

Abstract
Histone deacetylase 3 (HDAC3) is over-expressed in approximately half of all colon adenocarcinomas. We took an RNAi approach to determine how HDAC3 influenced chromatin modifications and the expression of growth regulatory genes in colon cancer cells. A survey of histone modifications revealed that HDAC3 knockdown in SW480 cells significantly increased histone H4-K12 acetylation, a modification present during chromatin assembly that has been implicated in imprinting. This modification was found to be most prominent in proliferating cells in the intestinal crypt and in APCMin tumors, but was less pronounced in the tumors that over-express HDAC3. Gene expression profiling of SW480 revealed that HDAC3 shRNA impacted the expression of genes in the Wnt and vitamin D signaling pathways. The impact of HDAC3 on Wnt signaling was complex, with both positive and negative effects observed. However, long-term knockdown of HDAC3 suppressed β-catenin translocation from the plasma membrane to the nucleus, and increased expression of Wnt inhibitors TLE1, TLE4 and SMO. HDAC3 knockdown also enhanced expression of the TLE1 and TLE4 repressors in HT-29 and HCT116 cells. HDAC3 shRNA enhanced expression of the vitamin D receptor in SW480 and HCT116 cells, and rendered SW480 cells sensitive to 1,25-dihydroxyvitamin D3. We propose that HDAC3 over-expression alters the epigenetic programming of colon cancer cells to impact intracellular Wnt signaling and their sensitivity to external growth regulation by vitamin D.
Keywords: HDAC3, Wnt signaling, β-catenin, VDR, TLE, c-MYC, colon cancer, butyrate, Acetyl histone H4K12, APCMin
Introduction
Carcinogenesis involves the mutation of key tumor suppressor genes and oncogenes, which impact cell proliferation, differentiation and death 1. It is becoming increasingly clear that epigenetic changes also alter the gene expression patterns of cells to promote their carcinogenic potential 2-6. Epigenetic modifications are manifested by inheritable alterations in chromatin structure that enforce the expression of specific genes in cell progeny. Present data indicate that epigenetic alterations can occur early in the carcinogenic sequence 7-10; a number of growth-regulatory genes have been found to be epigenetically silenced in colonic mucosa in older individuals, and this silencing may contribute to the increased risk of carcinogenesis associated with aging 11, 12. Understanding the protein complexes involved in regulating the epigenetic changes associated with aging and cancer could provide mechanistic insight into the carcinogenic process and potentially identify new targets for cancer treatment and prevention.
Inheritable changes in chromatin structure are frequently directed by DNA methylation. Methylated cytosines in CpG dinucleotides serve to recruit histone deacetylases (HDACs) and other transcription-repressing proteins through a family of methyl-cytosine binding proteins 13, 14. Epigenetic regulation may also occur in a manner that is independent of DNA methylation. In this case, specific chromatin markings (including histone acetylation) facilitate the inheritance of an “open” or “closed” chromatin structure 15. Oncogenic fusion proteins found in leukemia cells have been indentified that perform this function. Fusion of the AML1/RUNX protein with the transcriptional co-repressors TEL or ETO facilitates the recruitment of HDAC-containing co-repressors to specific genetic loci in acute lymphoblastic leukemia 16-19. Likewise, the fusion of RARα to the PML protein in acute myeloid leukemias suppresses the expression of RAR target genes 20-22. In addition to the formation of fusion proteins, the over-expression of chromatin-silencing proteins is another means of subverting the epigenetic program of a cell. The SNAIL repressor protein, for example, is frequently over-expressed in numerous cancer types where it can silence a number of growth regulatory genes 23-25. Interestingly, the expression of cellular HDACs themselves can be increased during cellular transformation 26. In colon cancer, evidence for increased expression of HDACs 1, 2 and 3 has been reported 27-30. The mechanism that controls HDAC expression in colon cancers is not entirely clear, but it may be linked to APC status and c-myc activation 29.
We have been studying the impact of HDAC3 on colon cancer cells. This HDAC is of particular interest since it is one of the most frequently up-regulated genes in human cancers 31. We and others have previously reported that reducing HDAC3 expression levels in colon cancer cells slows their proliferation and alters the expression of cell cycle regulatory proteins, including an increase in p21 expression 28, 30. In addition, we observed a change in the expression of cellular differentiation markers, with a notable decrease in the expression of the secretory proteins mucin 2 and intestinal trefoil factor 30. These findings suggested that reducing HDAC3 expression may generate a range of epigenetic changes that reprogram cell gene expression in colon cancer cells. To determine the extent and nature of the reprogramming events directed by HDAC3 over-expression, gene expression profiling was performed on a colon cancer cell line that expresses high levels of HDAC3 before and after HDAC3 knockdown with shRNA. Here we report that high HDAC3 expression alters the genetic programming of colon cancer cells, with both increases and decreases in gene expression observed. Interestingly, HDAC3 expression regulates genes in the Wnt, TGF-β, vitamin D and Interferon signaling pathways. A more detailed analysis indicates that HDAC3 expression levels can indeed impact the function of the Wnt and vitamin D signaling. These findings indicate that HDAC3 over-expression may play a critical role in colon carcinogenesis by influencing both endogenous oncogenic pathways (such as Wnt/β-catenin signaling) and the responsiveness of cells to exogenous growth-regulatory signals, such as 1,25-dihydroxyvitamin D3.
Materials and Methods
Cell culture and treatments
The SW480 cancer cell line was originally obtained from the American Type Culture Collection (Manassas, VA). They were transfected with a vector expressing shRNA targeting HDAC3 or a non-targeting control vector, as previously described 30. Two clones with reduced HDAC3 expression were utilized for the present studies. All cells were maintained at 37°C, 5% CO2 in McCoy's 5A media supplemented with 10% fetal bovine serum, 100 μm non-essential amino acids (Invitrogen) and antibiotic-antimycotic (Invitrogen). Butyrate (BA) was purchased from Sigma-Aldrich (St. Louis, MO) and used at a final concentration of 4 mM. Butyrate treatments were performed overnight (16 to 18 hours) unless otherwise indicated. 1,25 dihydroxyvitamin D3 was likewise obtained from Sigma-Aldrich, and used at the concentrations indicated in the text. Vitamin D3 treatments were performed for 48 hours, after which cells were trypsinized and counted by hemocytometer.
Cell cycle synchronization experiments were performed using the Lovastatin method, as previously described 32. Briefly, cells were grown in three 60 mm plates and treated with 60 μM Lovastatin (Mevinolin) for approximately 33 hours. One plate was taken for cells in G1 arrest, and the remaining two were passed at a 1:4 dilution into medium containing 5 mM mevalonic acid to release the G1 block. Protein was extracted from cells at various time points following release, and entrance into S phase was determined by immunoblotting for PCNA.
siRNA transient transfection
Transient transfection was performed as previously described 30. Briefly, cells were grown to confluency, trypsinized and resuspended in OptiMEM. Non-targeting control or HDAC3 siRNA (Dharmacon, Lafayette, CO) was combined with Lipofectamine 2000 reagent in OptiMEM at a 100 nM final concentration. Seventy-two hours post-transfection RNA was isolated and subjected to reverse-transcription and quantitative real-time PCR.
Immunoblotting
Proteins were extracted from cells as previously described by Spurling et al. 30. Cytosolic and nuclear extracts were denatured under reducing conditions, separated on a 12.5% or 15% SDS-polyacrylamide gel and then transferred in Tris-glycine buffer to a nitrocellulose membrane by voltage gradient transfer overnight. The blots were then dried and blocked with 5% non-fat dry milk in PBS + 1% Tween-20 and probed with primary antibodies at 1:500 to 1:1000 dilutions. Antibodies used in these studies were obtained from Santa Cruz Biotechnology: HDAC1 (H51), HDAC2 (H54), HDAC3 (H99), HDAC4 (H92), VDR (H81), β-catenin (C18), E-cadherin (G10) and actin (I19). HRP-conjugated secondary antibodies and enhanced chemiluminescence was used for detection as recommended by the manufacturer (Santa Cruz Biotechnology). Nucleosomes were extracted from cells as previously described 33, and transferred to 0.2 μm nitrocellulose in Tris-glycine buffer with 20% methanol and 0.1% SDS. Histone modifications were detected using modification-specific primary antibodies obtained from Upstate, and the chemiluminescent detection method.
Indirect immunofluorescence
Cells were treated as described in the text, and processed as previously described 34. Briefly, cells were washed with cold phosphate-buffered saline and fixed with 100% methanol for 30 min at −20 °C. Donkey serum (10% in phosphate-buffered saline) was used to block nonspecific binding for 30 min. After blocking, the cells were incubated with a goat anti-β-catenin antibody (H81, Santa Cruz Biotechnology) at a 1:75 dilution followed by incubation with a Cy3-conjugated anti-goat immunoglobulin (Jackson ImmunoResearch Laboratories, Inc.). Images were captured on an Olympus epifluorescent microscope.
RNA isolation and microarray data analysis
Total RNA was prepared using the Trizol reagent (Invitrogen, Carlsbad, CA). A microarray was run on a Sentrix ref-6 human beadchip (Illumina, San Diego, CA). The array was processed at the Molecular Genetics Core Facility at Children's Hospital Boston. Data were normalized using the “rank invariant” method in Illumina's BeadStudio software. This method normalizes with respect to a common reference sample based on a linear scale. To reduce the size of the gene list, only annotated genes (18,267 genes) were selected for further analysis. Pathway analysis was performed using Ingenuity Pathways Analysis software (Ingenuity Systems, Redwood City, CA).
Reverse-transcription PCR and quantitative real-time PCR
To synthesize cDNA, 2 μg aliquots of RNA were diluted in water and combined according to manufacturer's instructions with the High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA) reaction components. cDNA was stored at −20°C. Real-Time PCR was performed using an Applied Biosystem's 7500 Fast Real-Time PCR system and software. Reactions were run for 40 cycles with the TaqMan 2x PCR Master Mix in 10 μl volumes with cDNA corresponding to 2 μg RNA. TaqMan gene expression assays for TLE1, TLE4, c-MYC, VDR, and β-actin were purchased from Applied Biosystems (Foster City, CA).
Results
HDAC3 inhibition and global histone acetylation
To determine the impact of HDAC3 expression levels on gene expression patterns in colon cancer cells, we selected the SW480 colon cancer cell line that expresses relatively high levels of this HDAC 30. This cell line was then stably transfected with an shRNA expression vector to target HDAC3, or a non-targeting control vector (as previously described) 30. To validate the specific reduction in HDAC3, a western blot analysis was performed on both the control (shCNTL) and HDAC3 knockdown (shHD3) cell lines. Figure 1A shows the expression levels of HDACs 1, 2, 3 and 4 in these cell lines. A large reduction in HDAC3 is observed, whereas comparable levels of HDACs 1, 2, and 4 are present in both cell lines. The mRNA levels of other class I and class II HDACs were also determined (by microarray analysis), and were likewise found to be unaffected (although an increase in an HDAC7 transcript variant was observed). These findings indicate the shRNA is specific for HDAC3, and that compensatory increases in the expression of other HDACs are minimal.
Figure 1. HDAC3 expression is specifically reduced by HDAC3 shRNA.

[A] Western blotting of proteins from SW480 cells stably transfected with either a control vector (shCNTL) or a vector containing shRNA against HDAC3 (shHD3). Results from two independent cell lines are shown. Note the specific decrease in HDAC3 protein levels in comparison to HDACs 1, 2, and 4. [B] Effect of HDAC3 shRNA on histone modification. Chromatin was prepared from shCNTL and shHD3 cells, and analyzed for histone modification using specific antibodies (in duplicate). Optical density was determined using ImageJ software (NIH) and normalized to shCNTL levels. The bars shown are the average relative intensities of modifications from shCNTL cells (white bars) and shHD3 cells (gray bars).
HDACs have been reported to have a degree of enzymatic specificity for specific histone acetyl-lysine residues 35-38, and a previous report has described preferred substrates for HDAC3 complexes39. We determined the impact of HDAC3 knockdown on several different histone modifications by performing a western blot analysis (see figure in Ref. 30) and optical density scanning on nucleosomes extracted from SW480 cells expressing control or HDAC3 targeting shRNA (shCNTL and shHD3 cells, respectively; Figure 1B). HDAC3 inhibition resulted in specific alterations in acetylation, with the largest increase occurring on histone H4, particularly the H4-K12Ac modification. Analysis of additional samples indicates that this 2-fold increase is significant (p < 0.01).
To determine whether acetylation of histone H4 at lysine 12 is regulated in the intestine, we examined normal and cancerous intestinal tissue in APCMin mice for this modification. Tumors formed in APCMin mice are useful for this analysis since they express elevated levels of HDAC3 (Figure 2A)28. In the normal small intestine, positive staining for histone H4-K12 acetylation was observed in cells within the upper portion of the crypt, with lower levels of staining observed in cells of the villus (Figure 2B). This staining was typically observed in multiple, adjacent cells within the crypts. In contrast, a modification-independent histone H4 antibody showed even staining throughout the small intestinal mucosa (Figure 2B). When intestinal tumors were stained for histone H4 K12 acetylation, they showed less intense staining relative to normal crypt cells (Figure 2C). The tumor cells that did stain positively could also be distinguished from normal cells by the fact that they occurred more sporadically throughout the lesion.
Figure 2. Immunohistochemical analysis of intestinal tissue.
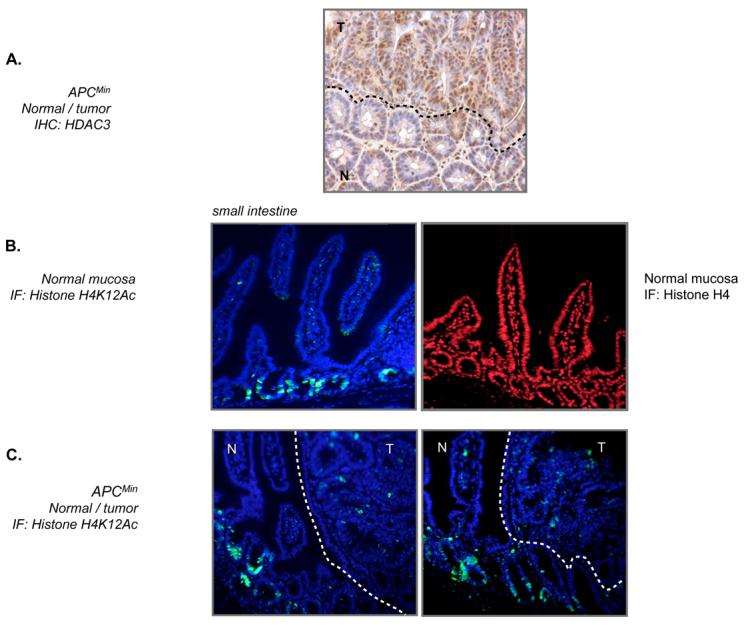
[A] Immunohistochemical staining of HDAC3 in an intestinal tumor of an APCMin mouse. Positive staining for HDAC3 in this section is brown. The line demarks normal and tumor regions of the tissue (labeled “N” and “T”, respectively). [B] Immunofluorescent staining of H4K12Ac and general histone H4 in normal small intestine. Specific antibody staining is shown in green (H4K12Ac) and red (H4) and the DNA is stained blue with DAPI. [B] Two sections of tumor and adjacent normal tissue within the APCMin mice were analyzed by immunofluorescence for histone H4-K12 (green) and DNA (blue). The line demarks the normal tissue (labeled “N”) and the tumor tissue (labeled “T”).
The staining of upper crypt cells for histone H4 acetylation at K12 suggested a potential overlap with the transiently amplifying population of crypt cells. To determine the extent of this overlap, intestinal tissue was dual-stained for histone H4 K12 acetylation and proliferating cell nuclear antigen (PCNA). There was a close association between PCNA expression and this histone modification, in both normal and tumor tissue (Figures 3A and 3B). This overlap was also observed in the colon, although additional cells may also stain in this tissue (data not shown). The linkage between cell proliferation and histone H4 K12 acetylation was also observed in cells that were synchronized in vitro. For these experiments, an SW480 cell line (expressing HDAC3 shRNA) was subjected to the lovastatin arrest and release procedure32 As shown in Figure 3C, histone H4 acetylation at K12 increased as cells entered S-phase, as shown by an increase in PCNA expression. As discussed in more detail below, histone H4 K12 acetylation is required for the proper loading of this histone onto newly synthesized chromatin, and might therefore provide a window of opportunity for adjusting chromatin programming, an opportunity that could be limited in cancer cells that express high levels of HDAC3.
Figure 3. H4K12Ac co-localizes with proliferating cells.
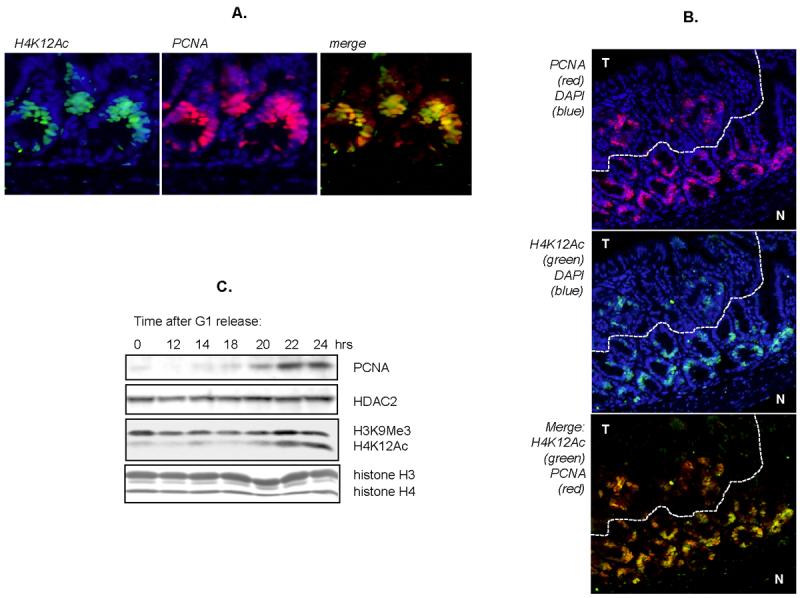
[A] APCMin small intestine normal tissue was co-stained for H4K12Ac (green), PCNA (red) and DAPI (blue). Note the high degree of overlap as shown in the merge panel. [B] Microadenomas (T) from APCMin mice show a similar pattern of overlap but a lower amount of cells staining positive for H4K12Ac compared to normal tissue (N). [C] Western blot analysis of synchronized SW480 cells show a co-ordinate increase in H4K12Ac and PCNA after release from G1 whereas histone H3K9Me does not change.
HDAC3 inhibition and gene expression changes
To determine the impact of HDAC3 expression levels and the corresponding histone acetylation changes on gene regulation in colon cancer cells, RNA was isolated from these cells and subjected to an Illumina microarray analysis. To ensure that the microarray data was consistent between biological replicates, scatterplots were constructed. Figures 4A and 4B illustrate that shCNTL and shHD3 cell line replicates have a high correlation as indicated by the regression line and r2 values (0.989 and 0.997 respectively). In contrast, when expression values for the shCNTL and shHD3 cell lines were compared, a broad scatter with an r2 of 0.849 was obtained, indicating numerous gene expression changes following HDAC3 inhibition (Figure 4C). Further analysis revealed that 18.2% of annotated genes are up-regulated 5-fold or more by HDAC3 inhibition, whereas 8.6% are down-regulated. A list of annotated genes altered by HDAC3 shRNA, and a gene ontology breakdown, are provided as supplemental data (supplemental Tables 1, 2 and 3).
Figure 4. Microarray gene expression analysis.

Expression values (rank-invariant normalization) were compared for similarity between shCNTL cells and shHD3 cells. [A] Comparison between biological duplicates for shCNTL cells shows that there is little variation (r2=0.989). [B] Similarly, there is little variation between the biological replicates for the shHD3 samples (r2 =0.997). [C] Comparison between shCNTL and shHD3 cells show increased and decreased gene expression in shHD3 cells (r2 =0.849). [D] Venn diagrams showing the number of genes that are up/down-regulated at least five-fold by butyrate (BA) and HDAC3 shRNA (shHD3). The number of commonly regulated genes is shown in the overlap region. A total of 18.2% of all annotated genes are up-regulated by shHD3 whereas only 8.6% were down-regulated.
Some of the gene expression changes on the array are likely due to a direct effect of decreased HDAC3 activity, whereas others may be secondary changes. To estimate the proportion of changes that may be due to the direct effects of HDAC3, we determined which HDAC3-sensitive genes could also be altered by a short-term treatment with the general HDAC inhibitor butyrate. An eighteen-hour butyrate exposure was selected for these experiments since this is the amount of time required to activate the highly butyrate-responsive p21 gene (data not shown). As shown in the Venn diagrams (Figure 4D), more than half of the genes activated by HDAC3 shRNA could also be activated by butyrate. Numerous genes were suppressed by both butyrate and HDAC3 shRNA, indicating that this transcriptional repressor may actually promote expression of some genes (directly or indirectly). The overlap between these genes was less extensive relative to the up-regulated genes, indicating that many of the down-regulated genes may not be direct targets of HDAC3.
To determine which cellular pathways are influenced by HDAC3 expression, genes that were altered 5-fold or more were analyzed by the IPA path-finder program. This program indicated potential effects on the TGF-β, Interferon and Wnt signaling pathways (Table 1). Semi-quantitative RT-PCR confirmed large changes in a subset of the genes shown in the table (data not shown). The effect of HDAC3 shRNA on the interferon and TGF-β signaling pathways was generally positive, with increased expression of both IFN and TGF-β target genes observed. In the case of the TGF-β pathway, this included an up-regulation of the vitamin-D receptor (VDR) and the VDR-target gene TGF-β2 40, 41. The influence on the Wnt pathway was complex, with both positive and negative effects observed.
Table 1. IPA pathway analysis.
Average gene expression levels for shHD3 cells were compared with the averages for shCNTL cells, and data were processed through the IPA pathway analyzer. Results show three major pathways affected by HDAC3 knockdown, including genes in the Wnt pathway [A], the Interferon pathway [B], and the TGFβ pathway [C].
| A | Gene Symbol | Role in Pathway |
|---|---|---|
| DKK3 | Wnt antagonist * | |
| FZD17 | Wnt receptor | |
| Up-regulated | SMO | Component of the competing hedgehog pathway * |
| TLE1 | TCF co-repressor * | |
| TLE4 | TCF co-repressor * | |
| WNT6 | Ligand | |
| WNT5A | Ligand | |
| DKK4 | Wnt antagonist | |
| FZD10 | Wnt receptor * | |
| Down-regulated | GJA1 | Wnt suppressed in development * |
| PPP2R2C | Wnt pathway enhancer * | |
| SOX8 | Wnt target gene * | |
| TCF1 | Binds beta-catenin * | |
| MYC | Wnt target gene * | |
| B | Gene Symbol | Role in Pathway |
| IFI35 | Interferon target gene | |
| IFITM1 | Interferon target gene | |
| Up-regulated | MX1 | Interferon-inducible protein |
| PSMB8 | Gamma-interferon induces expression | |
| SOCS1 | Gamma-interferon induces expression | |
| C | Gene Symbol | Role in Pathway |
| AXL | Receptor tyrosine kinase | |
| EGFR | Capable of binding TGF-beta | |
| HOXC8 | Binds SMAD proteins/transcription regulator | |
| INHA | Ligand | |
| Up-regulated | ROR2 | Receptor tyrosine kinase |
| ROR1 | Receptor tyrosine kinase | |
| SERPINE1 | Target gene | |
| TGFB2 | Ligand | |
| TGIF1 | Co-repressor | |
| VDR | Target gene | |
| INHBB | Ligand | |
| Down-regulated | NKX2-5 | Target gene |
Of the pathways identified, we decided to examine the HDAC3 shRNA-induced changes in Wnt and VDR signaling in more detail, since these pathways are critical for regulating the endogenous growth potential of colon cancer cells, and their responsiveness to exogenous vitamin D.
HDAC3 regulation of the Wnt pathway
The profile of genes in the Wnt pathway regulated by HDAC3 can both enhance and suppress Wnt signaling. For example, the FZD17 receptor is up-regulated whereas expression of the FZD10 receptor is reduced. Overall, however, the majority of changes are predicted to suppress Wnt signaling (as indicated in Table 1). One critical outcome of the Wnt pathway is the translocation of β-catenin from the cell membrane to the nucleus. We thus chose to analyze how HDAC3 suppression affected this step of the Wnt pathway. As shown in Figure 5A, the proportion of β-catenin in the nuclear fraction is lower in two clones expressing the HDAC3 shRNA than control cells. An indirect immuno-fluorescent analysis of β-catenin showed that cells expressing HDAC3 shRNA had more extensive plasma membrane localization relative to control cells (Figure 5B). Membrane association of β-catenin could also be stimulated by butyrate treatment of control cells, supporting a role for HDACs in regulating this localization (Figure 5B). These findings indicate that HDAC3 shRNA generally increases β-catenin mobility away from the nucleus and towards the plasma membrane of colon cancer cells.
Figure 5. Alterations in β-catenin localization by HDAC3 shRNA.

[A] Western blot analysis of β-catenin within nuclear and cytoplasmic/membrane fractions of shCNTL and shHD3 cell lines shows that shHD3 cells have less nuclear β-catenin. Actin was employed as a loading control. The results from two shHD3 cell lines are shown. [B] SW480 cells were plated onto glass coverslips for immunofluorescent analysis of β-catenin localization. shCNTL cells showed diffuse staining for β-catenin. shCNTL cells treated with butyrate showed a partial translocation of β-catenin to the plasma membrane. shHD3 cell lines show localization of β-catenin at the plasma membrane.
The expression of three genes in the Wnt signaling pathway was also examined by quantitative RT-PCR. Specifically, we quantified the expression of two inhibitors of the Wnt pathway, TLE1 and TLE4, which compete for β-catenin binding to TCF complexes in the nucleus. In addition, the expression level of the β-catenin target gene, c-MYC, was determined. As shown in Figure 6A, HDAC3 shRNA expression increases expression of both of the TLE genes, and decreases expression of c-MYC. These results are consistent with those found on the microarray. To determine the generality of this response, three colon cancer cell lines were transiently transfected with HDAC3 siRNA. In SW480, HCT116 and HT-29 cells, transfection with HDAC3 siRNA either increased the expression of a TLE, or potentiated the activation by butyrate treatment (Figure 6B, C and D; TLE4 was not expressed in HCT116 cells). Interestingly, although c-MYC expression was suppressed by butyrate, the HDAC3 siRNA enhanced its expression in each cell line (Figure 6B, C and D). This latter finding suggests a complex interaction between HDAC3 expression and Wnt signaling, as observed on the gene array. As discussed below, the impact of HDAC3 expression on Wnt signaling is therefore likely to depend on the relative expression of positive and negative Wnt effectors in individual cancers.
Figure 6. Real-time PCR in stable and transient HDAC3 knockdown cells for Wnt pathway components.


[A] RNA isolated from SW480 cells expressing control (shCNTL) or HDAC3 (shHD3) shRNA was used to synthesize cDNA and was analyzed for the expression of TLE1, TLE4 and c-MYC using quantitative real-time PCR. Real-time data is consistent with results from the microarray analysis. [B-D] TLE1, TLE4 and c-MYC mRNA expression following the transient knockdown of HDAC3 in SW480 [B], HT-29 [C] or HCT116 [D] cells. Cells were also treated with butyrate, as indicated. An ANOVA analysis was performed to determine significant differences; * indicates a significant difference between the indicated treatment and the control cells (p < 0.05).
HDAC3 expression and vitamin D responsiveness
One gene of interest within the TGF-β pathway was the vitamin D receptor (VDR). This receptor is important for mediating cellular differentiation and growth control by vitamin D metabolites, and may therefore play an important role in cancer prevention by vitamin D. As shown in Figure 7A, VDR mRNA levels were increased in SW480 cells either upon treatment with butyrate or following HDAC3 suppression with shRNA. HDAC3 inhibition by transfection of HCT116 cells with siRNA likewise stimulated VDR expression (Figure 7B). The increase in mRNA expression induced by HDAC3 shRNA was accompanied by an increase in VDR protein expression (Figure 7C). We also determined the impact of HDAC3 shRNA on the gene activation and growth control by the active vitamin D metabolite 1,25-dihydroxyvitamin D3. Cells expressing HDAC3 shRNA were able to activate E-cadherin following vitamin D3 treatment, and were more sensitive to growth inhibition by vitamin D3 (Figure 7D and 8). These findings indicate that HDAC3 can play an important role in mediating the sensitivity of cells to growth regulation by vitamin D.
Figure 7. HDAC3 regulation of VDR and vitamin D responses.

[A] VDR mRNA quantified by real-time PCR in shCNTL and siHD3 cells, treated with or without butyrate, as indicated. [B] VDR expression was also analyzed in the HCT116 cell line transiently transfected with control or HDAC3-targeting siRNA (siHD3), that were treated with or without butyrate (BA) as indicated. [C] VDR expression in shCNTL and shHD3 cells determined by immunoblotting. Two independent shHD3 cell lines were analyzed in duplicate. [D] shCNTL and shHD3 cells were treated with 1, 25 dihydroxyvitamin D3 for 24 hours and subjected to immunoblot analysis for the cell adhesion protein E-cadherin. Actin was employed as a loading control.
Figure 8. 1, 25 dihydroxyvitamin D3 decreases growth of cells with stable HDAC3 knockdown.
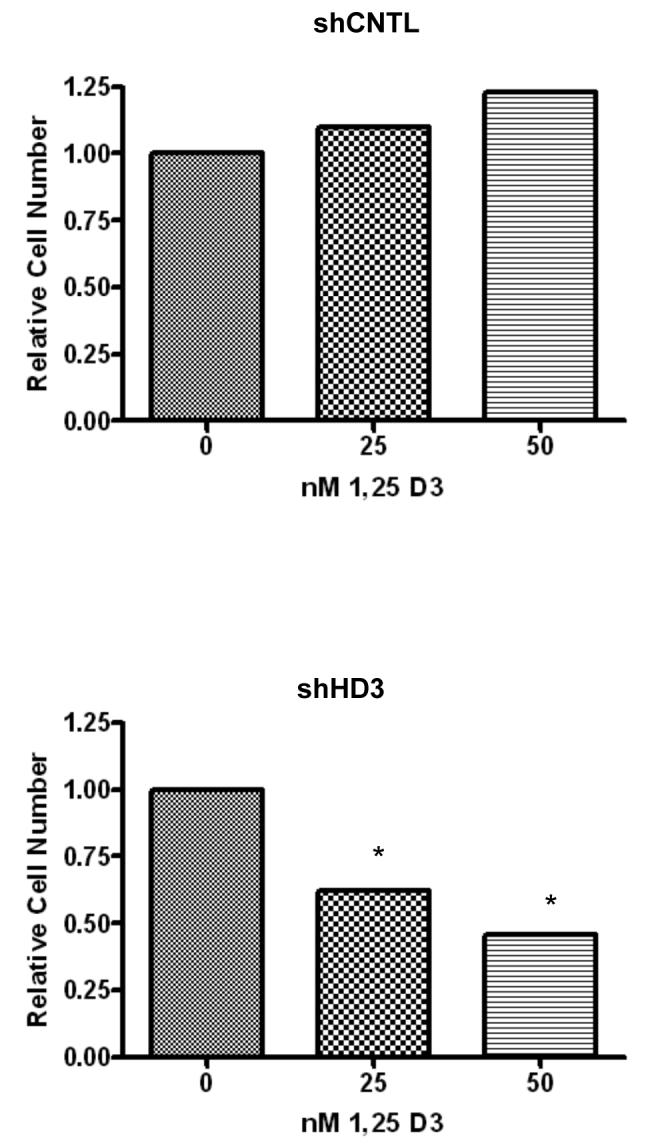
Treatment of cells with increasing amounts of 1, 25-dihydroxyvitamin D3 over a period of 2 days showed a dose dependent drop in cell number only in the cell line expressing lower amounts of HDAC3 (* indicates p < 0.05). Cells were treated at the indicated concentrations of 1,25 dihydroxyvitamin D3 in quadruplicate.
Discussion
The frequent over-expression of HDAC3 in human cancers suggests that this enzyme plays an important role in carcinogenesis. We took a gene array approach to further explore the potential role of HDAC3 in colon carcinogenesis. We found that the elevated expression of HDAC3 impacts three signaling pathways that are intimately involved in colon carcinogenesis: Wnt, TGF-β and Interferon. Our findings support a model in which HDAC3 expression plays a pivotal role in the epigenetic reprogramming of cancer cells to significantly enhance their growth potential and carcinogenicity. It is surprising that over-expression of a single HDAC impacts multiple cancer-related pathways, since there are other HDACs expressed in cancer cells. Interestingly, HDAC3 is also expressed at high levels in primordial gut tissues, raising the possibility that its expression in colon cancers serves to de-differentiate cells 42. In general, our findings indicate that increased HDAC3 expression may target this transcriptional regulatory protein to additional promoters and substrate proteins to alter the cell's phenotype. These findings suggest that HDAC3 may be a good target for highly-specific agents that treat or prevent colon cancer.
The Wnt signaling pathway is targeted for mutation in the majority of colorectal cancers 43, 44. The most frequent alteration is a truncating mutation in the APC gene, a negative regulator of the Wnt pathway 45. The truncated APC protein is no longer able to direct the degradation of cytosolic β-catenin, allowing β-catenin levels to increase and eventually enter the nucleus to activate transcription by associating with TCF proteins bound to target gene promoters 43. We found that HDAC3 had a complex effect on components of the Wnt pathway. However, the majority of gene expression changes induced by a stable shRNA knockdown indicated that HDAC3 expression generally enhances the Wnt pathway. The enhancement of Wnt signaling by HDAC3 was further supported by the finding that the stable HDAC3 knockdown enhanced the plasma membrane localization of β-catenin and reduced its nuclear accumulation. It should be noted that this alteration in nuclear β-catenin localization occurred even though the cells analyzed are APC mutants. The changes in β-catenin cellular localization may result from a number of gene expression changes induced by HDAC3 shRNA. For example, the HDAC3 shRNA reduced the expression of the TCF1 transcription factor in the colon cancer cells. Since TCF1 binds β-catenin in the nucleus 46 reducing its expression may eliminate an important nuclear “anchor” for β-catenin. A similar mechanism may also come into play with regard to the HDAC3 shRNA-induced increase in TLE1 and TLE4 expression. TLEs compete with β-catenin for TCF binding47, so increased TLE1 and TLE4 expression may also reduce the nuclear binding sites for β-catenin. In addition to these potential nuclear anchor effects, activation of the competitive hedgehog pathway may also contribute to β-catenin inhibition; HDAC3 shRNA increased SMO expression, which has been shown to inhibit TCF4-β-catenin complex formation by the hedgehog pathway 48.
Although the stable reduction in HDAC3 expression by shRNA increased expression of the TCF co-repressors TLE1 and TLE4, and decreased expression of the Wnt target gene c-MYC, the influence of a transient decrease in HDAC3 expression by siRNA was more complex. Although HDAC3 siRNA increased TLE expression (either alone or in combination with butyrate), an increase in c-MYC expression was also observed. It has been reported that the HDAC inhibition by butyrate can activate the Wnt pathway, and that hyper-activation of this pathway can lead to cell death49, 50. We propose that HDAC3 inhibition has a similar effect on Wnt signaling. In the short term, HDAC3 shRNA may promote Wnt signaling. However, only cells with a robust negative feedback response, resulting for example from a strong TLE activation, survive51. This model predicts that the expression of HDAC3 in colon cancers may depend on their ability to modulate Wnt signaling through the altered expression of Wnt inhibitory proteins.
Other pathways found to be affected by HDAC3 include the interferon-regulated genes and genes in the TGF-β signaling pathway. Although the interferon response may be initiated non-specifically through double stranded siRNA molecules, it is unlikely that this is why we see activation of these genes: we do not observe increases in interferon expression or apoptosis in cells expressing HDAC3 shRNA30 (and data not shown). In addition, most of the classical dsRNA response genes are not activated in cells expressing HDAC3 shRNA52, 53. Interferon (IFN) signaling can influence carcinogenesis through a number of mechanisms 54, 55, including a direct influence on cell proliferation 56 and by enhancing the immunogenicity of cancer cells 57. We found genes in both of these categories activated by HDAC3 shRNA. The mechanism by which HDAC3 inhibition activates expression of these IFN-regulated genes is not presently clear, but it is unlikely to occur through the STAT1 transcription factor, since activation of this transcription factor is inhibited by HDAC inhibitors58. With regard to TGF-β signaling, activation of this pathway may result in part from increased TGFβ2 expression. TGFβ2 is one of several members of a superfamily of cytokines that play a critical role in controlling cellular growth, differentiation and development 59. In early colon tumorigenesis, TGFβ signaling is thought to block tumor development by inducing cell cycle arrest and apoptosis and cancer cells may become resistant to TGFβ by carrying a mutation of the type II receptor (TGFβ-RII)60, or through SMAD4 mutation 60, 61. However, TGFβ signaling is a double-edged sword as it can also promote cancer growth and metastasis 62. In fact, a number of genes in the TGFβ pathway enhanced by HDAC3 shRNA are associated with tumor progression (AXL, EGFR, and SERPINE1).
One interesting gene in the TGFβ pathway that was activated by HDAC3 shRNA was the VDR gene. HDAC3 knockdown not only restored VDR expression, but also restored the responsiveness of the cells to growth inhibition by 1,25-dihydroxyvitamin D3. HDAC3 expression has also been found to regulate the cellular response to another dietary factor; previous work indicated that growth regulation by butyrate (derived from dietary fiber) is also sensitive to HDAC3 expression levels 30. Since butyrate has been reported to increase VDR expression 63, a picture emerges of a web of interactions between HDAC3 expression and multiple dietary agents. Understanding these interactions could ultimately enhance our understanding of how dietary agents interact with each other and specific cellular genotypes to prevent cancer development.
The changes in gene expression induced by HDAC3 knockdown are likely to be mediated in part by the changes in histone acetylation. Acetylation of histone H4 at lysine 12 (H4K12Ac) appears to be particularly sensitive to HDAC3 expression. Although purified HDAC3 can deacetylate both histone H3 and histone H4 64-66, HDAC3 complexes isolated from cells display a higher degree of specificity 38. HDAC3 complexes have been found to completely deacetylate H2A, H4K5 and H4K12, while only partially deacetylating other residues 39. Acetylated histone H4K12 may therefore be a preferred substrate for HDAC3 complexes 39. Although the “histone code” involves multiple modifications functioning in concert, histone H4K12 acetylation may play a particularly important role in regulating chromatin structure. Evidence has been obtained that acetylation of histone H4K12 plays a critical role in propagating chromatin structure following DNA replication. For example, the ability of yeast to maintain newly synthesized chromatin in an “open” state is dramatically reduced when histone H4 is mutated to suppress K12 acetylation 67. It should also be noted that new chromatin assembled following DNA replication utilizes histone H4 acetylated at lysine 12. Our finding that histone H4K12 acetylation is highest in proliferating cells is consistent with the usage of H4K12Ac in new chromatin assembly. We propose that the loading of acetylated histone H4K12 on the DNA following replication provides a window of opportunity to open the chromatin on repressed genes, with the ultimate chromatin status determined by the presence of specific trans-acting factors. This window of open chromatin may be significantly limited in tumor cells that over-express HDAC3. In APCMin mice, we find that histone H4K12 acetylation is generally lower in tumors relative to normal mucosa. Additional experimentation will be required to determine whether this modification relates to different patterns of gene expression within the tumor, and whether the frequency of this modification relates to the differentiation and invasiveness of the cancers.
Supplementary Material
Acknowledgements
This work was supported in part by NIH grant R01 CA81428 and by the University of Connecticut Research Foundation. We extend our gratitude to Advanced Turbine Services for the donation of equipment used in these studies.
Abbreviations
- HDAC
Histone deacetylase
- VDR
vitamin D receptor
- TGFβ
transforming growth factor beta
- IFN
interferon
- siRNA
short interfering RNA
- SDS
sodium dodecyl sulfate
- shRNA
short hairpin RNA
- DAPI
4′,6-diamidino-2-phenylindole
References
- 1.Spandidos DA. Oncogenes and tumor suppressor genes as paradigms in oncogenesis. J Buon. 2007;12(Suppl 1):S9–12. [PubMed] [Google Scholar]
- 2.Clark SJ. Action at a distance: epigenetic silencing of large chromosomal regions in carcinogenesis. Hum Mol Genet. 2007;16(Spec No 1):R88–95. doi: 10.1093/hmg/ddm051. [DOI] [PubMed] [Google Scholar]
- 3.Herranz M, Esteller M. DNA methylation and histone modifications in patients with cancer: potential prognostic and therapeutic targets. Methods Mol Biol. 2007;361:25–62. doi: 10.1385/1-59745-208-4:25. [DOI] [PubMed] [Google Scholar]
- 4.Kondo Y, Issa JP. Epigenetic changes in colorectal cancer. Cancer Metastasis Rev. 2004;23:29–39. doi: 10.1023/a:1025806911782. [DOI] [PubMed] [Google Scholar]
- 5.Stebbing J, Bower M, Syed N, Smith P, Yu V, Crook T. Epigenetics: an emerging technology in the diagnosis and treatment of cancer. Pharmacogenomics. 2006;7:747–57. doi: 10.2217/14622416.7.5.747. [DOI] [PubMed] [Google Scholar]
- 6.Ting AH, McGarvey KM, Baylin SB. The cancer epigenome--components and functional correlates. Genes Dev. 2006;20:3215–31. doi: 10.1101/gad.1464906. [DOI] [PubMed] [Google Scholar]
- 7.Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6:107–16. doi: 10.1038/nrc1799. [DOI] [PubMed] [Google Scholar]
- 8.Herman JG. Epigenetic changes in cancer and preneoplasia. Cold Spring Harb Symp Quant Biol. 2005;70:329–33. doi: 10.1101/sqb.2005.70.036. [DOI] [PubMed] [Google Scholar]
- 9.Balch C, Nephew KP, Huang TH, Bapat SA. Epigenetic “bivalently marked” process of cancer stem cell-driven tumorigenesis. Bioessays. 2007;29:842–5. doi: 10.1002/bies.20619. [DOI] [PubMed] [Google Scholar]
- 10.Hake SB, Xiao A, Allis CD. Linking the epigenetic ‘language’ of covalent histone modifications to cancer. Br J Cancer. 2004;90:761–9. doi: 10.1038/sj.bjc.6601575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahuja N, Li Q, Mohan AL, Baylin SB, Issa JP. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res. 1998;58:5489–94. [PubMed] [Google Scholar]
- 12.Ahuja N, Issa JP. Aging, methylation and cancer. Histol Histopathol. 2000;15:835–42. doi: 10.14670/HH-15.835. [DOI] [PubMed] [Google Scholar]
- 13.Rountree MR, Bachman KE, Herman JG, Baylin SB. DNA methylation, chromatin inheritance, and cancer. Oncogene. 2001;20:3156–65. doi: 10.1038/sj.onc.1204339. [DOI] [PubMed] [Google Scholar]
- 14.Ballestar E, Esteller M. Methyl-CpG-binding proteins in cancer: blaming the DNA methylation messenger. Biochem Cell Biol. 2005;83:374–84. doi: 10.1139/o05-035. [DOI] [PubMed] [Google Scholar]
- 15.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–12. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 16.Karagianni P, Wong J. HDAC3: taking the SMRT-N-CoRrect road to repression. Oncogene. 2007;26:5439–49. doi: 10.1038/sj.onc.1210612. [DOI] [PubMed] [Google Scholar]
- 17.Chakrabarti SR, Nucifora G. The leukemia-associated gene TEL encodes a transcription repressor which associates with SMRT and mSin3A. Biochem Biophys Res Commun. 1999;264:871–7. doi: 10.1006/bbrc.1999.1605. [DOI] [PubMed] [Google Scholar]
- 18.Fenrick R, Amann JM, Lutterbach B, Wang L, Westendorf JJ, Downing JR, Hiebert SW. Both TEL and AML-1 contribute repression domains to the t(12;21) fusion protein. Mol Cell Biol. 1999;19:6566–74. doi: 10.1128/mcb.19.10.6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guidez F, Petrie K, Ford AM, Lu H, Bennett CA, MacGregor A, Hannemann J, Ito Y, Ghysdael J, Greaves M, Wiedemann LM, Zelent A. Recruitment of the nuclear receptor corepressor N-CoR by the TEL moiety of the childhood leukemia-associated TEL-AML1 oncoprotein. Blood. 2000;96:2557–61. [PubMed] [Google Scholar]
- 20.Park DJ, Vuong PT, de Vos S, Douer D, Koeffler HP. Comparative analysis of genes regulated by PML/RAR alpha and PLZF/RAR alpha in response to retinoic acid using oligonucleotide arrays. Blood. 2003;102:3727–36. doi: 10.1182/blood-2003-02-0412. [DOI] [PubMed] [Google Scholar]
- 21.Atsumi A, Tomita A, Kiyoi H, Naoe T. Histone deacetylase 3 (HDAC3) is recruited to target promoters by PML-RARalpha as a component of the N-CoR co-repressor complex to repress transcription in vivo. Biochem Biophys Res Commun. 2006;345:1471–80. doi: 10.1016/j.bbrc.2006.05.047. [DOI] [PubMed] [Google Scholar]
- 22.Villa R, Morey L, Raker VA, Buschbeck M, Gutierrez A, De Santis F, Corsaro M, Varas F, Bossi D, Minucci S, Pelicci PG, Di Croce L. The methyl-CpG binding protein MBD1 is required for PML-RARalpha function. Proc Natl Acad Sci U S A. 2006;103:1400–5. doi: 10.1073/pnas.0509343103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olmeda D, Jorda M, Peinado H, Fabra A, Cano A. Snail silencing effectively suppresses tumour growth and invasiveness. Oncogene. 2007;26:1862–74. doi: 10.1038/sj.onc.1209997. [DOI] [PubMed] [Google Scholar]
- 24.Blechschmidt K, Sassen S, Schmalfeldt B, Schuster T, Hofler H, Becker KF. The E-cadherin repressor Snail is associated with lower overall survival of ovarian cancer patients. Br J Cancer. 2007;16:222–8. doi: 10.1038/sj.bjc.6604115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Larriba MJ, Munoz A. SNAIL vs vitamin D receptor expression in colon cancer: therapeutics implications. Br J Cancer. 2005;92:985–9. doi: 10.1038/sj.bjc.6602484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patra SK, Patra A, Dahiya R. Histone deacetylase and DNA methyltransferase in human prostate cancer. Biochem Biophys Res Commun. 2001;287:705–13. doi: 10.1006/bbrc.2001.5639. [DOI] [PubMed] [Google Scholar]
- 27.Huang BH, Laban M, Leung CH, Lee L, Lee CK, Salto-Tellez M, Raju GC, Hooi SC. Inhibition of histone deacetylase 2 increases apoptosis and p21Cip1/WAF1 expression, independent of histone deacetylase 1. Cell Death Differ. 2005;12:395–404. doi: 10.1038/sj.cdd.4401567. [DOI] [PubMed] [Google Scholar]
- 28.Wilson AJ, Byun DS, Popova N, Murray LB, L'Italien K, Sowa Y, Arango D, Velcich A, Augenlicht LH, Mariadason JM. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem. 2006;281:13548–58. doi: 10.1074/jbc.M510023200. [DOI] [PubMed] [Google Scholar]
- 29.Zhu P, Martin E, Mengwasser J, Schlag P, Janssen KP, Gottlicher M. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell. 2004;5:455–63. doi: 10.1016/s1535-6108(04)00114-x. [DOI] [PubMed] [Google Scholar]
- 30.Spurling CC, Godman CA, Noonan EJ, Rasmussen TP, Rosenberg DW, Giardina C. HDAC3 overexpression and colon cancer cell proliferation and differentiation. Mol Carcinog. 2007;47:137–47. doi: 10.1002/mc.20373. [DOI] [PubMed] [Google Scholar]
- 31.Pilarsky C, Wenzig M, Specht T, Saeger HD, Grutzmann R. Identification and validation of commonly overexpressed genes in solid tumors by comparison of microarray data. Neoplasia. 2004;6:744–50. doi: 10.1593/neo.04277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keyomarsi K, Sandoval L, Band V, Pardee AB. Synchronization of tumor and normal cells from G1 to multiple cell cycles by lovastatin. Cancer Res. 1991;51:3602–9. [PubMed] [Google Scholar]
- 33.Dai B, Rasmussen TP. Global epiproteomic signatures distinguish embryonic stem cells from differentiated cells. Stem Cells. 2007;25:2567–74. doi: 10.1634/stemcells.2007-0131. [DOI] [PubMed] [Google Scholar]
- 34.Yin L, Laevsky G, Giardina C. Butyrate suppression of colonocyte NF-kappa B activation and cellular proteasome activity. J Biol Chem. 2001;276:44641–6. doi: 10.1074/jbc.M105170200. [DOI] [PubMed] [Google Scholar]
- 35.Bjerling P, Silverstein RA, Thon G, Caudy A, Grewal S, Ekwall K. Functional divergence between histone deacetylases in fission yeast by distinct cellular localization and in vivo specificity. Mol Cell Biol. 2002;22:2170–81. doi: 10.1128/MCB.22.7.2170-2181.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 37.Suka N, Luo K, Grunstein M. Sir2p and Sas2p opposingly regulate acetylation of yeast histone H4 lysine16 and spreading of heterochromatin. Nat Genet. 2002;32:378–83. doi: 10.1038/ng1017. [DOI] [PubMed] [Google Scholar]
- 38.Hartman HB, Yu J, Alenghat T, Ishizuka T, Lazar MA. The histone-binding code of nuclear receptor co-repressors matches the substrate specificity of histone deacetylase 3. EMBO Rep. 2005;6:445–51. doi: 10.1038/sj.embor.7400391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson CA, White DA, Lavender JS, O'Neill LP, Turner BM. Human class I histone deacetylase complexes show enhanced catalytic activity in the presence of ATP and co-immunoprecipitate with the ATP-dependent chaperone protein Hsp70. J Biol Chem. 2002;277:9590–7. doi: 10.1074/jbc.M107942200. [DOI] [PubMed] [Google Scholar]
- 40.Towsend K, Trevino V, Falciani F, Stewart PM, Hewison M, Campbell MJ. Identification of VDR-responsive gene signatures in breast cancer cells. Oncology. 2006;71:111–23. doi: 10.1159/000100989. [DOI] [PubMed] [Google Scholar]
- 41.Wu Y, Craig TA, Lutz WH, Kumar R. Identification of 1 alpha,25-dihydroxyvitamin D3 response elements in the human transforming growth factor beta 2 gene. Biochemistry. 1999;38:2654–60. doi: 10.1021/bi981944s. [DOI] [PubMed] [Google Scholar]
- 42.Tou L, Liu Q, Shivdasani RA. Regulation of mammalian epithelial differentiation and intestine development by class I histone deacetylases. Mol Cell Biol. 2004;24:3132–9. doi: 10.1128/MCB.24.8.3132-3139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schneikert J, Behrens J. The canonical Wnt signalling pathway and its APC partner in colon cancer development. Gut. 2007;56:417–25. doi: 10.1136/gut.2006.093310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gmeiner WH, Hellmann GM, Shen P. Tissue-dependent and -independent gene expression changes in metastatic colon cancer. Oncol Rep. 2008;19:245–51. [PubMed] [Google Scholar]
- 45.Segditsas S, Tomlinson I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene. 2006;25:7531–7. doi: 10.1038/sj.onc.1210059. [DOI] [PubMed] [Google Scholar]
- 46.Roose J, Clevers H. TCF transcription factors: molecular switches in carcinogenesis. Biochim Biophys Acta. 1999;1424:M23–37. doi: 10.1016/s0304-419x(99)00026-8. [DOI] [PubMed] [Google Scholar]
- 47.Daniels DL, Weis WI. Beta-catenin directly displaces Groucho/TLE repressors from Tcf/Lef in Wnt-mediated transcription activation. Nat Struct Mol Biol. 2005;12:364–71. doi: 10.1038/nsmb912. [DOI] [PubMed] [Google Scholar]
- 48.van den Brink GR, Bleuming SA, Hardwick JC, Schepman BL, Offerhaus GJ, Keller JJ, Nielsen C, Gaffield W, van Deventer SJ, Roberts DJ, Peppelenbosch MP. Indian Hedgehog is an antagonist of Wnt signaling in colonic epithelial cell differentiation. Nat Genet. 2004;36:277–82. doi: 10.1038/ng1304. [DOI] [PubMed] [Google Scholar]
- 49.Bordonaro M, Lazarova DL, Sartorelli AC. Butyrate and Wnt signaling: a possible solution to the puzzle of dietary fiber and colon cancer risk? Cell Cycle. 2008;7:1178–83. doi: 10.4161/cc.7.9.5818. [DOI] [PubMed] [Google Scholar]
- 50.Lazarova DL, Bordonaro M, Carbone R, Sartorelli AC. Linear relationship between Wnt activity levels and apoptosis in colorectal carcinoma cells exposed to butyrate. Int J Cancer. 2004;110:523–31. doi: 10.1002/ijc.20152. [DOI] [PubMed] [Google Scholar]
- 51.Willert J, Epping M, Pollack JR, Brown PO, Nusse R. A transcriptional response to Wnt protein in human embryonic carcinoma cells. BMC Dev Biol. 2002;2:8. doi: 10.1186/1471-213x-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scacheri PC, Rozenblatt-Rosen O, Caplen NJ, Wolfsberg TG, Umayam L, Lee JC, Hughes CM, Shanmugam KS, Bhattacharjee A, Meyerson M, Collins FS. Short interfering RNAs can induce unexpected and divergent changes in the levels of untargeted proteins in mammalian cells. Proc Natl Acad Sci U S A. 2004;101:1892–7. doi: 10.1073/pnas.0308698100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sledz CA, Williams BR. RNA interference and double-stranded-RNA-activated pathways. Biochem Soc Trans. 2004;32:952–6. doi: 10.1042/BST0320952. [DOI] [PubMed] [Google Scholar]
- 54.Dighe AS, Richards E, Old LJ, Schreiber RD. Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFN gamma receptors. Immunity. 1994;1:447–56. doi: 10.1016/1074-7613(94)90087-6. [DOI] [PubMed] [Google Scholar]
- 55.Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, Old LJ, Schreiber RD. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci U S A. 1998;95:7556–61. doi: 10.1073/pnas.95.13.7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang G, Xu Y, Chen X, Hu G. IFITM1 plays an essential role in the antiproliferative action of interferon-gamma. Oncogene. 2007;26:594–603. doi: 10.1038/sj.onc.1209807. [DOI] [PubMed] [Google Scholar]
- 57.Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137–48. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 58.Klampfer L, Huang J, Swaby LA, Augenlicht L. Requirement of histone deacetylase activity for signaling by STAT1. J Biol Chem. 2004;279:30358–68. doi: 10.1074/jbc.M401359200. [DOI] [PubMed] [Google Scholar]
- 59.Jakowlew SB. Transforming growth factor-beta in cancer and metastasis. Cancer Metastasis Rev. 2006;25:435–57. doi: 10.1007/s10555-006-9006-2. [DOI] [PubMed] [Google Scholar]
- 60.Lynch JP, Hoops TC. The genetic pathogenesis of colorectal cancer. Hematol Oncol Clin North Am. 2002;16:775–810. doi: 10.1016/s0889-8588(02)00029-1. [DOI] [PubMed] [Google Scholar]
- 61.Thiagalingam S, Lengauer C, Leach FS, Schutte M, Hahn SA, Overhauser J, Willson JK, Markowitz S, Hamilton SR, Kern SE, Kinzler KW, Vogelstein B. Evaluation of candidate tumour suppressor genes on chromosome 18 in colorectal cancers. Nat Genet. 1996;13:343–6. doi: 10.1038/ng0796-343. [DOI] [PubMed] [Google Scholar]
- 62.Reiss M. TGF-beta and cancer. Microbes Infect. 1999;1:1327–47. doi: 10.1016/s1286-4579(99)00251-8. [DOI] [PubMed] [Google Scholar]
- 63.Gaschott T, Stein J. Short-chain fatty acids and colon cancer cells: the vitamin D receptor--butyrate connection. Recent Results Cancer Res. 2003;164:247–57. doi: 10.1007/978-3-642-55580-0_18. [DOI] [PubMed] [Google Scholar]
- 64.Dangond F, Hafler DA, Tong JK, Randall J, Kojima R, Utku N, Gullans SR. Differential display cloning of a novel human histone deacetylase (HDAC3) cDNA from PHA-activated immune cells. Biochem Biophys Res Commun. 1998;242:648–52. doi: 10.1006/bbrc.1997.8033. [DOI] [PubMed] [Google Scholar]
- 65.Emiliani S, Fischle W, Van Lint C, Al-Abed Y, Verdin E. Characterization of a human RPD3 ortholog, HDAC3. Proc Natl Acad Sci U S A. 1998;95:2795–800. doi: 10.1073/pnas.95.6.2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang WM, Yao YL, Sun JM, Davie JR, Seto E. Isolation and characterization of cDNAs corresponding to an additional member of the human histone deacetylase gene family. J Biol Chem. 1997;272:28001–7. doi: 10.1074/jbc.272.44.28001. [DOI] [PubMed] [Google Scholar]
- 67.Smith CM, Haimberger ZW, Johnson CO, Wolf AJ, Gafken PR, Zhang Z, Parthun MR, Gottschling DE. Heritable chromatin structure: mapping “memory” in histones H3 and H4. Proc Natl Acad Sci U S A. 2002;99(Suppl 4):16454–61. doi: 10.1073/pnas.182424999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.