

Abstract
DNA gyrase is unique among topoisomerases in its ability to introduce negative supercoils into closed-circular DNA. We have demonstrated that deletion of the C-terminal DNA-binding domain of the A subunit of gyrase gives rise to an enzyme that cannot supercoil DNA but relaxes DNA in an ATP-dependent manner. Novobiocin, a competitive inhibitor of ATP binding by gyrase, inhibits this reaction. The truncated enzyme, unlike gyrase, does not introduce a right-handed wrap when bound to DNA and stabilizes DNA crossovers; characteristics reminiscent of conventional type II topoisomerases. This new enzyme form can decatenate DNA circles with increased efficiency compared with intact gyrase and, as a result, can complement the temperature-sensitive phenotype of a parCts mutant. Thus these results suggest that the unique properties of DNA gyrase are attributable to the wrapping of DNA around the C-terminal DNA-binding domains of the A subunits and provide an insight into the mechanism of type II topoisomerases.
Keywords: supercoiling, decatenation, quinolone, coumarin
DNA topoisomerases are a ubiquitous class of enzymes responsible for the alteration of the topological state of DNA (1). As they are essential to all cells, topoisomerases are potential targets of antibacterial and antineoplastic drugs (2). The topoisomerization reaction is achieved by the cleavage of DNA and the formation of a transient DNA gate through which a part of the same or another DNA molecule is passed. Mechanistic differences in this reaction distinguish topoisomerases into two types. Type I enzymes introduce a single-stranded break in DNA and facilitate the passage of a second strand through this gate. Type II topoisomerases create a double-stranded break and allow the passage of a double-stranded DNA segment. Covalent linkage between type II enzymes and DNA occurs by a transesterification reaction between two tyrosine residues and a pair of phosphates 4 bp apart in the DNA. This results in the attachment of the 5′ ends of each cleaved DNA strand to the enzyme and the formation of a double-stranded DNA gate. Another DNA segment is then passed through this gate and the break is resealed. Turnover of type II enzymes in topoisomerization reactions generally requires the hydrolysis of ATP.
Type II topoisomerases are able to catalyze a number of reactions, including the relaxation of negative and positive supercoils, the catenation and decatenation of DNA circles, and the knotting and unknotting of DNA (3). DNA gyrase is the only topoisomerase able to actively introduce negative supercoils into DNA molecules, in a reaction dependent upon ATP hydrolysis (4). In the absence of ATP, gyrase can relax supercoiled DNA (5, 6). Although gyrase can decatenate DNA (7), this reaction is not as efficient as with other type II enzymes (8). In the presence of quinolone drugs, gyrase can introduce double-stranded breaks in DNA after treatment with SDS and protease (5, 6). In addition to DNA gyrase, a second type II enzyme, topoisomerase IV (topo IV), exists in prokaryotes. Topo IV is unable to supercoil DNA but, unlike gyrase, can, through its decatenating activity, support the final stages of replication during in vitro oriC DNA replication (8); in vivo, topo IV mutants are deficient in chromosomal partitioning (9). Eukaryotic cells contain at least one type II enzyme; topo II in lower eukaryotes (e.g., Saccharomyces cerevisiae and Drosophila melanogaster) and the two isoforms topo IIα and topo IIβ in human cells (10). All type II enzymes share a degree of sequence similarity, but tend to differ at their C termini.
DNA gyrase is composed of two subunits, GyrA (97 kDa) and GyrB (90 kDa); the active form being an A2B2 heterotetramer (3), which binds ≈130 bp of DNA (11) in a positive-superhelical sense around its core (12). Topo IV is a heterotetramer like gyrase, with ParC and ParE being the equivalents of GyrA and GyrB (13). Eukaryotic topo II molecules are homodimers where the monomer is equivalent to a fusion of the A and B subunits of gyrase such that the B protein corresponds approximately to the N terminus of the eukaryotic proteins while the A subunit is equivalent to the C terminus (14). The latter enzymes bind DNA without wrapping it and protect only 25–35 bp from nuclease digestion (15, 16).
When GyrA is treated with either trypsin or chymotrypsin, two large fragments are generated with approximate masses of 64 and 33 kDa (17). The 64-kDa fragment makes up the N-terminal 571 amino acids of the intact protein and contains both the active site tyrosine and the sites in GyrA to which quinolone resistance has been mapped (18). In vitro experiments in the presence of GyrB have shown that this fragment performs quinolone-induced DNA cleavage as efficiently as the intact protein. Subsequent deletion experiments showed that the smallest fragment able to catalyze DNA cleavage was a 58-kDa protein (residues 7–523) (19). The C-terminal 33-kDa fragment (residues 572–875) is unable to catalyze any of the reactions of gyrase but has been shown to bind DNA in a Mg2+-independent manner, in contrast to the binding of the holoenzyme or the A protein. When the 33-kDa domain is allowed to bind nicked DNA and the nick is resealed by Escherichia coli ligase, the DNA becomes positively supercoiled (20). It is likely that this domain provides a nonspecific DNA-binding function responsible for the positive wrapping of DNA around the enzyme. This mode of binding could present the transport DNA segment to the DNA gate in an orientation such that subsequent strand passage will favor the introduction of negative supercoils. The fact that the C terminus of gyrase bears little sequence similarity to that of the other type II enzymes, when compared with other parts of the protein (21), could account for the differences in mechanism between gyrase and other type II topoisomerases. In this case, removal of the C-terminal domain of GyrA may give rise to an enzyme that relaxes instead of supercoiling DNA and catalyzes intermolecular strand passage more efficiently than intact gyrase; i.e., this new enzyme form may behave like a conventional type II topoisomerase. In this paper, we have tested this proposal using a 59-kDa N-terminal fragment of GyrA.
MATERIALS AND METHODS
Enzymes and DNA.
GyrA, GyrB, the 59-kDa fragment (gift of C. V. Smith, University of Leicester), and the 64- and 33-kDa fragments (gifts of S. E. Critchlow, University of Leicester) were purified as described (19, 20). Chicken erythrocyte topo I and negatively supercoiled and relaxed forms of plasmid pBR322 were provided by A. J. Howells, University of Leicester. Linear pBR322 was prepared by digestion of the supercoiled form with EcoRI. Singly nicked pBR322 was prepared by digestion of the supercoiled form with DNase I in the presence of EtBr (22). Positively and highly negatively supercoiled DNA (σ ≈ −0.13) was prepared as described (23).
Enzyme Assays.
DNA supercoiling, ATP-independent relaxation, and cleavage assays were performed as described (17). ATP-dependent relaxation was carried out in 35 mM Tris·HCl, pH 7.5/12.5 mM KCl/8 mM MgCl2/BSA (0.36 mg/ml)/tRNA (9 μg/ml)/6.5% glycerol/5 mM DTT/1.4 mM ATP/negatively supercoiled pBR322 (10 μg/ml). The extent of DNA wrapping around GyrA and the 59-kDa domain was determined by incubating nicked pBR322 (26.6 μg/ml) with different concentrations of the enzyme and resealing the nick with E. coli ligase as described (20). Decatenation reactions were performed using kinetoplast DNA (Topogen, Columbus; 13.3 μg/ml) under ATP-dependent relaxation conditions at 37°C. Catenation reactions were performed in 25 mM Tris·HCl, pH 7.5/15 mM KCl/2 mM MgCl2/5 mM spermidine/BSA (0.36 mg/ml)/tRNA (9 μg/ml)/6.5% glycerol/5 mM DTT/0.5 mM ATP/negatively supercoiled pBR322 (6.7 μg/ml), at 37°C. Relaxation of positively and highly negatively supercoiled DNA was performed under ATP-dependent relaxation conditions. Adenosine 5′-[β,γ-imido]triphosphate (ADPNP) single-strand-passage experiments were performed as described elsewhere (23). ATPase rates were determined by a linked enzyme assay (24) in the presence of excess of GyrA or the 59-kDa protein and various forms of pBR322 DNA. Binding to DNA was measured by retention by nitrocellulose filters as described (25).
RESULTS
The A592B2 Complex Can Relax Supercoiled DNA in the Presence of ATP.
The N-terminal domain of GyrA has been identified as being responsible for the DNA breakage-reunion reaction of DNA gyrase (17). When expressed as a 64-kDa fragment [GyrA-(1–572)] and complexed with GyrB (A642B2), it was found to support low levels of DNA supercoiling and ATP-independent relaxation [≈0.1% compared with A2B2 (19)]. When expressed as a 59-kDa fragment [GyrA-(1–523)] and complexed with GyrB (A592B2), it was again found to exhibit low levels of relaxation activity but undetectable supercoiling activity (19). Subsequently the supercoiling activity of A642B2 has been reevaluated and found to be undetectable (26). We assume that the low-level supercoiling activity previously found (19) is attributable to contamination by GyrA. Both the A642B2 and A592B2 complexes show levels of quinolone-induced DNA cleavage comparable to that of intact gyrase (19, 26).
We have confirmed that the A592B2 complex is unable to supercoil relaxed DNA, even at high protein concentrations, and supports only low levels of ATP-independent relaxation (less than 10% compared with intact gyrase, data not shown); this again may be affected by contamination by GyrA. However, we have found that the A592B2 complex is able to efficiently relax negatively supercoiled DNA in the presence of ATP (Fig. 1A). Neither the 59-kDa protein nor GyrB alone can support this reaction. The relaxation reaction supported by the A592B2 complex is distributive and not processive as is the case for supercoiling by gyrase. The time dependence of relaxation clearly suggests a catalytic reaction (Fig. 1A). To rule out the possibility of a slow stoichiometric reaction, time course experiments of relaxation at low protein/DNA ratios were performed (Fig. 1A). Under these conditions, each A592B2 complex increased the linking number of the DNA by at least 25. We also found that A642B2 is capable of ATP-dependent relaxation of DNA.
Figure 1.
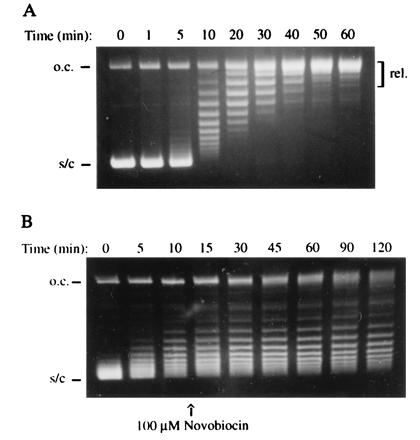
(A) Time course of ATP-dependent relaxation supported by A592B2. The reaction contained 3.1 nM enzyme and 3.5 nM supercoiled pBR322 and was incubated at 25°C for the times indicated. Samples were analyzed on a 1% agarose gel (o.c., open circular; s/c, negatively supercoiled; rel., relaxed). (B) Inhibition of the relaxation of supercoiled pBR322 (3.5 nM) with A592B2 (62 nM) by the addition of 100 μM novobiocin after 10 min.
Novobiocin is a specific inhibitor of DNA gyrase that has recently been shown (27) to act by competitively inhibiting the binding of ATP. Addition of novobiocin completely inhibited DNA relaxation by the A592B2 complex (Fig. 1B). Moreover, the nonhydrolyzable ATP analog, ADPNP, failed to support the reaction. These results demonstrate the ATP-dependent nature of the relaxation reaction. The A592B2 complex is also able to relax positively and highly negatively supercoiled DNA in ATP-dependent reactions. Gyrase, by contrast, converts positively supercoiled DNA to the negatively supercoiled form in the presence of ATP. The efficiency of the ATP-dependent relaxation reaction of A592B2 was compared with that of the supercoiling reaction of gyrase. The rate at which A592B2 removes supecoils is not constant throughout the reaction (Fig. 1A and see Fig. 4C). Under the same conditions, in the first steps of the reaction, the rate of relaxation by A592B2 is approximately 20% of the rate of supercoiling by gyrase during the same time period. At the later stages of the reaction, this rate drops to less than 10% that of the initial steps of the gyrase supercoiling reaction (see Fig. 4 B and C). Control experiments have shown that this is not due to loss of enzyme activity and can be explained in terms of the decreasing concentration of DNA crossovers (see below).
Figure 4.
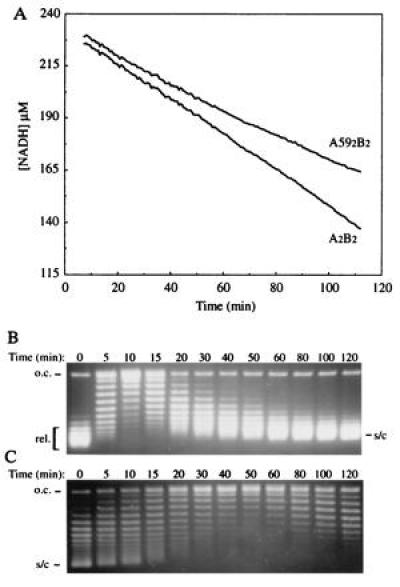
(A) ATPase of A592B2 and A2B2. The experiment was performed using an enzyme-linked assay (24) and the decrease in NADH concentration reflects the amount of ATP hydrolyzed. (B and C) Reactions contained 100 nM pBR322 DNA (supercoiled for the A592B2 reaction and relaxed for the A2B2 reaction) and were simultaneously monitored for topo activity by analyzing samples on 1% agarose gels containing chloroquine at 1 μg/ml: (B) Supercoiling by 10 nM A2B2. (C) Relaxation by 50 nM A592B2. The rate of hydrolysis by A2B2 was stable at 1.04 s−1 throughout the experiment while the rate of the A592B2-supported reaction dropped from 0.24 s−1 in the first 5 min to 0.09 s−1 in the last stages of the reaction.
To test whether the supercoiling ability of the A592B2 complex could be restored, we tried adding back the 33-kDa C-terminal DNA-binding domain of GyrA. In the presence of high concentrations of the 33-kDa domain only weak positive supercoiling was observed. This was probably caused by relaxation by the A592B2 complex of the negative writhe introduced into relaxed DNA upon the binding of the 33-kDa domains. This result is in contrast to the N-terminal 64-kDa domain of GyrA that shows relatively efficient supercoiling activity in the presence of the 33-kDa domain (20, 26). It is likely that the ≈5 kDa missing between the C terminus of the 59-kDa domain and the N terminus of the 33-kDa protein is important for complex stability.
The A592B2 Complex Is a Potent Decatenating Enzyme.
The ability of A592B2 to decatenate DNA circles was investigated by examining its ability to release minicircles from kinetoplast DNA (k-DNA) networks. The size of the network is so large that when subjected to electrophoresis on an agarose gel only the released minicircles will enter the gel while the network remains in the wells. The decatenation of k-DNA was performed under ATP-dependent relaxation conditions and the ability of A592B2 to perform the reaction was compared with that of gyrase (Fig. 2). Comparing the enzyme concentration required for the release of the same number of minicircles, it was estimated that the truncated enzyme is ≈30 times more efficient in decatenation than gyrase. These levels of decatenation are approximately one order of magnitude lower than those exhibited by human topo IIα using k-DNA as substrate (T. R. Hammonds, personal communication). Control experiments have shown that the failure of previous studies to detect decatenation by the N-terminal domain of GyrA (19) can largely been attributed to the different reaction conditions employed; the earlier studies were conducted at a lower temperature (25°C compared with 37°C) and lower salt concentration. Enzyme concentrations that, under the conditions used in this work, yielded full decatenation of k-DNA, produced only low levels of free minicircles under the conditions employed previously (19). Differences could also be due to the different catenated substrates used: a pair of interlinked circles in the previous work (19) and k-DNA in the present experiments; k-DNA may present a better substrate due to the high local concentration of crossovers (see below).
Figure 2.
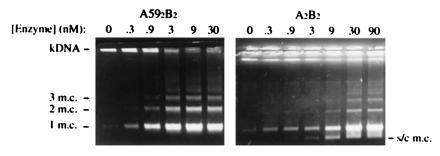
Decatenation of k-DNA by A592B2 and A2B2. Reactions contained k-DNA at 13.3 μg/ml and were incubated for 1 hr at 37°C. Samples were analyzed on a 1% agarose gel (1 m.c., free monomeric minicircle; 2 m.c., two interlinked minicircles; 3 m.c., three interlinked minicircles; s/c m.c., supercoiled minicircle). The unresolved k-DNA network remains in the wells.
DNA gyrase can catalyze the catenation of plasmid DNA under conditions of high spermidine and low Mg2+ concentration (A. P. Tingey, personal communication). Under these conditions, we were unable to detect catenation by the A592B2 complex. An extensive investigation of alternative reaction conditions failed to detect any catenation activity with the truncated enzyme.
The Interaction of A592B2 with DNA Is Characterized by Binding to Crossovers.
Using either supercoiled or relaxed radiolabeled pBR322, the amount of DNA retained on nitrocellulose filters by the A592B2 complex was found to be less than 10% of that retained by intact gyrase (A2B2). It has previously been shown that gyrase shows a preference for binding to relaxed as compared with negatively supercoiled DNA (28); we found that the truncated enzyme exhibits a preference for the supercoiled form.
When gyrase binds to nicked-circular DNA and the nick is resealed by E. coli ligase the DNA is found to be positively supercoiled (12). We performed this experiment with the A592B2 complex and found that, unlike gyrase, the truncated enzyme does not wrap DNA (Fig. 3A); on the contrary, the A592B2 complex reduces the linking number of DNA by less than 0.03 per enzyme molecule. This is likely to be a manifestation of the absence of the C-terminal DNA-binding domain, which has been shown to wrap DNA in a positive sense even when incubated with DNA on its own (20). This experiment was performed both in the absence and the presence of ciprofloxacin (CFX) and the results were found to be identical; CFX stabilizes the relatively weak enzyme–DNA complex.
Figure 3.
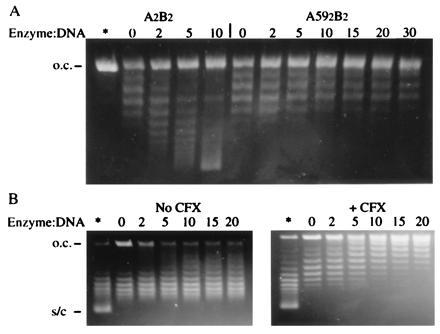
(A) Determination of the extent of DNA wrapping. Open-circular pBR322 (400 ng) was incubated for 30 min at 25°C with the enzyme/DNA ratios indicated and then the nick was resealed with E. coli ligase. The asterisk indicates the starting open-circular material. Samples were analyzed on an 1% agarose gel containing chloroquine at 0.4 μg/ml. (B) Stabilization of crossovers by A592B2. Negatively supercoiled pBR322 (400 ng) was relaxed by topo I in the presence of the indicated ratios of enzyme/DNA, in the absence and presence of CFX. The tracks labeled with the asterisk contain the starting supercoiled material while those labeled 0 (zero) contain no enzyme and are fully relaxed by topo I. Samples were analyzed on an agarose gel containing chloroquine at 0.8 μg/ml.
The ATPase activity of DNA gyrase is stimulated by the binding of DNA (25). We found that linear pBR322 failed to stimulate the ATPase of the A592B2 complex. Investigation of ATP hydrolysis in the presence of a large excess of negatively supercoiled DNA showed a rate that decreased during the course of the reaction (Fig. 4). Samples were removed from the reaction and analyzed by electrophoresis to monitor the progress of relaxation. As the reaction proceeded the rate of hydrolysis gradually decreased (Fig. 4), whereas intact gyrase exhibits a stable ATPase rate during DNA supercoiling even when all the substrate is fully supercoiled (Fig. 4 and also ref. 23). The rate of hydrolysis by A2B2 was stable at 1.04 s−1 throughout the experiment while the rate of the A592B2-supported reaction dropped from 0.24 s−1 in the first 5 min to 0.09 s−1 in the last stages of the reaction. A similar trend can be observed also in the rate of supercoil relaxation and DNA supercoiling (Figs. 4 B and C and 1A). The rate of ATP hydrolysis in the presence of linear DNA remains constant throughout the course of this experiment, indicating that this phenomenon is not due to loss of enzyme activity. It is evident that the binding of the A592B2 complex to linear or relaxed DNA does not efficiently stimulate the ATPase. In the presence of the quinolone drug CFX, the ATPase activity of the A592B2 complex in the presence of linear and relaxed DNA was stimulated (data not shown); consistent with the stabilization of the DNA–protein complex in the presence of the drug (28). These results can be interpreted as preferential binding to DNA crossovers stabilizing the complex and increasing the rate of hydrolysis; subsequent strand passage would remove the crossovers and, thus, decrease the rate. Close investigation of a time course of relaxation (Fig. 1A) reveals that the rate of the reaction slows down with time, again consistent with preferential binding to crossovers.
To investigate the binding of the A592B2 complex to DNA crossovers further, supercoiled DNA was relaxed by chicken erythrocyte topo I in the presence of different concentrations of the truncated enzyme. Topo I does not require ATP and, thus, allows the experiment to be carried out in the absence of nucleotide (1). As the interaction between A592B2 and DNA is relatively weak, this experiment was performed in parallel in the presence of CFX to stabilize the A592B2–DNA complex and obtain a clearer result. As seen previously with topo II (29), A592B2 inhibits full relaxation by topo I (Fig. 3B). This is a clear indication of the truncated enzyme binding to DNA crossovers, thus stabilising them and preventing them from being removed by topo I. This interaction is consistent with the higher affinity of the truncated enzyme for supercoiled DNA and the effects of different forms of DNA on the ATPase rate of the A592B2 complex. Control experiments have shown that the slight unwinding of relaxed DNA observed in the experiment containing CFX is due to the well-documented interaction between quinolones and DNA (30) and does not interfere with the results of this experiment.
Complementation of a parC Temperature-Sensitive Mutant.
So far the truncated enzyme seems to behave in vitro like a typical type II topoisomerase, being able to support all the reactions catalyzed by topo IV or its eukaryotic counterpart topo II. We tested whether this enzyme can behave the same way in vivo and complement a topo IV-deficient strain. For this purpose, a temperature-sensitive parCts E. coli mutant was used (gift of K. Marians, Memorial Sloan–Kettering Cancer Center, New York). The ts strain and its parent strain E. coli C600 were transformed with plasmids encoding wild-type ParC (pET-3c-parC), the 59-kDa fragment (pRJR10.18), GyrA (pPH3), GyrB (pAG111), and the vector pTTQ18* (20). The various strains were incubated at the permissive (30°C) and nonpermissive (42°C) temperatures (Fig. 5). The 59-kDa protein could clearly complement the ParC deficiency at 42°C in contrast to GyrA, which failed to do so at any inducer concentration. The strains transformed with the wild-type ParC-expressing plasmid showed the highest levels of complementation, while the A59 protein was less effective (≈5%). On the other hand, the strains transformed with the GyrA- or the GyrB-expressing plasmids or the vector alone failed to survive at the nonpermissive temperature (Fig. 5).
Figure 5.
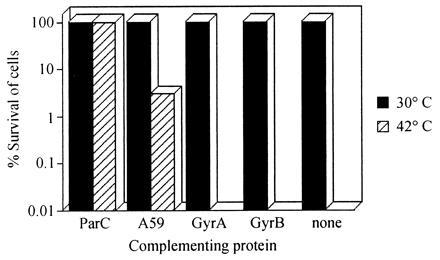
Complementation of a parCts strain. Histogram showing the relative efficiency with which plasmids expressing different proteins complemented the parCts phenotype. For the bar labeled none, the cells were transformed with plasmid pTTQ18*, which is the vector used in the construction of the gyrase plasmids (20).
DISCUSSION
DNA gyrase is an atypical type II topoisomerase. While the other type II enzymes (topo IV and topo II) carry out relaxation and decatenation reactions, gyrase is so far the only topoisomerase able also to negatively supercoil DNA. The type II enzymes exhibit a great degree of sequence similarity but diverge significantly at their C termini (21). One of the principal differences between gyrase and the conventional type II enzymes is in the mode of interaction with the DNA. The relaxing and decatenating enzymes bind 25–35 bp of DNA without wrapping it around their core (15, 16), while gyrase binds ≈130 bp in a positive-superhelical manner (11, 12); this interaction is probably the basis of their mechanistic differences. Topo II does not prescribe the orientation for the DNA segment to be translocated. The enzyme will perform strand passage with any transport segment presented to the DNA gate, thus permitting both intra- and intermolecular strand-translocation events. By contrast, the mechanism of supercoiling by gyrase requires the enzyme to distinguish between different DNA segments, so that subsequent strand passage will result in the introduction of two negative turns. For this to be achieved, a positive node on the DNA has to be inverted. In the case of gyrase, this node is introduced by the wrapping of DNA around the enzyme. The C-terminal domain of the A protein of DNA gyrase, which exhibits significant sequence diversity from the other topoisomerases, has been shown to be responsible for the right-handed wrapping of the DNA (20). We proposed that removal of this domain will give rise to an enzyme that will function by a similar mechanism to that of other type II enzymes.
For the purpose of these experiments, we have used a 59-kDa N-terminal fragment that contains the first 523 residues of GyrA, thus removing the C-terminal DNA-binding domain. This fragment contains all the residues required for topoisomerase activity since it can perform quinolone-induced DNA cleavage when complexed with GyrB. The complex between the 59-kDa protein and GyrB (A592B2) is unable to catalyze the supercoiling of DNA and can only support low levels of ATP-independent relaxation. The mode of binding of this enzyme to DNA is distinctly different to that of gyrase and is very similar to the other type II enzymes. With A592B2 no significant wrapping of DNA occurs and the binding of the enzyme appears to cause a slight unwinding. The extent of this change in the linking number is very small and was estimated to be less than −0.03 turns per enzyme molecule. Most DNA-binding proteins cause some distortion of the helix upon binding (e.g., polymerases) and such a small unwinding is not unexpected. The inability to introduce a positive node on the DNA is consistent with the failure of this enzyme to supercoil DNA. Hydroxyl-radical footprinting of A642B2 bound to DNA (in the presence of CFX, to stabilize the protein–DNA complex) shows that this complex protects 20–25 bp around the cleavage site (G. Orphanides, personal communication). The 59-kDa protein is not much smaller than the 64-kDa domain and is likely to give a similar protection pattern. Similar experiments involving nuclease digestion with topo IV or eukaryotic topo II have shown a protection of 30–34 and 25–28 bp, respectively (15, 16). Thus, these results indicate that the mode of DNA binding by A592B2 is very similar to that of topo II and topo IV.
Given the similarities in the DNA binding between A592B2 and the typical type II enzymes, the ATP-dependent relaxation of supercoiled DNA by the A592B2 complex does not come as a surprise. The enzyme seems not to distinguish between transport segments since it is able to relax both negatively and positively supercoiled DNA. By contrast, gyrase clearly dictates the direction of strand passage since it converts the positively supercoiled substrate to the negatively supercoiled form. The reaction of A592B2 is catalytic in nature as indicated by the number of superhelical turns removed by each molecule. The ATP dependency of this reaction was shown by the lack of activity in the absence of ATP or in the presence of ADPNP. Moreover, the reaction was completely stopped upon the addition of novobiocin, a competitive inhibitor of ATP hydrolysis by gyrase.
The affinity of A592B2 for DNA has been significantly affected by the removal of the C-terminal domains. Nitrocellulose filter binding experiments showed that the binding of A592B2 to DNA is much weaker than that of gyrase and exhibits a preference for supercoiled DNA, unlike gyrase. This preference can be explained on the basis of its preferential binding to DNA crossovers. This was supported by the observation that topo I is unable to fully relax the supercoiled DNA–A592B2 complex in the absence of ATP; i.e., the enzyme stabilizes DNA crossovers and prevents them from being removed by topo I. When the complex was stabilized by quinolones, the effect was even more pronounced. The small unwinding observed upon binding of the complex to DNA cannot account for this change since it can only result in less than one turn at the highest enzyme concentration used. A similar mode of binding was observed in the case of yeast topo II (29). Binding at crossovers stabilizes the weak binding of the enzyme to the DNA and stimulates ATP hydrolysis. Removal of the crossovers during relaxation destabilizes the enzyme–DNA complex and reduces the rate of hydrolysis (Fig. 4). Binding of the enzyme to crossovers gives an insight into the mechanism of DNA relaxation. Such a mode of binding presents one of the two helixes to the DNA gate formed on the other. Translocation of this segment through the gate will invert the node and change the linking number by 2. In a negatively supercoiled substrate most nodes are negative while the opposite applies to a positively supercoiled molecule. Inversion of these nodes will result in substrate relaxation. Destabilization of the enzyme–DNA complex subsequent to strand passage favors dissociation and binding to another crossover, as is indicated by the distributive nature of relaxation.
If the crossover is between a pair of helices that are part of different molecules, as in the case of catenated circles, then the relaxing enzyme will preferably bind to the crossover point and unlink the two molecules by translocating one helix. This was found to be the case with the A592B2 complex, which has an approximately 30-fold higher efficiency in decatenating k-DNA than gyrase. When gyrase is presented with this substrate, it will mainly perform DNA supercoiling. The wrapping of the DNA in the case of gyrase favors the translocation of a segment coming from the same DNA molecule, close to or within the wrapped segment, instead of an interlinked DNA helix. Gyrase has been shown to support catenation of DNA circles under conditions of DNA condensation (31). The inability of A592B2 to perform this reaction is probably a result of its lower affinity for DNA. The equilibrium between the catenation and decatenation reactions should significantly favor unlinking since crossovers between interlinked molecules are clearly more probable than points of transient intermolecular contact in unlinked circles.
Deletion of the C-terminal domain of gyrase results in an enzyme with significantly different characteristics. On the basis of its altered mode of DNA binding, this enzyme is able to perform in vitro like topo II or IV. To test this possibility in vivo, the ability of the 59-kDa protein to complement a ParC-deficient strain was investigated. The temperature-sensitive phenotype was successfully complemented by the 59-kDa domain while expression of GyrA failed to maintain cell viability. The action of the 59-kDa protein is likely to be through its association with endogenous GyrB rather than ParE, since the addition of ParE to GyrA failed to show in vitro activity (13).
These results give an insight into the mechanism of action of topoisomerases. The mechanism of ATP-dependent reactions catalyzed by the conventional type II enzymes and the A592B2 complex is characterized by the undiscriminating nature of strand passage. By contrast, gyrase dictates the direction of ATP-dependent strand passage by means of its mode of binding to DNA that creates a node and presents the transport segment to the enzyme gate. This difference in the mechanism is reflected both in the intra- and intermolecular reactions. Conventional type II topoisomerases are better decatenating enzymes, while DNA gyrase favors the intramolecular reaction because of the high probability of the transport segment to be part of the same molecule as the gate segment.
Gyrase also catalyzes the relaxation of negative supercoils in the absence of nucleotide cofactor (5, 6), a reaction also carried out by topo II′, the form of gyrase that results after proteolytic removal of the ATPase domains (32). This reaction must proceed through a distinct mechanism from that of ATP-dependent topoisomerization. The limit of the supercoiling reaction catalyzed by gyrase has been shown to be thermodynamic rather than steric (33, 34). In this case, the enzyme must be able to prevent translocation of the DNA segment through the DNA gate if the increase in free energy of the resulting topoisomer is higher than that released by ATP hydrolysis. This could be accomplished if the transport segment was able to translocate back through the gate when the ATP-released energy is not sufficient. It is possible that the ATP-independent relaxation is a result of such a backward reaction. Incubation of highly negatively supercoiled DNA (σ < −0.11) with gyrase in the presence of ATP results in relaxation of the substrate until it reaches a superhelical density of approximately −0.11, the limit of supercoiling by gyrase (A.M., unpublished observations). Similarly, when a supercoiling reaction catalyzed by gyrase is allowed to reach the limit of supercoiling and then ADP is added so that the sum of [ATP] and [ADP] is constant but the ratio of [ATP] to [ADP] is varied, the reaction reaches another limit that is characterized by lower absolute superhelical density (35). Given that the free energy of ATP hydrolysis depends on the ratio of [ATP] to [ADP], in these experiments, a thermodynamic equilibrium seems to have been reached between two opposing reactions, ATP-dependent supercoiling and ATP-independent relaxation.
If the two-gate model for the mechanism of type II enzymes (36, 37) is considered, the ATP-dependent reactions proceed by the translocation of the transport segment first through the ATP gate and then through the DNA gate. However, ATP-independent strand passage is likely to proceed in the opposite direction. Gyrase cannot relax positively supercoiled DNA in the absence of nucleotide (23), and it has recently been shown that the probability of ADPNP-driven strand passage is high for positively supercoiled DNA but gradually decreases with decreasing linking number (23). On the basis of the mode of binding of gyrase, it is evident that positively supercoiled DNA would facilitate an orientation of the transport segment such that it presents itself to the ATP gate. Negatively supercoiled DNA will not as readily wrap itself around the enzyme and present itself to the ATP gate, therefore, the probability of strand passage with this substrate is significantly lower (23). At the same time, this substrate will have a higher possibility of assuming a conformation that presents a DNA segment to be translocated through the DNA gate from the other direction. Transportation of this segment through the DNA gate first and then through the ATP gate will result in one round of relaxation. This reaction is feasible by the escape mechanism described above since the supercoiled substrate has higher free energy than the relaxed form. The ATP-independent reaction does not occur in the case of the conventional type II enzymes and could be due to an intrinsic feature of gyrase, maybe reflecting a weaker interface between the two A subunits that can now become transiently open, thus, allowing the admission of a DNA segment through the bottom gate. Alternatively, the possibility also exists that such a backward reaction could proceed via a one-gate mechanism.
These experiments are a demonstration of the common evolutionary origins of type II topoisomerases. It is likely that all type II topoisomerases share a common ancestor but have evolved to perform more specialized roles in different organisms. In the case of DNA gyrase, the C terminus is involved in the wrapping of the DNA around the enzyme, a mode of binding that creates a positive node and strongly favors strand passage in the direction of reduction of the linking number. In the case of eukaryotic topo II, the C terminus performs a different function, being thought to have a regulatory role (10).
Acknowledgments
We thank Drs. A. D. Bates and N. A. Gormley for reading the manuscript; Ms. C. V. Smith, Dr. S. E. Critchlow, and Mrs. A. J. Howells for gifts of protein and DNA; Dr. K. J. Marians for providing the parCts strain; and Prof. J. C. Wang for suggesting the complementation experiment. S.C.K. is supported by a studentship funded by the Biotechnology and Biological Sciences Research Council and Glaxo–Wellcome, and a grant from the Alexander S. Onassis Public Benefit Foundation. A.M. is a Lister-Institute Jenner Fellow.
Footnotes
Abbreviations: GyrA, DNA gyrase A protein; GyrB, DNA gyrase B protein; A59, 59-kDa fragment of the A protein; topo, topoisomerase; ADPNP, adenosine 5′-[β,γ-imido]triphosphate;k-DNA, kinetoplast DNA; CFX, ciprofloxacin.
References
- 1.Wang J C. Annu Rev Biochem. 1996;65:635–692. doi: 10.1146/annurev.bi.65.070196.003223. [DOI] [PubMed] [Google Scholar]
- 2.Liu L F. Annu Rev Biochem. 1989;58:351–375. doi: 10.1146/annurev.bi.58.070189.002031. [DOI] [PubMed] [Google Scholar]
- 3.Reece R J, Maxwell A. CRC Crit Rev Biochem Mol Biol. 1991;26:335–375. doi: 10.3109/10409239109114072. [DOI] [PubMed] [Google Scholar]
- 4.Gellert M, Mizuuchi K, O’Dea M H, Nash H A. Proc Natl Acad Sci USA. 1976;73:3872–3876. doi: 10.1073/pnas.73.11.3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gellert M, Mizuuchi K, O’Dea M H, Itoh T, Tomizawa J. Proc Natl Acad Sci USA. 1977;74:4772–4776. doi: 10.1073/pnas.74.11.4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sugino A, Peebles C L, Kruezer K N, Cozzarelli N R. Proc Natl Acad Sci USA. 1977;74:4767–4771. doi: 10.1073/pnas.74.11.4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu L F, Liu C-C, Alberts B M. Cell. 1980;19:697–707. doi: 10.1016/s0092-8674(80)80046-8. [DOI] [PubMed] [Google Scholar]
- 8.Peng H, Marians K J. Proc Natl Acad Sci USA. 1993;90:8571–8575. doi: 10.1073/pnas.90.18.8571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adams D E, Shekhtman E M, Zechiedrich E L, Schmid M B, Cozzarelli N R. Cell. 1992;71:277–288. doi: 10.1016/0092-8674(92)90356-h. [DOI] [PubMed] [Google Scholar]
- 10.Watt P M, Hickson I D. Biochem J. 1994;303:681–695. doi: 10.1042/bj3030681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orphanides G, Maxwell A. Nucleic Acids Res. 1994;22:1567–1575. doi: 10.1093/nar/22.9.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu L F, Wang J C. Proc Natl Acad Sci USA. 1978;75:2098–2102. doi: 10.1073/pnas.75.5.2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kato J, Suzuki H, Ikeda H. J Biol Chem. 1992;267:25676–25684. [PubMed] [Google Scholar]
- 14.Lynn R, Giaever G, Swanberg S, Wang J C. Science. 1986;233:647–648. doi: 10.1126/science.3014661. [DOI] [PubMed] [Google Scholar]
- 15.Peng H, Marians K J. J Biol Chem. 1995;270:25286–25290. doi: 10.1074/jbc.270.42.25286. [DOI] [PubMed] [Google Scholar]
- 16.Lee M P, Sander M, Hsieh T. J Biol Chem. 1989;264:21779–21787. [PubMed] [Google Scholar]
- 17.Reece R J, Maxwell A. J Biol Chem. 1989;264:19648–19653. [PubMed] [Google Scholar]
- 18.Maxwell A. J Antimicrob Chemother. 1992;30:409–416. doi: 10.1093/jac/30.4.409. [DOI] [PubMed] [Google Scholar]
- 19.Reece R J, Maxwell A. J Biol Chem. 1991;266:3540–3546. [PubMed] [Google Scholar]
- 20.Reece R J, Maxwell A. Nucleic Acids Res. 1991;19:1399–1405. doi: 10.1093/nar/19.7.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caron P R, Wang J C. In: Molecular Biology of DNA Topoisomerases and Its Application to Chemotherapy. Andoh T, Ikeda H, Oguro M, editors. Boca Raton, FL: CRC; 1993. pp. 1–18. [Google Scholar]
- 22.Greenfield L, Simpson L, Kaplan D. Biochim Biophys Acta. 1975;407:365–375. doi: 10.1016/0005-2787(75)90104-5. [DOI] [PubMed] [Google Scholar]
- 23.Bates A D, O’Dea M H, Gellert M. Biochemistry. 1996;35:1408–1416. doi: 10.1021/bi952433y. [DOI] [PubMed] [Google Scholar]
- 24.Ali J A, Jackson A P, Howells A J, Maxwell A. Biochemistry. 1993;32:2717–2724. doi: 10.1021/bi00061a033. [DOI] [PubMed] [Google Scholar]
- 25.Maxwell A, Gellert M. J Biol Chem. 1984;259:14472–14480. [PubMed] [Google Scholar]
- 26.Critchlow S E, Maxwell A. Biochemistry. 1996;35:7387–7393. doi: 10.1021/bi9603175. [DOI] [PubMed] [Google Scholar]
- 27.Lewis R J, Singh O M P, Smith C V, Skarynski T, Maxwell A, Wonacott A J, Wigley D B. EMBO J. 1996;15:1412–1420. [PMC free article] [PubMed] [Google Scholar]
- 28.Higgins N P, Cozzarelli N R. Nucleic Acids Res. 1982;10:6833–6847. doi: 10.1093/nar/10.21.6833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roca J, Berger J M, Wang J C. J Biol Chem. 1993;268:14250–14255. [PubMed] [Google Scholar]
- 30.Tornaletti S, Pedrini A M. Biochim Biophys Acta. 1988;949:279–287. doi: 10.1016/0167-4781(88)90153-4. [DOI] [PubMed] [Google Scholar]
- 31.Kreuzer K N, Cozzarelli N R. Cell. 1980;20:245–254. doi: 10.1016/0092-8674(80)90252-4. [DOI] [PubMed] [Google Scholar]
- 32.Gellert M, Fisher L M, O’Dea M H. Proc Natl Acad Sci USA. 1979;76:6289–6293. doi: 10.1073/pnas.76.12.6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cullis P M, Maxwell A, Weiner D P. Biochemistry. 1992;31:9642–9646. doi: 10.1021/bi00155a017. [DOI] [PubMed] [Google Scholar]
- 34.Bates A D, Maxwell A. EMBO J. 1989;8:1861–1866. doi: 10.1002/j.1460-2075.1989.tb03582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Westerhoff V H, O’Dea M H, Maxwell A, Gellert M. Cell Biophys. 1988;12:157–181. doi: 10.1007/BF02918357. [DOI] [PubMed] [Google Scholar]
- 36.Berger J M, Gamblin S J, Harrison S C, Wang J C. Nature (London) 1996;379:225–232. doi: 10.1038/379225a0. [DOI] [PubMed] [Google Scholar]
- 37.Roca J, Wang J C. Cell. 1994;77:609–616. doi: 10.1016/0092-8674(94)90222-4. [DOI] [PubMed] [Google Scholar]