and IK1 density. APD is significantly lengthened with a corresponding reduction in Iss (but not IK1) in paced Cx43+/− cells compared with unpaced myocytes, whereas these parameters are unchanged in wild-type myocytes with pacing. Thus, although short-term pacing in the wild-type murine heart is not associated with measurable electrophysiological effects, pacing in the setting of reduced baseline Cx43 expression results in significant prolongation of refractoriness and altered repolarization.
Our results are consistent with prior data showing shortening of the APD90 in cultured strands of neonatal ventricular myocytes isolated from heterozygous Cx43 knockout hearts (35). In another model of loss of Cx43 expression, the heart-specific conditional knockout of Cx43, we observed a reduction in APD with correspondingly increased outward potassium currents (9). However, this is the first study to demonstrate that pacing a Cx43-deficient mouse heart alters indexes of repolarization and refractoriness, whereas no such changes occur with pacing in the wild-type mouse.
An alteration in repolarization after short- or long-term pacing is termed cardiac memory, a phenomenon described in larger animals such as dogs and humans (29, 31, 32). Cardiac memory has not been reported previously in the mouse model and in our study was not evident in wild-type mice after short-term pacing. The mouse is not an ideal species to study repolarization, however, since repolarizing currents in the mouse heart differ substantially from those of larger mammals (27). Nonetheless, because pacing-induced alterations in repolarization consistent with cardiac memory were seen only in the Cx43-deficient mice, it appears that Cx43 may influence processes that are responsible for memory.
A potential link between Cx43 expression and the remodeling of active ion channels has been suggested by experiments involving the infarct border zone in the canine model. GJR in this region was found to correspond spatially to the central common pathway of figure-of-eight reentrant ventricular tachycardia circuits (30). Furthermore, significant alterations in sodium, L-type calcium and potassium currents, and kinetics, as well as associated protein subunits, were observed in the same region of the infarct border zone (4). Thus, in combination with the data presented in this study, it is reasonable to conclude that reduced Cx43 expression, as observed in the infarct border zone and in paced Cx43+/− mice, may potentiate remodeling of repolarizing currents.
Dyssynchrony of cardiac activation is a predictor of poor outcome in patients with systolic dysfunction (5, 17, 34, 37). Patients with heart failure and dyssynchrony have been shown to benefit from cardiac resynchronization therapy (CRT) with a biventricular pacemaker (1, 3, 6, 7, 38). However, a substantial proportion of treated patients fail to respond to CRT (1, 3). How dyssynchrony interacts deleteriously with other factors in the failing heart is not fully understood (34). Our data suggest that decreased expression of Cx43, which has been described in heart disease (20, 21, 24, 30), can influence the expression and/or function of active ion channels and, in combination with pacing, may lead to increased dispersion of repolarization properties.
It is important to note that although APD is shortened in the unpaced Cx43+/− myocytes and lengthens to wild-type levels in the paced Cx43+/− myocytes, refractoriness is unchanged from wild-types in unpaced Cx43+/− myocytes and prolongs significantly with pacing. This implies that there is an additional factor responsible for prolonging refractoriness in the Cx43+/− hearts, which is not influenced by pacing. Possibilities include altered kinetics of active ion channels other than potassium channels. Sodium channel alterations are unlikely to contribute to changes in repolarization in the Cx43+/− hearts, since parameters of sodium channel function in Cx43-deficient myocytes are reportedly unchanged compared with wild-type controls (18). We are currently investigating calcium channel kinetics in myocytes isolated from Cx43+/− hearts to determine whether alterations may contribute to prolonged refractoriness.
In conclusion, the present findings suggest that myocardial tissue with reduced baseline expression of Cx43 responds quite differently to pacing than does myocardium with wild-type levels of Cx43. In the setting of decreased Cx43 expression, short-term pacing results in significant prolongation of refractoriness, as well as changes in APD and Iss. Prior studies have indicated that in the diseased heart Cx43 abundance can be variable, with focal areas of reduced expression corresponding to hibernating and ischemic zones (20). Our results suggest that the imposition of pacing in such a clinical scenario could lead to the remodeling of repolarization currents in regions with reduced Cx43 expression, thus greatly enhancing dispersion of refractoriness around hibernating or ischemic zones and potentially creating a substrate for reentry.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-081336 (to D. E. Gutstein) and HL-066140 (to A. L. Wit), a Grant-in-Aid from the American Heart Association (to D. E. Gutstein), British Heart Foundation Grant RG/05/009, and support from the National Institute for Health Research Biomedical Research Centre Funding Scheme (to N. S. Peters).
Acknowledgments
We acknowledge consultative input from Drs. Michael R. Rosen and Eric A. Sobie.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Abraham WT, Fisher WG, Smith AL, Delurgio DB, Leon AR, Loh E, Kocovic DZ, Packer M, Clavell AL, Hayes DL, Ellestad M, Trupp RJ, Underwood J, Pickering F, Truex C, McAtee P, Messenger J. Cardiac resynchronization in chronic heart failure. N Engl J Med 346: 1845–1853, 2002. [DOI] [PubMed] [Google Scholar]
- 2.Apkon M, Nerbonne JM. Characterization of two distinct depolarization-activated K+ currents in isolated adult rat ventricular myocytes. J Gen Physiol 97: 973–1011, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Auricchio A, Stellbrink C, Sack S, Block M, Vogt J, Bakker P, Huth C, Schondube F, Wolfhard U, Bocker D, Krahnefeld O, Kirkels H. Long-term clinical effect of hemodynamically optimized cardiac resynchronization therapy in patients with heart failure and ventricular conduction delay. J Am Coll Cardiol 39: 2026–2033, 2002. [DOI] [PubMed] [Google Scholar]
- 4.Baba S, Dun W, Cabo C, Boyden PA. Remodeling in cells from different regions of the reentrant circuit during ventricular tachycardia. Circulation 112: 2386–2396, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baldasseroni S, Opasich C, Gorini M, Lucci D, Marchionni N, Marini M, Campana C, Perini G, Deorsola A, Masotti G, Tavazzi L, Maggioni AP. Left bundle-branch block is associated with increased 1-year sudden and total mortality rate in 5517 outpatients with congestive heart failure: a report from the Italian network on congestive heart failure. Am Heart J 143: 398–405, 2002. [DOI] [PubMed] [Google Scholar]
- 6.Bristow MR, Saxon LA, Boehmer J, Krueger S, Kass DA, De Marco T, Carson P, DiCarlo L, DeMets D, White BG, DeVries DW, Feldman AM. Cardiac-resynchronization therapy with or without an implantable defibrillator in advanced chronic heart failure. N Engl J Med 350: 2140–2150, 2004. [DOI] [PubMed] [Google Scholar]
- 7.Cleland JG, Daubert JC, Erdmann E, Freemantle N, Gras D, Kappenberger L, Tavazzi L. The effect of cardiac resynchronization on morbidity and mortality in heart failure. N Engl J Med 352: 1539–1549, 2005. [DOI] [PubMed] [Google Scholar]
- 8.Danik SB, Liu F, Zhang J, Suk HJ, Morley GE, Fishman GI, Gutstein DE. Modulation of cardiac gap junction expression and arrhythmic susceptibility. Circ Res 95: 1035–1041, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Danik SB, Rosner G, Lader J, Gutstein DE, Fishman GI, Morley GE. Electrical remodeling contributes to complex tachyarrhythmias in connexin43-deficient mouse hearts. FASEB J 22: 1204–1212, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eloff BC, Lerner DL, Yamada KA, Schuessler RB, Saffitz JE, Rosenbaum DS. High resolution optical mapping reveals conduction slowing in connexin43 deficient mice. Cardiovasc Res 51: 681–690, 2001. [DOI] [PubMed] [Google Scholar]
- 11.Guerrero PA, Schuessler RB, Davis LM, Beyer EC, Johnson CM, Yamada KA, Saffitz JE. Slow ventricular conduction in mice heterozygous for a connexin43 null mutation. J Clin Invest 99: 1991–1998, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gutstein DE, Danik SB, Lewitton S, France D, Liu F, Chen FL, Zhang J, Ghodsi N, Morley GE, Fishman GI. Focal gap junction uncoupling and spontaneous ventricular ectopy. Am J Physiol Heart Circ Physiol 289: H1091–H1098, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gutstein DE, Danik SB, Sereysky JB, Morley GE, Fishman GI. Subdiaphragmatic murine electrophysiological studies: sequential determination of ventricular refractoriness and arrhythmia induction. Am J Physiol Heart Circ Physiol 285: H1091–H1096, 2003. [DOI] [PubMed] [Google Scholar]
- 14.Gutstein DE, Liu FY, Meyers MB, Choo A, Fishman GI. The organization of adherens junctions and desmosomes at the cardiac intercalated disc is independent of gap junctions. J Cell Sci 116: 875–885, 2003. [DOI] [PubMed] [Google Scholar]
- 15.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch 391: 85–100, 1981. [DOI] [PubMed] [Google Scholar]
- 16.Ibarra J, Morley GE, Delmar M. Dynamics of the inward rectifier K+ current during the action potential of guinea pig ventricular myocytes. Biophys J 60: 1534–1539, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iuliano S, Fisher SG, Karasik PE, Fletcher RD, Singh SN. QRS duration and mortality in patients with congestive heart failure. Am Heart J 143: 1085–1091, 2002. [DOI] [PubMed] [Google Scholar]
- 18.Johnson CM, Green KG, Kanter EM, Bou-Abboud E, Saffitz JE, Yamada KA. Voltage-gated Na+ channel activity and connexin expression in Cx43-deficient cardiac myocytes. J Cardiovasc Electrophysiol 10: 1390–1401, 1999. [DOI] [PubMed] [Google Scholar]
- 19.Kanno S, Saffitz JE. The role of myocardial gap junctions in electrical conduction and arrhythmogenesis. Cardiovasc Pathol 10: 169–177, 2001. [DOI] [PubMed] [Google Scholar]
- 20.Kaprielian RR, Gunning M, Dupont E, Sheppard MN, Rothery SM, Underwood R, Pennell DJ, Fox K, Pepper J, Poole-Wilson PA, Severs NJ. Downregulation of immunodetectable connexin43 and decreased gap junction size in the pathogenesis of chronic hibernation in the human left ventricle. Circulation 97: 651–660, 1998. [DOI] [PubMed] [Google Scholar]
- 21.Kitamura H, Ohnishi Y, Yoshida A, Okajima K, Azumi H, Ishida A, Galeano EJ, Kubo S, Hayashi Y, Itoh H, Yokoyama M. Heterogeneous loss of connexin43 protein in nonischemic dilated cardiomyopathy with ventricular tachycardia. J Cardiovasc Electrophysiol 13: 865–870, 2002. [DOI] [PubMed] [Google Scholar]
- 22.Kontogeorgis A, Kaba RA, Kang E, Feig JE, Gupta PP, Ponzio M, Liu F, Rindler MJ, Wit AL, Fisher EA, Peters NS, Gutstein DE. Short-term pacing in the mouse alters cardiac expression of connexin43. BMC Physiol 8: 8, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu S, Liu F, Schneider AE, St Amand T, Epstein JA, Gutstein DE. Distinct cardiac malformations caused by absence of connexin 43 in the neural crest and in the non-crest neural tube. Development 133: 2063–2073, 2006. [DOI] [PubMed] [Google Scholar]
- 24.Matsushita T, Oyamada M, Fujimoto K, Yasuda Y, Masuda S, Wada Y, Oka T, Takamatsu T. Remodeling of cell-cell and cell-extracellular matrix interactions at the border zone of rat myocardial infarcts. Circ Res 85: 1046–1055, 1999. [DOI] [PubMed] [Google Scholar]
- 25.Morley GE, Vaidya D, Samie FH, Lo C, Delmar M, Jalife J. Characterization of conduction in the ventricles of normal and heterozygous Cx43 knockout mice using optical mapping. J Cardiovasc Electrophysiol 10: 1361–1375, 1999. [DOI] [PubMed] [Google Scholar]
- 26.Nakamura TY, Coetzee WA, Vega-Saenz De Miera E, Artman M, Rudy B. Modulation of Kv4 channels, key components of rat ventricular transient outward K+ current, by PKC. Am J Physiol Heart Circ Physiol 273: H1775–H1786, 1997. [DOI] [PubMed] [Google Scholar]
- 27.Nerbonne JM Studying cardiac arrhythmias in the mouse—a reasonable model for probing mechanisms? Trends Cardiovasc Med 14: 83–93, 2004. [DOI] [PubMed] [Google Scholar]
- 28.Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev 85: 1205–1253, 2005. [DOI] [PubMed] [Google Scholar]
- 29.Patberg KW, Shvilkin A, Plotnikov AN, Chandra P, Josephson ME, Rosen MR. Cardiac memory: mechanisms and clinical implications. Heart Rhythm 2: 1376–1382, 2005. [DOI] [PubMed] [Google Scholar]
- 30.Peters NS, Coromilas J, Severs NJ, Wit AL. Disturbed connexin43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia. Circulation 95: 988–996, 1997. [DOI] [PubMed] [Google Scholar]
- 31.Rosen MR, Cohen IS. Cardiac memory…new insights into molecular mechanisms. J Physiol 570: 209–218, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosenbaum MB, Blanco HH, Elizari MV, Lazzari JO, Davidenko JM. Electrotonic modulation of the T wave and cardiac memory. Am J Cardiol 50: 213–222, 1982. [DOI] [PubMed] [Google Scholar]
- 33.Severs NJ, Coppen SR, Dupont E, Yeh HI, Ko YS, Matsushita T. Gap junction alterations in human cardiac disease. Cardiovasc Res 62: 368–377, 2004. [DOI] [PubMed] [Google Scholar]
- 34.Spragg DD, Kass DA. Pathobiology of left ventricular dyssynchrony and resynchronization. Prog Cardiovasc Dis 49: 26–41, 2006. [DOI] [PubMed] [Google Scholar]
- 35.Thomas SP, Kucera JP, Bircher-Lehmann L, Rudy Y, Saffitz JE, Kleber AG. Impulse propagation in synthetic strands of neonatal cardiac myocytes with genetically reduced levels of connexin43. Circ Res 92: 1209–1216, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang L, Duff HJ. Developmental changes in transient outward current in mouse ventricle. Circ Res 81: 120–127, 1997. [DOI] [PubMed] [Google Scholar]
- 37.Wilkoff BL, Cook JR, Epstein AE, Greene HL, Hallstrom AP, Hsia H, Kutalek SP, Sharma A. Dual-chamber pacing or ventricular backup pacing in patients with an implantable defibrillator: the Dual Chamber and VVI Implantable Defibrillator (DAVID) Trial. JAMA 288: 3115–3123, 2002. [DOI] [PubMed] [Google Scholar]
- 38.Young JB, Abraham WT, Smith AL, Leon AR, Lieberman R, Wilkoff B, Canby RC, Schroeder JS, Liem LB, Hall S, Wheelan K. Combined cardiac resynchronization and implantable cardioversion defibrillation in advanced chronic heart failure: the MIRACLE ICD Trial. JAMA 289: 2685–2694, 2003. [DOI] [PubMed] [Google Scholar]