

Abstract
The PI3 kinase (PI3K) family plays a complex role in cell biology and metabolism. Signaling through the PI3Ks is frequently activated in many human cancers, including glioblastoma, because of gain‐of‐function mutations in PIK3CA or loss of PTEN. Experiments involving genetic mouse models and small molecule inhibitors have helped to elucidate the roles of the regulatory and catalytic subunits of PI3K in metabolism and cancer. Downstream of PI3K is Akt, a critical effector of growth, proliferation and survival. The suggested dependence of glioblastoma tumors on PI3K signaling implies that PI3K inhibitors should lead to effective killing of these cancer cells, but that has been shown not to be the case. The engagement of other survival pathways in response to PI3K inhibition prompts the need to develop combination therapies that promote cytotoxicity in cancer cells.
Keywords: glioblastoma, PI3 kinase, kinase inhibitors, Akt, metabolism, apoptosis, combination therapy
INTRODUCTION
The phosphatidylinositol‐3′kinase (PI3K) family plays complex and extensive roles in essentially all aspects of cell biology and metabolism (63). Signaling through the PI3Ks is central to cell survival, growth and proliferation and, as a consequence, is frequently activated in many human cancers. PI3K is regulated in part by growth factors that signal as ligands to receptor tyrosine kinases (RTKs). Through coupling to adaptor proteins, PI3K binds to phosphotyrosine residues on activated RTKs, linking PI3K to its substrates. The PI3Ks are lipid kinases. Activated PI3Ks phosphorylate the lipid phosphatidylinositol (4,5)‐bisphosphate (PIP2) to generate phosphatidylinositol(3,4,5)‐trisphosphate (PIP3). A number of cancers show gain‐of‐function mutations in the PI3K catalytic gene PIK3CA, leading to constitutive kinase activity. Independently of upstream regulators, PI3Ks can also be activated through mutation of the lipid phosphatase PTEN. Loss of PTEN function leads to failure to convert PIP3 back to PIP2, resulting in deregulation of PI3K in the absence of upstream signals from RTKs. One of the most frequently mutated tumor suppressor genes in human cancer, PTEN, shows a very high frequency of mutations, estimated at 50%–80% in sporadic tumors (including glioblastoma and prostate cancer), and 30%–50% in breast, colon and lung tumors (52).
Once activated by PI3K, PIP3 recruits the plekstrin‐homology (PH) domain protein Akt to the cell membrane where it is phosphorylated and regulates a complex and growing number of downstream targets and pathways (41). The Akt family of kinases is a critical regulator of cell growth and survival. In fact, activating mutations in the RTK–PI3K axis occur in over 80% of glioblastomas (60). However, Akt kinases have been challenging to inhibit pharmacologically. As such, PI3K is an attractive target for small molecule inhibitor therapy, with the expectation that blocking the activity of this lipid kinase and downstream survival kinase targets should lead to effective killing of cancer cells dependent on these pathways.
DISCOVERY AND CLASSIFICATION OF PI3 KINASES
Phosphatidylinositol kinase activity was first discovered in the purified Rous sarcoma virus transforming gene product, pp60v‐src, which had the ability to form mono‐ and diphosphorylated derivatives of phosphatidylinositol, as well as phosphorylating diacylglycerol to form phosphatidic acid (57). PI kinase activity was also found in immunoprecipitates containing polyoma middle‐T antigen, due to its association with pp60c‐src, the cellular homologue of pp60v‐src (67). This lipid kinase activity was reduced in polyoma mutants defective in transformation, suggesting a role for phosphatidylinositol metabolism in transformation by the polyoma virus (31).
In both normal and transformed murine fibroblasts, distinct type I and II PIKs were found. Type I specifically associated with middle‐T/pp60c‐src and also co‐precipitated with partially purified PDGF (platelet‐derived growth factor) receptor (68). An increase in PIK activity was common to both growth factor stimulation and oncogenic transformation. An 85 kDa phosphoprotein also appeared in immunoprecipitates from both type I and type II PIKs correlating with the increase in PIK activity (32). Type II PIKs (later classified into type II and type III) phosphorylate the 4‐OH position of the inositol ring on phosphatidylinositol to produce phosphatidylinositol 4‐monophosphate. Although these kinases associate with growth factor receptors, their roles in both normal cell biology and in cancer are still being clarified. In contrast, type I PIK was characterized to phosphorylate the 3′‐hydroxyl group on the inositol ring of PIP2 to generate a new phospholipid, PIP3. Thus, the enzyme was named PI3K (69). PI3K was eventually purified in rat liver and found to consist of a complex of an 85 kDa and 110 kDa protein with the ability to phosphorylate phosphatidylinositol (PI), phosphatidylinositol 4‐phosphate (PI(4)P) and PIP2 (7).
CLASSIFICATION AND SUBUNIT CHARACTERIZATION WITHIN THE PI3K FAMILY
In mammals, the PI3K family (now distinct from the PIK family) is also divided into different classes based on specific lipid substrates, structure and mode of regulation. Of particular importance to this review because of their roles in cancer, the class I PI3Ks, phosphorylate PI, PI(4)P and PIP2 in vitro, preferring PIP2 in cells, generating PIP3. Activated by RTKs, these enzymes consist of a 110 kDa catalytic subunit (p110) and an adaptor/regulatory subunit complexed as a heterodimer. Class I PI3Ks are further subdivided into class IA and IB. Mammals have three isoforms of the class IA catalytic subunit (alpha, beta and delta). The regulatory subunits, collectively known as p85s, are generated by three genes, PIK3R1, PIK3R2 and PIK3R3, which encode the p85alpha, p85beta and p55gamma isoforms, respectively. The PIK3R1 gene also encodes two shorter isoforms, p55alpha and p50alpha (12). P110beta has also been found to be stimulated by G‐protein‐coupled receptors (GPCR) (45). While the alpha and beta isoforms are found in most tissues, the delta isoform is mainly found in leukocytes. Class IB PI3Ks consist of a single catalytic isoform, p110gamma, complexed with a 101‐kDa regulatory protein (p101). They are exclusively activated by heterotrimeric G‐proteins and are abundant in white blood cells (64) (Figure 1). The structure and function of the other classes of PI3Ks have been discussed in recent reviews 1, 42; therefore, our discussion will be limited to the class IA PI3Ks.
Figure 1.
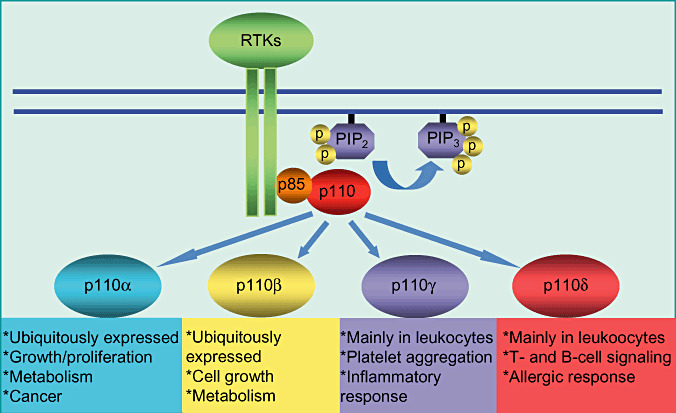
The roles of P110 isoforms. The isoforms of the class I p110 catalytic subunit serve different cellular functions. Of the four, p110 alpha has been most widely implicated in human cancers, in addition to its role in cell proliferation and metabolism. P110 beta has also been shown to have a role in growth and metabolism, while p110 gamma and p110 delta have immune functions. RTK = receptor tyrosine kinase; PIP2 = phosphatidylinositol (4,5)‐bisphosphate; PIP3 = phosphatidylinositol(3,4,5)‐trisphosphate.
Class IA PI3Ks are regulated through the inhibitory action of the p85 subunit on the p110 catalytic subunit. P85 subunits contain two Src homology 2 (SH2) domains. The region between these two domains binds to the catalytic subunit. In resting cells, p85 stabilizes the overall confirmation of p110 and protects it from thermal inactivation in vitro (70). P85‐p110 heterodimers are activated when the p85 SH2 domains bind to phosphorylated tyrosine residues on activated tyrosine kinase receptors, resulting in a conformational change in p85 that releases inhibition of p110. As a result, cytosolic PI3Ks are translocated to the cell membrane where they are brought into the proximity of their lipid substrates. In the case of class IA PI3Ks, the active p110 catalytic subunit catalyzes the conversion of PIP2 to PIP3. This reaction is regulated negatively by PTEN, which converts PIP3 back to PIP2 (Figure 2).
Figure 2.

Activation of class I PI3Ks. Class I PI3 kinases are activated by upstream signals from receptor tyrosine kinases (RTKs). Activated PI3 kinase catalyzes the production of the second messenger (phosphatidylinositol(3,4,5)‐trisphosphate) PIP3 from phosphatidylinositol (4,5)‐bisphosphate (PIP2), which recruits the Akt protein kinase to the membrane. The phosphatase and tensin homolog deleted from chromosome 10 (PTEN) acts as a critical negative regulator of PI3K signaling by removing the D3 phosphate from PIP3 to produce PIP2.
PI3 KINASES AS EXECUTORS OF RTK SIGNALING: LESSONS FROM MOUSE GENETICS
PI3Ks exist as heterodimers of catalytic and regulatory subunits, with genetic complexity in each family allowing the formation of multiple different heterodimers. Perhaps not surprisingly therefore, interpreting the phenotypes of mice deleted for specific subunits has been challenging because the potential for compensatory heterodimerization of the unmanipulated p110 catalytic subunits and their regulatory partners, the p85, p55 and p50 proteins. So what have we learned from mouse models of PI3K signaling? Insulin receptor signaling is a fundamental RTK and PI3K‐driven cellular process and remains the best studied system in which to study the impact of PI3K signaling. The first surprise was that mice deleted either homozygously or heterozygously for p85 subunits showed increased insulin signaling. In addition, the p110 catalytic subunit was found to have a critical role in insulin signaling.
Studies involving the impairment of p85 subunits demonstrate the difficulty in defining the roles of specific PI3K subunits, as deleting p85 causes changes in the activity of other regulatory subunits or the catalytic subunits. For example, mice homozygous for the deletion of p85alpha (PIK3R1) alone showed increased insulin sensitivity and hypoglycemia which correlated with up‐regulation of the p55alpha and p50alpha regulatory subunits (59). Loss of all isoforms of PIK3R1 (including p50alpha and p55alpha) resulted in perinatal lethality and caused a decrease in the expression and activity of class IA PI3K catalytic subunits. Outbreeding to different strains generated PIK3R1 mutant mice on mixed backgrounds. Some of these outbred mice, when homozygous for deletion of all PIK3R1 isoforms, could survive 3–7 weeks, manifesting hypoglycemia and increased glucose tolerance (17). Heterozygous disruption of PIK3R1 improved insulin signaling and glucose homeostasis, while PI3K activation in response to insulin stimulation remained normal (43). P85beta knockout mice showed hypoinsulinemia, hypoglycemia and improved insulin sensitivity, with preserved PI3K activity despite the reduction in the level of regulatory subunits (61).
Controversy exists in explaining why deletion of these PI3K regulatory subunits resulted in improvement in insulin signaling. Enhanced PI3K signaling in insulin‐responsive cells was at first attributed to the pool of unbound p85 in cells that competed with p85/p110 complexes for binding to phosphotyrosine sites. The p85 knockout mice were therefore thought to preferentially reduce this inhibitory population of free p85 monomers, resulting in enhanced signaling by the remaining p110:p85 heterodimers (38). More recently, however, monomeric p85 has been shown to be unstable (5), loss of p110alpha or beta was shown to reduce overall p85 levels 5, 71, and introduction of free p85alpha in pan‐p85alpha knockout adipocytes did not increase PI3K activity (62). Recently, Geering and coworkers used affinity and ion exchange chromatography and quantitative mass spectrometry to demonstrate that p85 and p110 were present in equimolar amounts in mouse cell lines and existed as obligate heterodimers, finding no evidence for the presence of free p85 or p110 subunits (18). Thus, the free p85 hypothesis, while providing a reasonable explanation for the observation that p85 knockout mice show enhanced signaling through the insulin receptor, does not provide a compelling explanation for the unexpected phenotype of these mice.
REFINING THE GENETIC INTERROGATION: KNOCKIN, TISSUE SPECIFIC DELETION AND CHEMICAL GENETICS
Initial experiments in mice with homozygous deletions of either p110alpha or p110beta were unrevealing as these genetic alterations were found to be embryonic lethal 2, 3. As deletion of PI3K genes in mice often altered the expression of nontargeted PIK proteins, many subsequent experiments to elucidate the function of p110 isoforms were employed using small molecule inhibitors, kinase‐dead knockin mutations and tissue‐specific inactivation of PI3K genes. Knockin mice that expressed kinase‐dead proteins allow preservation of signaling complex stoichiometry. Similarly, small molecule inhibitors make it possible to interrogate the consequences of inhibiting kinase activity without altering protein levels. Inhibiting the p110alpha subunit of PI3K genetically in mouse models and pharmacologically in cells and in mice led to the same result—that p110alpha plays a critical role in insulin signaling 16, 33.
Mice heterozygous for a knockin mutation expressing a kinase‐dead p110alpha protein had 50% loss of p110alpha function, with levels of p110alpha, beta, delta and p85 subunits that were similar to wild type. These mice exhibited growth retardation, and fibroblasts derived from mutant animals showed impaired insulin‐like growth factor (IGF)‐1‐stimulated proliferation. Although levels of insulin receptors and recruitment of p85 to insulin receptor substrate (IRS) complexes were unaffected by inactivation of p110alpha, insulin‐stimulated PI3K activity and phosphorylation of Akt were both reduced. These results suggested a role for p110alpha in both insulin sensitivity and glucose metabolism because of reduced insulin‐stimulated p110alpha activity and diminished activation of downstream effector pathways (16).
Using small molecule inhibitors of class IA PI3Ks, Knight and coworkers studied the effects of PI3K inhibitors in adipocytes and myotubes. Inhibitors of p110alpha blocked insulin‐stimulated phosphorylation of Akt, while inhibitors of p110beta had no effect on insulin‐stimulated phosphorylation. In addition, inhibitors of p110alpha blocked insulin‐stimulated glucose uptake in adipocytes, with inhibitors of p110beta again having no effect on glucose transport. These results were aligned with the mouse studies above, both suggesting p110alpha as the primary PI3K required for insulin signaling (33).
A ROLE FOR P110BETA IN RTK SIGNALING: FINE‐TUNING OF AKT REGULATION
Studies reviewed in the preceding paragraphs showed that p110alpha and p110beta were activated in response to insulin receptor stimulation; however, only p110alpha was needed to regulate glucose transport. What then is the consequence of p110beta activation or blockade? Two recent studies show that the p110beta subunit of PI3K also has a role in glucose metabolism. In conditional knockout mice carrying a conditional PIK3CB allele, liver‐specific deletion of p110beta led to impaired insulin sensitivity and glucose homeostasis, with no significant change in Akt phosphorylation. Mouse embryonic fibroblasts (MEFs) obtained from embryos with a p110beta deletion also showed no effect on Akt phosphorylation in response to stimulation by IGF, epidermal growth factor (EGF) or PDGF, suggesting a kinase‐independent role for p110beta in insulin metabolism. However, a decrease in pAkt observed in response to lysophosphatidic acid (LPA) was restored by wild‐type p110beta, suggesting a catalytic function of p110beta in LPA signaling and supporting p110beta's role in GPCR signaling (27). These data were consistent with the previous use of small molecule inhibitors of p110beta which did not affect insulin‐stimulated phosphorylation of proteins in the PI3K pathway, while Akt phosphorylation induced by LPA was inhibited (33).
Using a different approach, Ciraolo et al generated mice carrying a kinase‐dead p110beta mutant and found homozygous mice developed mild insulin resistance detectable from 6 months of age. Comparison of wild‐type MEFs and MEFs homozygous for the inactive p110beta kinase revealed that the catalytic function of beta was not required for Akt activation shortly after growth factor stimulation but was required for signaling via GPCRs. While p110alpha was crucial for short‐term insulin receptor signaling, p110beta was required to sustain long‐term insulin signaling, as insulin‐stimulated activation of Akt declined rapidly in the absence of p110beta catalytic activity. This result was also confirmed pharmacologically using a small molecule p110beta inhibitor (10).
PI3K IN ONCOGENIC TRANSFORMATION AND IN CANCER THERAPY
In addition to their roles in insulin signaling, both p110alpha and p110beta are also critical to oncogenic transformation. In one publication, MEFs were derived from the embryos of mice containing a conditional inactivating mutation of the PIK3CA gene (71). Inactivation of p110alpha was then driven in vitro using Cre‐loxP mediated recombination, and the MEFs were immortalized by stably introducing dominant negative p53. Ablation of p110alpha in these cells impeded oncogenic transformation induced by IGF‐1, insulin or EGF stimulation (71). The role of p110beta in oncogenic transformation was explored in mice with conditional and prostate‐specific mutations in PTEN and p110beta. In prostatic epithelium, ablation of p110beta, and not p110alpha, blocked tumorigenesis caused by loss of PTEN with an accompanying diminution of pAkt levels, suggesting a role for p110beta catalytic activity in tumorigenesis (27). In addition, the role of p110beta was further explored in mammary gland cancer, where a critical role was found for p110beta catalytic activity in ERBB2‐driven tumor growth (10).
PI3Ks represent an attractive therapeutic target based on their prevalence of activation in many human cancer types. Mutations in PTEN result in activation of PI3K, and are detected at considerable frequencies in glioblastoma, prostate and breast cancer 35, 50, 65. A high frequency of mutations in PIK3CA, the gene encoding the p110alpha subunit of PI3K, was found in colorectal, gastric, breast and lung cancers, although more rarely in brain cancers such as glioblastoma 22, 34, 44, 53, 60. The most common of the PI3K activating mutations are represented by single amino acid substitutions in the helical or kinase domains of these proteins. Expression of both types of mutants in chicken embryo fibroblasts induced oncogenic transformation with high efficiency, which correlated with enhanced lipid kinase activity and constitutive activation of Akt signaling (29). Recent studies show that gain of function induced by helical domain mutations is independent of binding to p85 but requires interaction with Ras, a small GTPase that can also activate PI3K. In contrast, the kinase domain mutations are active in the absence of Ras binding but show critical dependence on interaction with p85 (72).
Downstream of PI3K, Akt is a critical effector of growth, proliferation and survival (Figure 3). Upon activation of PI3K and the generation of PIP3, Akt is recruited to the cell membrane via a PH domain, where it is phosphorylated on Thr308 and Ser473. Akt activation is of interest in cancer because of its effects on growth, survival and proliferation, processes that enhance cancer cell growth and blocking these processes would presumably inhibit cancer cell growth.
Figure 3.

Downstream of PI3 kinase. Phosphorylation of Akt by PDK1 and a feedback loop involving the mammalian target of rapamycin (mTOR)‐rictor complex resulting in the activation of Akt leads to the phosphorylation of multiple downstream substrates that regulate various cellular processes. RTK = receptor tyrosine kinase; GSK = glycogen synthase kinase; TSC = tuberous sclerosis complex; PIP2 = phosphatidylinositol (4,5)‐bisphosphate; PIP3 = phosphatidylinositol(3,4,5)‐trisphosphate.
An abundance of substrates exist for Akt. Among the key targets is glycogen synthase kinase‐3 (GSK‐3), a protein kinase that is inhibited by Akt phosphorylation. GSK‐3 drives phosphorylation of cell cycle regulators such as c‐Myc, cyclin D1 and cyclin E, orchestrating their degradation in the proteosome, and thereby preventing progression through the cell cycle. Akt phosphorylation and subsequent inactivation of GSK‐3 stabilizes these cell cycle regulatory proteins and drives proliferation (55). PI3K and Akt are also involved in the activation of mammalian target of rapamycin (mTOR), a protein kinase with homology to PI3Ks. mTOR plays a central role in regulating protein synthesis and cell growth in response to the availability of nutrients and growth factors, and also feeds back as a negative regulator of PI3K signaling. Consistent with the critical roles of PI3K and Akt in regulating growth and proliferation, a screen of glioma cell lines using a panel of small molecule PI3K inhibitors showed that inhibitors of the p110alpha subunit were effective in blocking proliferation (13). A dual inhibitor of p110alpha and mTOR was found to be more effective than any individual inhibitor of p110alpha or mTOR in inducing proliferative arrest in glioma cell lines, an activity traced in part to interruption of the feedback loop between mTOR and PI3K.
Amplification of epidermal growth factor receptor (EGFR) is common in glioblastoma leading to consequent activation of PI3K. Loss of PTEN or mutation in PIK3CA leads to persistent activation of the PI3K signaling pathway in the absence of upstream stimulation. Activation of PI3K correlates with decreased apoptosis in tumor samples (8), suggesting a critical dependence on PI3K. While the efficacy of EGFR inhibitors has been disappointing in glioma, patients with low levels of phosphorylated Akt showed better responses than those with high levels (20), supporting the idea that survival of glioma cells is dependent on aberrant PI3K signaling. In further support of the importance of EGFR and PI3K as targets for glioma therapy, a dual p110alpha/mTOR inhibitor was shown to cooperate with inhibition of EGFR to further block proliferation of EGFR‐driven, PTEN‐mutant glioma cells (15). Importantly however, proliferative arrest in response to inhibition of EGFR, PI3K and mTOR (either individually or in combination) occurred in the absence of appreciable apoptosis 14, 15.
DO GLIOBLASTOMA TUMORS SHOW A CRITICAL DEPENDENCE ON PI3K SIGNALING?
The fact that PI3K inhibitors do not induce cell death is surprising considering Akt's role in cell survival. Akt directly phosphorylates several members of the apoptotic pathway. Bad is a proapoptotic member of the Bcl‐2 family that promotes cell death by binding with anti‐apoptotic Bcl‐xL. Phosphorylation of Bad by Akt prevents this interaction with Bcl‐xL, allowing the anti‐apoptotic protein to promote cell survival. Akt phosphorylation also inhibits the catalytic activity of caspase‐9, an initiator caspase that cleaves and activates downstream effector caspases. Phosphorylation of Forkhead Box O transcription factors by Akt also prevents their nuclear translocation, inhibiting the translation of target genes such as Bim and Fas ligand, two proapoptotic proteins. Akt also indirectly influences two regulators of cell death, NF‐kappaB and p53 (47).
So how does the complex interplay between PI3K, Akt and mTOR play into cancer? Activation of PI3K occurs commonly in cancers including glioblastoma, the most common primary brain tumor. Activation of PI3K is associated with increased metabolism, suggesting potential dependence of cancer cells on PI3K signaling, and raising the possibility that blockade of PI3K signaling in glioma should effectively kill these cells. Is there evidence that malignancies that show frequent activation of PI3K are particularly dependent on aberrant signaling through lipid kinases, suggesting unique vulnerability?
Cancer cells contain multiple genetic and epigenetic mutations that impact gene expression. Recent studies in glioblastoma suggest an average of ∼60 mutations in most of these cancers, suggesting a daunting degree of genetic complexity 48, 60. Yet despite this fact, it has been shown for some cancers that reversing only one of these changes is enough to inhibit cancer cell growth, a phenomenon coined “oncogene addiction”23, 66. The concept of oncogene addiction gives a label to a phenomenological observation, without providing true mechanistic insight. The concept stems from the idea that cancers consist of critical regulators (driver mutations) along with less central passenger mutations that are present but that may not be critical to malignant progression. Cancer may be exclusively dependent on certain driver mutations to maintain their malignant phenotype. In human cancers, a number of drugs that target individual oncogenes have been shown to have therapeutic efficacy—e.g. BCR/ABL inhibitors in chronic myeloid leukemia (25), Herceptin in HER2‐driven breast cancers (56), and EGFR inhibitors in a subset of EGFR‐mutant non‐small cell lung cancers (39). A strong selective pressure for cells to develop de novo mutations in these respective oncogenes further suggests a central role for these mutations as driver mutations. The observation that therapies targeting these mutations are highly effective suggests a critical dependence of cancer cells on specific driver oncogenes or on oncogenic pathways regulated by these drivers.
THE WARBURG EFFECT IN GLIOBLASTOMA, FURTHER EVIDENCE FOR DEPENDENCE ON PI3K SIGNALING
A hallmark of cancer cells is the preferential metabolism of glucose via anaerobic glycolysis over oxidative phosphorylation even in the presence of oxygen, a phenomenon first described by Otto Warburg and now known as the Warburg effect. This shift to glycolytic metabolism allows use of fluorodeoxyglucose positron emission tomography imaging to identify tissues with a high rate of glucose uptake. As previously mentioned, PI3K plays a key role in insulin signaling and glucose metabolism. Elstrom and coworkers showed that Akt directly influences glucose metabolism in transformed cells and that Akt activation renders cancer cells dependent on glucose availability for survival (11). In human glioblastoma cells, the rate of glucose metabolism correlated with the activity of Akt and Akt‐expressing cells were more susceptible to death after glucose withdrawal (11). Oxidative stress was identified as the cause of selective susceptibility to glucose withdrawal‐induced apoptosis in malignant cells (26). Activation of AMP‐activated protein kinase suppresses anabolic and stimulates catabolic pathways in non‐transformed cells to maintain the energy balance within cells during times of stress (21). However, cancer cells cannot adapt to glucose deprivation by metabolizing other carbon sources. As shown in Akt‐expressing glioblastoma cells, fatty acid oxidation was impaired in response to glucose withdrawal, causing the cells to die. However, pharmacological stimulation of fatty acid oxidation with 5‐aminoimidazole‐4‐carboxamide ribonucleoside was sufficient to maintain cell survival, suggesting that Akt activation blocked the ability of cancer cells to metabolize non‐glucose substrates, leading to addiction (6).
Akt helps to regulate glycolysis by regulating localization of the glucose transporter, Glut1, to the cell surface and stimulating hexokinase activity, an enzyme in the glycolytic pathway. Normally, after withdrawal of growth factors, cytochrome c release is triggered from the mitochondria and the caspase cascade is activated (36). The proapoptotic protein Bax shows a conformation that is sensitive to glucose metabolism, and increased glycolysis prevents activation of Bax. Thus, in cancer cells with activated PI3K/Akt signaling, glucose dependence allows the cells to avoid apoptosis in the face of growth factor withdrawal (51). As such, we would expect that inhibiting PI3K and thus Akt would prevent glucose‐addicted cancer cells from performing glycolysis, and Bax activation and apoptosis could resume.
WHY PI3K INHIBITORS DO NOT LEAD TO APOPTOSIS IN GLIOMA
Despite the prominence and suggested dependence on PI3K signaling in glioma, PI3K inhibitors have been shown to be cytostatic rather than cytotoxic in glioma cell lines and xenografts (14) even when used in combination with EGFR and mTOR inhibitors (15). NVP‐BEZ235, a dual PI3K/mTOR inhibitor currently in phase I clinical trials, also possesses strong anti‐proliferative activity with no obvious decline in cell viability (40). A possible explanation is that PI3K inhibition is inducing other processes and pathways that promote cell survival. An obvious way to circumvent this problem is to combine inhibitors of PI3K with inhibitors of other pathways that augment apoptosis in this setting.
What are the possible pathways that might promote survival in the setting of PI3K and PI3K/mTOR blockade? Autophagy is a cellular process that allows cells to survive periods of catabolic stress by cannibalizing themselves to preserve adenosine triphosphate (ATP) stores. During autophagy, cells degrade their own proteins and organelles, through formation of a double‐membraned vesicle (autophagosome) that fuses with a lysosome, leading to the degradation of the vesicle contents. Because cells can die despite inducing autophagy, this process has also been proposed as a mechanism of caspase‐independent cell death. However, induction of autophagy in response to changing environmental conditions, such as nutrient deprivation, allows the cell to recycle proteins and organelles as a source of new macromolecules.
Under conditions of nutrient deprivation, autophagy can suppress cell death. Cells at the center of tumors are metabolically stressed in this manner and these regions utilize autophagy to support tumor cell survival (28). While many anticancer drugs have been shown to induce autophagy and eventually lead to cell death, it is unclear whether autophagy directly causes cell death, or serves as a mechanism for cell survival before the cells die because of some alternative mechanism (24).
In HeLa cells, inhibiting autophagy in the setting of growth factor withdrawal caused cells to die, suggesting a protective role for autophagy (4). In addition, in cells lacking the intrinsic apoptotic pathway, growth factor withdrawal induced autophagosome formation and inhibition of this process led to cell death, again suggesting that autophagy enabled cells to prolong their survival (37). Hippert and coworkers propose that in the case of tumors, autophagy may promote tumor cell survival in the setting of limited angiogenesis, nutrient deprivation and hypoxia (24). Because the p110alpha isoform of PI3K is essential to angiogenesis (19), PI3K inhibitors should be able to inhibit angiogenesis and promote tumor cell death. However, if tumor cells utilize autophagy as a survival mechanism, then cells treated with PI3K inhibitors might be able to circumvent the expected cytotoxicity induced by this class of agents. Whether blockade of autophagy will augment cytotoxicity in the setting of PI3K or PI3K/mTOR blockade remains an important area for future investigation.
COMBINATION THERAPIES WITH PI3K INHIBITORS TO PROMOTE APOPTOSIS IN GLIOMA
Prosurvival signaling by PI3K contributes to therapeutic resistance in the setting of established anticancer therapies. Therefore, combining cytotoxic or radiation therapy with inhibitors of PI3K represents an alternative approach to enhance the cytotoxicity of either monotherapy in glioma. As an example, activation of the PI3K pathway plays a role in radioresistance in glioma (54). Several studies have shown that PI3K inhibition sensitizes glioma cells to radiation therapy. That induction of PTEN or treatment with a PI3K inhibitor can improve the efficacy of radiation therapy is attributed to down‐regulation of PI3K signaling, which blocks the cell's ability to repair DNA damage following ionizing radiation (30). Class I PI3K inhibitors in particular enhance the effects of radiotherapy in cancer. The dual PI3Kalpha/mTOR inhibitor, PI‐103, enhanced radiosensitivity in various human cancer lines (49). The additive effects of PI3K inhibition and radiation were most prominent in glioma cell lines with mutant PTEN. In this setting, inhibitors of p110alpha specifically potentiated the effects of radiation, whereas inhibitors of p110beta did not (9).
PI3K inhibition has also been shown to sensitize cells to chemically induced cell death. Apoptotic cell death proceeds through one of two pathways: the extrinsic—death ligand/receptor‐mediated pathway, and the intrinsic—mitochondrial‐mediated pathway (58). Sensitivity of glioma cells to apoptosis induced through either of these pathways was shown to be enhanced by PI3K inhibition. Extrinsic apoptosis induced by stimulation of the tumor necrosis factor receptor family, such as tumor necrosis factor‐related apoptosis‐inducing ligand (TRAIL) and CD95, was synergistically enhanced by broad‐spectrum PI3K inhibitors, LY294002 and wortmannin. LY294002 also synergized with several chemotherapeutic drugs, such as doxorubicin and etoposide, to trigger activation of the mitochondrial apoptotic pathway (46). Together, these results suggest that PI3K inhibition can be used in concert with traditional therapies that have been ineffective towards glioblastoma to restore the cell's ability to undergo apoptosis.
FUTURE DIRECTIONS IN DEVELOPING CYTOTOXIC THERAPIES
In the war against cancer, it is reasonable to find strategies that prevent cancer growth and proliferation. However, the reservoir of malignant cells remaining after such approaches raises risk of recurrence. As recurrent cancer is generally associated with therapy‐resistance, it is critical to identify therapeutic approaches that actively trigger cell death. One potential strategy is to identify those subsets of specific cancers that are truly dependent on PI3K signaling for survival, with the expectation that PI3K blockade in this setting would induce cell death. An alterative approach is to inhibit autophagy in addition to PI3K. If autophagy is indeed serving as a survival mechanism to oppose blockade of other survival pathways in glioma, combination therapy targeting both PI3K and critical targets regulating the autophagic process could render cancer cells defenseless to these multiple assaults, resulting in cell death. A complementary focus is to find additional signaling or survival pathways to which glioma cells are critically dependent. Finally, therapeutic index is an issue that must be addressed when developing cancer therapies. Broad‐spectrum PI3K inhibitors, such as LY294002, are unsuitable for patient use because of their toxicity, while the isoform‐specific inhibitors previously mentioned are generally cytostatic rather than cytotoxic. As promise has been shown using combination therapies of PI3K inhibitors with other cytotoxic treatments, the challenge for the immediate future is to determine effective combinations of targets that yield cytotoxicity specifically in cancer cells without causing general toxicity.
ACKNOWLEDGMENTS
We are grateful to Theo Nicolaides for the critical review of this manuscript. We acknowledge support from the NIH, Burroughs Wellcome Fund, American Brain Tumor Association, The Brain Tumor Society; Alex's Lemonade Stand, Children's National Brain Tumor, Wallace H. Coulter, Katie Dougherty, Pediatric Brain Tumor, Samuel G. Waxman and V Foundations and a UC‐Genentech Discovery Grant.
REFERENCES
- 1. Andrews S, Stephens LR, Hawkins PT (2007) PI3K class IB pathway. Sci STKE 2007:cm2. [DOI] [PubMed] [Google Scholar]
- 2. Bi L, Okabe I, Bernard DJ, Wynshaw‐Boris A, Nussbaum RL (1999) Proliferative defect and embryonic lethality in mice homozygous for a deletion in the p110alpha subunit of phosphoinositide 3‐kinase. J Biol Chem 274:10963–10968. [DOI] [PubMed] [Google Scholar]
- 3. Bi L, Okabe I, Bernard DJ, Nussbaum RL (2002) Early embryonic lethality in mice deficient in the p110beta catalytic subunit of PI 3‐kinase. Mamm Genome 13:169–172. [DOI] [PubMed] [Google Scholar]
- 4. Boya P, Gonzalez‐Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N et al (2005) Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 25:1025–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brachmann SM, Ueki K, Engelman JA, Kahn RC, Cantley LC (2005) Phosphoinositide 3‐kinase catalytic subunit deletion and regulatory subunit deletion have opposite effects on insulin sensitivity in mice. Mol Cell Biol 25:1596–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Buzzai M, Bauer DE, Jones RG, Deberardinis RJ, Hatzivassiliou G, Elstrom RL, Thompson CB (2005) The glucose dependence of Akt‐transformed cells can be reversed by pharmacologic activation of fatty acid beta‐oxidation. Oncogene 24:4165–4173. [DOI] [PubMed] [Google Scholar]
- 7. Carpenter CL, Duckworth BC, Auger KR, Cohen B, Schaffhausen BS, Cantley LC (1990) Purification and characterization of phosphoinositide 3‐kinase from rat liver. J Biol Chem 265:19704–19711. [PubMed] [Google Scholar]
- 8. Chakravarti A, Zhai G, Suzuki Y, Sarkesh S, Black PM, Muzikansky A, Loeffler JS (2004) The prognostic significance of phosphatidylinositol 3‐kinase pathway activation in human gliomas. J Clin Oncol 22:1926–1933. [DOI] [PubMed] [Google Scholar]
- 9. Chen JS, Zhou LJ, Entin‐Meer M, Yang X, Donker M, Knight ZA et al (2008) Characterization of structurally distinct, isoform‐selective phosphoinositide 3′‐kinase inhibitors in combination with radiation in the treatment of glioblastoma. Mol Cancer Ther 7:841–850. [DOI] [PubMed] [Google Scholar]
- 10. Ciraolo E, Iezzi M, Marone R, Marengo S, Curcio C, Costa C et al (2008) Phosphoinositide 3‐kinase p110beta activity: key role in metabolism and mammary gland cancer but not development. Sci Signal 1:ra3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR et al (2004) Akt stimulates aerobic glycolysis in cancer cells. Cancer Res 64:3892–3899. [DOI] [PubMed] [Google Scholar]
- 12. Engelman JA, Luo J, Cantley LC (2006) The evolution of phosphatidylinositol 3‐kinases as regulators of growth and metabolism. Nat Rev Genet 7:606–619. [DOI] [PubMed] [Google Scholar]
- 13. Fan QW Weiss WA (2006) Isoform specific inhibitors of PI3 kinase in glioma. Cell Cycle 5:2301–2305. [DOI] [PubMed] [Google Scholar]
- 14. Fan QW, Knight ZA, Goldenberg DD, Yu W, Mostov KE, Stokoe D et al (2006) A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell 9:341–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fan QW, Cheng CK, Nicolaides TP, Hackett CS, Knight ZA, Shokat KM, Weiss WA (2007) A dual phosphoinositide‐3‐kinase alpha/mTOR inhibitor cooperates with blockade of epidermal growth factor receptor in PTEN‐mutant glioma. Cancer Res 67:7960–7965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Foukas LC, Claret M, Pearce W, Okkenhaug K, Meek S, Peskett E et al (2006) Critical role for the p110alpha phosphoinositide‐3‐OH kinase in growth and metabolic regulation. Nature 441:366–370. [DOI] [PubMed] [Google Scholar]
- 17. Fruman DA, Mauvais‐Jarvis F, Pollard DA, Yballe CM, Brazil D, Bronson RT et al (2000) Hypoglycaemia, liver necrosis and perinatal death in mice lacking all isoforms of phosphoinositide 3‐kinase p85 alpha. Nat Genet 26:379–382. [DOI] [PubMed] [Google Scholar]
- 18. Geering B, Cutillas PR, Nock G, Gharbi SI, Vanhaesebroeck B (2007) Class IA phosphoinositide 3‐kinases are obligate p85‐p110 heterodimers. Proc Natl Acad Sci USA 104:7809–7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Graupera M, Guillermet‐Guibert J, Foukas LC, Phng LK, Cain RJ, Salpekar A et al (2008) Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature 453:662–666. [DOI] [PubMed] [Google Scholar]
- 20. Haas‐Kogan DA, Prados MD, Tihan T, Eberhard DA, Jelluma N, Arvold ND et al (2005) Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J Natl Cancer Inst 97:880–887. [DOI] [PubMed] [Google Scholar]
- 21. Hardie DG (2003) Minireview: the AMP‐activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology 144:5179–5183. [DOI] [PubMed] [Google Scholar]
- 22. Hartmann C, Bartels G, Gehlhaar C, Holtkamp N, Von Deimling A (2005) PIK3CA mutations in glioblastoma multiforme. Acta Neuropathol 109:639–642. [DOI] [PubMed] [Google Scholar]
- 23. Hingorani SR, Tuveson DA (2003) Targeting oncogene dependence and resistance. Cancer Cell 3:414–417. [DOI] [PubMed] [Google Scholar]
- 24. Hippert MM, O'Toole PS, Thorburn A (2006) Autophagy in cancer: good, bad, or both? Cancer Res 66:9349–9351. [DOI] [PubMed] [Google Scholar]
- 25. Hughes TP, Kaeda J, Branford S, Rudzki Z, Hochhaus A, Hensley ML et al (2003) Frequency of major molecular responses to imatinib or interferon alfa plus cytarabine in newly diagnosed chronic myeloid leukemia. N Engl J Med 349:1423–1432. [DOI] [PubMed] [Google Scholar]
- 26. Jelluma N, Yang X, Stokoe D, Evan GI, Dansen TB, Haas‐Kogan DA (2006) Glucose withdrawal induces oxidative stress followed by apoptosis in glioblastoma cells but not in normal human astrocytes. Mol Cancer Res 4:319–330. [DOI] [PubMed] [Google Scholar]
- 27. Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH et al (2008) Essential roles of PI(3)K‐p110beta in cell growth, metabolism and tumorigenesis. Nature 454:776–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jin S, White E (2008) Tumor suppression by autophagy through the management of metabolic stress. Autophagy 4:563–566. [PMC free article] [PubMed] [Google Scholar]
- 29. Kang S, Bader AG, Vogt PK (2005) Phosphatidylinositol 3‐kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci USA 102:802–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kao GD, Jiang Z, Fernandes AM, Gupta AK, Maity A (2007) Inhibition of phosphatidylinositol‐3‐OH kinase/Akt signaling impairs DNA repair in glioblastoma cells following ionizing radiation. J Biol Chem 282:21206–21212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kaplan DR, Whitman M, Schaffhausen B, Raptis L, Garcea RL, Pallas D et al (1986) Phosphatidylinositol metabolism and polyoma‐mediated transformation. Proc Natl Acad Sci USA 83:3624–3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kaplan DR, Whitman M, Schaffhausen B, Pallas DC, White M, Cantley L, Roberts TM (1987) Common elements in growth factor stimulation and oncogenic transformation: 85 kd phosphoprotein and phosphatidylinositol kinase activity. Cell 50:1021–1029. [DOI] [PubMed] [Google Scholar]
- 33. Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O et al (2006) A pharmacological map of the PI3‐K family defines a role for p110alpha in insulin signaling. Cell 125:733–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Knobbe CB, Trampe‐Kieslich A, Reifenberger G (2005) Genetic alteration and expression of the phosphoinositol‐3‐kinase/Akt pathway genes PIK3CA and PIKE in human glioblastomas. Neuropathol Appl Neurobiol 31:486–490. [DOI] [PubMed] [Google Scholar]
- 35. Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI et al (1997) PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275:1943–1947. [DOI] [PubMed] [Google Scholar]
- 36. Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X (1997) Cytochrome c and dATP‐dependent formation of Apaf‐1/caspase‐9 complex initiates an apoptotic protease cascade. Cell 91:479–489. [DOI] [PubMed] [Google Scholar]
- 37. Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB (2005) Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 120:237–248. [DOI] [PubMed] [Google Scholar]
- 38. Luo J, Cantley LC (2005) The negative regulation of phosphoinositide 3‐kinase signaling by p85 and its implication in cancer. Cell Cycle 4:1309–1312. [DOI] [PubMed] [Google Scholar]
- 39. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW et al (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med 350:2129–2139. [DOI] [PubMed] [Google Scholar]
- 40. Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C et al (2008) Identification and characterization of NVP‐BEZ235, a new orally available dual phosphatidylinositol 3‐kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther 7:1851–1863. [DOI] [PubMed] [Google Scholar]
- 41. Manning BD, Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129:1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Marone R, Cmiljanovic V, Giese B, Wymann MP (2008) Targeting phosphoinositide 3‐kinase: moving towards therapy. Biochim Biophys Acta 1784:159–185. [DOI] [PubMed] [Google Scholar]
- 43. Mauvais‐Jarvis F, Ueki K, Fruman DA, Hirshman MF, Sakamoto K, Goodyear LJ et al (2002) Reduced expression of the murine p85alpha subunit of phosphoinositide 3‐kinase improves insulin signaling and ameliorates diabetes. J Clin Invest 109:141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mueller W, Mizoguchi M, Silen E, D'Amore K, Nutt CL, Louis DN (2005) Mutations of the PIK3CA gene are rare in human glioblastoma. Acta Neuropathol 109:654–655. [DOI] [PubMed] [Google Scholar]
- 45. Murga C, Fukuhara S, Gutkind JS (2000) A novel role for phosphatidylinositol 3‐kinase beta in signaling from G protein‐coupled receptors to Akt. J Biol Chem 275:12069–12073. [DOI] [PubMed] [Google Scholar]
- 46. Opel D, Westhoff MA, Bender A, Braun V, Debatin KM, Fulda S (2008) Phosphatidylinositol 3‐kinase inhibition broadly sensitizes glioblastoma cells to death receptor‐ and drug‐induced apoptosis. Cancer Res 68:6271–6280. [DOI] [PubMed] [Google Scholar]
- 47. Parcellier A, Tintignac LA, Zhuravleva E, Hemmings BA (2008) PKB and the mitochondria: AKTing on apoptosis. Cell Signal 20:21–30. [DOI] [PubMed] [Google Scholar]
- 48. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P et al (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Prevo R, Deutsch E, Sampson O, Diplexcito J, Cengel K, Harper J et al (2008) Class I PI3 kinase inhibition by the pyridinylfuranopyrimidine inhibitor PI‐103 enhances tumor radiosensitivity. Cancer Res 68:5915–5923. [DOI] [PubMed] [Google Scholar]
- 50. Rasheed BK, Stenzel TT, McLendon RE, Parsons R, Friedman AH, Friedman HS et al (1997) PTEN gene mutations are seen in high‐grade but not in low‐grade gliomas. Cancer Res 57:4187–4190. [PubMed] [Google Scholar]
- 51. Rathmell JC, Fox CJ, Plas DR, Hammerman PS, Cinalli RM, Thompson CB (2003) Akt‐directed glucose metabolism can prevent Bax conformation change and promote growth factor‐independent survival. Mol Cell Biol 23:7315–7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Salmena L, Carracedo A, Pandolfi PP (2008) Tenets of PTEN tumor suppression. Cell 133:403–414. [DOI] [PubMed] [Google Scholar]
- 53. Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S et al (2004) High frequency of mutations of the PIK3CA gene in human cancers. Science 304:554. [DOI] [PubMed] [Google Scholar]
- 54. Schlegel J, Durchschlag G, Piontek G, Grosu AL (2002) Activation of the phosphatidylinositol‐3′‐kinase/protein kinase B‐dependent antiapoptotic pathway plays an important role in the development of radioresistance of human glioma cells. Ann NY Acad Sci 973:224–227. [DOI] [PubMed] [Google Scholar]
- 55. Shaw RJ, Cantley LC (2006) Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 441:424–430. [DOI] [PubMed] [Google Scholar]
- 56. Slamon DJ, Leyland‐Jones B, Shak S, Fuchs H, Paton V, Bajamonde A et al (2001) Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 344:783–792. [DOI] [PubMed] [Google Scholar]
- 57. Sugimoto Y, Whitman M, Cantley LC, Erikson RL (1984) Evidence that the Rous sarcoma virus transforming gene product phosphorylates phosphatidylinositol and diacylglycerol. Proc Natl Acad Sci USA 81:2117–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Taylor RC, Cullen SP, Martin SJ (2008) Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol 9:231–241. [DOI] [PubMed] [Google Scholar]
- 59. Terauchi Y, Tsuji Y, Satoh S, Minoura H, Murakami K, Okuno A et al (1999) Increased insulin sensitivity and hypoglycaemia in mice lacking the p85 alpha subunit of phosphoinositide 3‐kinase. Nat Genet 21:230–235. [DOI] [PubMed] [Google Scholar]
- 60. The Cancer Genome Atlas Research Network (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455:1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ueki K, Yballe CM, Brachmann SM, Vicent D, Watt JM, Kahn CR, Cantley LC (2002) Increased insulin sensitivity in mice lacking p85beta subunit of phosphoinositide 3‐kinase. Proc Natl Acad Sci USA 99:419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ueki K, Fruman DA, Yballe CM, Fasshauer M, Klein J, Asano T et al (2003) Positive and negative roles of p85 alpha and p85 beta regulatory subunits of phosphoinositide 3‐kinase in insulin signaling. J Biol Chem 278:48453–48466. [DOI] [PubMed] [Google Scholar]
- 63. Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC et al (2001) Synthesis and function of 3‐phosphorylated inositol lipids. Annu Rev Biochem 70:535–602. [DOI] [PubMed] [Google Scholar]
- 64. Vanhaesebroeck B, Ali K, Bilancio A, Geering B, Foukas LC (2005) Signalling by PI3K isoforms: insights from gene‐targeted mice. Trends Biochem Sci 30:194–204. [DOI] [PubMed] [Google Scholar]
- 65. Wang SI, Puc J, Li J, Bruce JN, Cairns P, Sidransky D, Parsons R (1997) Somatic mutations of PTEN in glioblastoma multiforme. Cancer Res 57:4183–4186. [PubMed] [Google Scholar]
- 66. Weinstein IB (2002) Cancer. Addiction to oncogenes—the Achilles heal of cancer. Science 297:63–64. [DOI] [PubMed] [Google Scholar]
- 67. Whitman M, Kaplan DR, Schaffhausen B, Cantley L, Roberts TM (1985) Association of phosphatidylinositol kinase activity with polyoma middle‐T competent for transformation. Nature 315:239–242. [DOI] [PubMed] [Google Scholar]
- 68. Whitman M, Kaplan D, Roberts T, Cantley L (1987) Evidence for two distinct phosphatidylinositol kinases in fibroblasts. Implications for cellular regulation. Biochem J 247:165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Whitman M, Downes CP, Keeler M, Keller T, Cantley L (1988) Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol‐3‐phosphate. Nature 332:644–646. [DOI] [PubMed] [Google Scholar]
- 70. Yu J, Zhang Y, McIlroy J, Rordorf‐Nikolic T, Orr GA, Backer JM (1998) Regulation of the p85/p110 phosphatidylinositol 3′‐kinase: stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol Cell Biol 18:1379–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhao JJ, Cheng H, Jia S, Wang L, Gjoerup OV, Mikami A, Roberts TM (2006) The p110alpha isoform of PI3K is essential for proper growth factor signaling and oncogenic transformation. Proc Natl Acad Sci USA 103:16296–16300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhao L, Vogt PK (2008) Class I PI3K in oncogenic cellular transformation. Oncogene 27:5486–5496. [DOI] [PMC free article] [PubMed] [Google Scholar]