

Abstract
Although phase I to III trials represent the standard for introducing new drugs to clinical therapy, there has been increasing demand for translational research in oncology over the past decade. Thus, for most novel therapies such as 'targeted agents', a critical aspect for drug development in oncology has been to select the right patients for therapy. Translational research plays a pivotal role, not only in phase II trials but also in phase I and III and even in phase IV trials. The importance of distinguishing between our translational 'aims' in phase II and phase III trials is emphasized. Although translational research in phase III trials aims to identify optimal markers for clinical use, phase II studies may represent an optimal setting to explore tumour biology and the mechanisms of drug resistance in depth.
Introduction
The roles of phase I to IV trials are now well defined. However, the concept of translational research is not part of this progression, simply because the need for translational research – as part of therapy development – became evident long after the concepts of phase I to III trials were defined. For many anticancer therapies, such as targeted therapies, identification of predictive markers for proper patient selection is mandatory. Use of therapies that are directed at well defined targets, such as trastuzumab and lapatinib, by definition require measurement of specific molecular targets. Although no proof for such a concept exists at present, we can envision that the efficacy of other therapies – such as anti-angiogenic treatment – may partly depend on molecular markers such as vascular growth receptors [1].
Considering chemotherapy in general, we lack key information about the mechanisms of drug resistance in vivo [2]. Yet, the clinical observation of lack of cross-resistance between different compounds such as taxanes and anthracyclines [3] indicates that different mechanisms are involved. Hence, identification of these mechanisms through predictive testing may help to define the optimal therapy in the individual patient. Turning our attention to endocrine therapy, substantial therapeutic opportunities may result from uncovering the mechanisms of acquired resistance among receptor-positive tumours. Over the past decade we have witnessed a dramatic increase in our fundamental biological knowledge, as well as significant improvements in analytical methods. Thus, the potential for improved predictive testing has never been better. This being so, we must examine how translational research can best be fitted into our general clinical trial programmes.
Phase I studies
The aim of phase I studies is to explore drug toxicity. Traditionally, such studies may involve about 12 to 20 patients who are treated with the drug under investigation at escalating doses. This may be achieved either through dose escalation in the same individuals or by increasing doses between patient cohorts, for instance by using a Fibonacci approach [4]. Often, such studies involve a pharmacokinetic component, in which pharmacokinetic parameters such as plasma half-life and drug clearance rate are assessed. For many drugs the 95% confidence intervals for throughput levels may span an interval in the order of about 1:4, but for others (for example, medroxyprogesterone acetate administered to breast cancer patients at high doses [500 to 1,000 mg] orally) throughput level 95% confidence intervals may span an order of 1:10 [5]. This is probably due to inter-individual variations in absorption as well as metabolism. Such variations may have clinical implications for therapeutic efficacy, questioning the need for routine plasma drug level assessment [5].
Of most concern with respect to variations in metabolism is atypical metabolism in some patients. The issue of polymorphic metabolism of tamoxifen related to therapy efficacy is briefly commented upon in relation to phase III trials here and in detail in another paper in this supplemental issue. Of importance to toxicity is the fact that some patients, because of allelic polymorphisms, may suffer from parent drug or metabolite accumulation, generating toxic side effects. Serious problems in general are associated with homozygosity for a poor metabolizing (or 'PM') allele. A classical example is that of the antihypertensive agent debrisoquine; some patients experience hypotension together with high plasma levels of unmetabolized drug in concert with low excretion of the major 4-hydroxylated metabolite in the urine [6]. In 1980, Evans and coworkers [7] reported that 8% to 9% of UK Caucasians inherit PM status for the antihypertensive agent debrisoquine in a recessive manner. Another example is that of the oral anti-diabetic tolbutamide. This compound is mainly metabolized through cytochrome P450 (CYP)2C, which is also the metabolic pathway for other important compounds, including the active S-enantiomer of warfarin [8]. Among Caucasians, the incidence of the PM *3 allele is about 6%. About 0.3% of the population are poor metabolizers of these compounds because of homozygosity for this allele, which may cause serious side effects during therapy despite normal drug dosing. Notably, for some CYPs, such as CYP2C19, the incidence of PM alleles may vary between ethnic groups [8], supporting the need to determine drug metabolism in different populations.
Although novel endocrine agents recently incorporated into clinical practice have been associated with low toxicity, we should not automatically infer that this will always be the case. The first-generation aromatase inhibitor aminoglutethimide exhibited significant toxicity, resulting in neurological side effects and, in some cases, bone marrow toxicity [9]. Although the pharmacokinetics of the compound were duly explored [10], whether side effects such as drowsiness could be related to variation in drug metabolism was never properly examined.
In summary, atypical metabolism caused by gene polymorphisms can be difficult to detect because of the low incidence of many PM alleles, but side effects that may be related to individual overdosing should provoke pharmacogenetic investigations to identify potential PM alleles that can cause toxic effects in some patients.
Phase II studies
There are three main reasons why phase II studies represent a key scenario in translational research. First, for many drugs, identification of predictive markers is critical to deciding whether the compound should move forward into a phase III study. Second, because a limited number of patients is enrolled in phase II protocols, dose selection for phase III studies based on differences in responsiveness to different drug doses in phase II trials may be suboptimal. Thus, translational research with identification of suitable biomarkers may add important information to the decision process. Third, phase II studies conducted in dedicated centres may offer a unique opportunity to collect tissue samples and conduct molecular biological analyses aimed at identifying mechanisms regulating drug sensitivity and resistance.
Helpful examples when considering the first issue include targeted therapies such as endocrine treatment in general and use of trastuzumab. None of these therapies achieved their current clinical status without identification of target proteins as predictive markers. For novel compounds, a response rate of, say, 20% in general may not favour further development. In addition to patient welfare, health authorities currently mandate consideration of issues such as cost/utility before they will approve therapies, be they 'low cost' [11] or 'high cost' [12]. Thus, eliminating, say, 50% of patients predicted upfront to be nonresponders from inclusion in a phase II trial may increase the response rate among remaining patients from 20% to 40%. This is an outcome that may potentially be critical in determining whether a compound should advance into phase III studies.
Second, a critical part of phase II studies is proper dose selection for further processing into phase III studies. For instance, the efficacy of aromatase inhibitors may be determined though direct in vivo assessment of the key end-point, aromatase inhibition, with use of radiolabelled steroid injections [13-15]. Applying this technique, we found anastrozole 1 mg/day to be as effective at inhibiting in vivo aromatization and plasma oestrogen suppression as 10 mg/day [16]. In contrast, when the selective oestrogen receptor (ER) modulator droloxifene was compared with tamoxifen in an international phase III study [17], a drug dose of 40 mg was selected. The reason for this was that clinical results from a phase II study [18] had revealed that droloxifene at 40 mg/day and 100 mg/day yielded similar response rates. The phase III study reported that droloxifene 40 mg/day and tamoxifen 20 mg/day yielded similar response rates in postmenopausal women. In contrast, droloxifene was found inferior in premenopausal women. This finding actually leads one to question whether droloxifene was administered at a suboptimal dose. This hypothesis is indirectly supported by the finding that droloxifene 100 mg/day causes alterations in plasma levels of sex hormone binding globulin, insulin-like growth factor I and its major binding proteins, resembling what we see during treatment with tamoxifen, whereas droloxifene 40 mg/day exhibited only moderate effects [19-23]. In this case, dose selection based on biomarkers probably had been superior to dose selection on the basis of response rates.
Regarding the third aim of phase II studies, we must discriminate between biological end-points in phase II and phase III studies. As outlined below, translational research in phase III studies should aim to confirm predictive factors that may be suitable for routine application. In contrast, in phase II studies our biological research aims are much wider, namely to identify the biological mechanisms underlying the observations recorded. Although in recent years pharmaceutical companies have tended to try to get drugs through phase II into phase III trials as quickly as possible, spending some additional time on phase II studies and recruiting patients suitable for translational research may be rewarding. Although not every patient harbouring breast cancer metastases has deposits that are suitable for a biopsy, lesions located in the liver or locoregionally are, in general, suitable for Tru-Cut tissue collection, and thus for snap-frozen tissue biobanking.
Predicting responsiveness to endocrine therapy or trastuzumab, expression of the ER or amplification of the human epidermal growth factor receptor (HER)-2 proto-oncogene are good predictive factors. However, not every patient with an ER-positive tumour responds to endocrine manipulation, and not every patient exhibiting HER-2 amplification benefits from targeted therapy with, for example, trastuzumab. Most likely, there is no single mechanism of growth factor inactivation; apart from simple over-expression or under-expression of a protein product, we may be dealing with proteins that are modified by single base non-sense or mis-sense mutations, truncations or deletions [2]. Although chemo-resistance has been linked to mutations in particular genes that execute apoptosis or DNA repair, there is emerging evidence that we need to focus on gene 'pathways' and not single gene products if we are to understand these functions [24]. Furthermore, although non-sense mutations eliminate the function of a gene product, the biological effect of a mis-sense mutation (or a polymorphism) may be difficult to predict upfront. For example, considering the key gene TP53, whose protein product p53 is involved in growth arrest as well as apoptosis and DNA repair, some mis-sense single base substitutions (affecting DNA binding) may be deleterious whereas other mutations appear to generate normal functioning protein [25]. Finally, because various drugs, including anthracyclines and taxanes, exhibit lack of cross-resistance, clearly the mechanism of resistance must differ. Thus, to explore drug resistance the mechanisms of chemo-resistance should be explored in relation to drug monotherapy whenever possible [26].
This approach may be illustrated with some examples. Previously, we discovered mutations in TP53 to be associated with resistance to anthracycline therapy in breast cancer [25]. Looking for mutations in other genes that are involved in the same pathway to account for resistance in tumours harbouring wild-type TP53, we were able to exclude mutations as well as polymorphisms that affect the p53 downstream target p21 [27,28]. In a recent phase II study in which patients with primary, locally advanced breast cancers were randomly assigned to epirubicin or taxol monotherapy (Figure 1), we discovered non-sense mutations in the CHEK2 gene (which encodes the chk2 protein), one of the genes acting upstream of p53 [29], to predict resistance to epirubicin, similar to our observation regarding TP53 mutations. In the same study, we excluded mutations in another p53 activator, namely p19 (Figure 2). These discoveries confirmed the importance of CHEK2 activating TP53 in response to DNA damage in vivo and provide information key to our understanding of the apoptotic response in breast cancer. However, with a minority of epirubicin-resistant patients harbouring CHEK2 mutations, there is not yet any clinical rationale for recommending CHEK2 gene sequencing as a routine test in breast cancer patients. On the other hand, if we are able to confirm that most patients exhibiting anthra-cycline resistance harbour disturbances in the p53 'pathway', then much effort should be channelled toward developing a predictive marker to identify general defects in this pathway.
Figure 1.
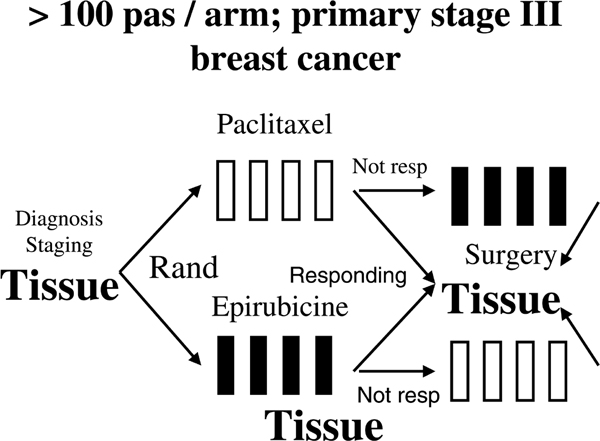
Example of a phase II study aiming at identifying mechanisms of drug resistance. Presented is an example of a phase II study designed with the primary aim of exploring mechanisms of resistance to epirubicin and paclitaxel; each drug was administered as a monotherapy regimen [33].
Figure 2.
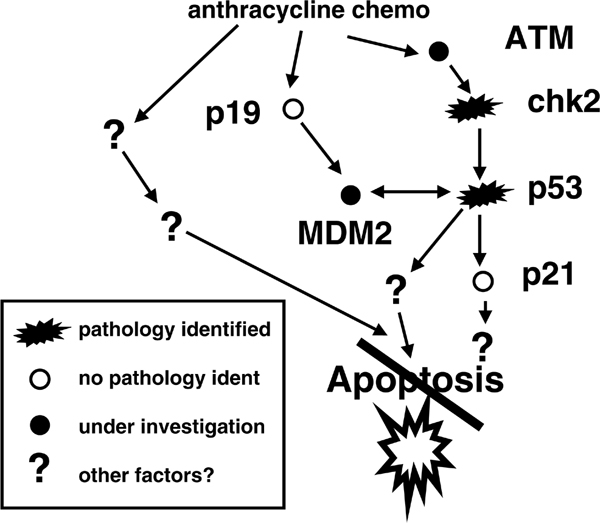
Exploring disturbances in molecular pathways as a cause of drug resistance. Presented are results from studies 'dissecting' the p53 pathway with respect to anthracycline resistance in breast cancer. Thus far, we have identified mutations affecting TP53 (encoding the p53 protein) as well as CHEK2 (encoding the chk2 protein) to be associated with drug resistance; in contrast, we have not identified mutations associated with resistance in the genes encoding p21, p21b or p19 [26-28,33-35].
Phase III studies
Predictive factors may serve two ultimate goals with respect to phase III trials. In a similar manner to phase II studies, they are needed to select the proper patient population for trial enrolment. Second, they may identify subgroups of patients who may benefit from one therapy as opposed to another.
Regarding the selection of patient populations for inclusion in phase III trials, we may often benefit from the findings of previous studies conducted to identify predictive markers. Thus, the general opinion is that only patients harbouring ER and/or progesterone receptor positive tumours are eligible for trials evaluating endocrine agents. Similarly, HER-2 over-expression and/or amplification is considered a requirement for anti-HER-2 strategies to work.
Considering the second issue, the finding of a lack of cross-resistance between cytotoxic compounds such as the anthracyclines and taxanes supports the view that different mechanisms of resistance are involved. Thus, a better understanding of such mechanisms may allow proper selection of patients for each particular therapy. The question becomes not whether the one drug may be superior for breast cancer therapy in general, but rather which patients may benefit from which treatment option, and how should we identify them?
A second example relates to pharmacogenomics. The CYP2D6 enzyme (responsible for converting tamoxifen into its active metabolite 4-hydroxy-n-desmethyltamoxifen [endoxifen]) has a polymorphic allele *4, which is associated with low enzymatic activity. 4-Hydroxy-n-desmethyltamoxifen was initially described by Lien and colleagues [30] in 1989. According to Goetz and coworkers [31], who were first to describe the *4 allele, about 20% of people exhibit heterogeneity for this variant, whereas about 7% were found to be homozygous. Importantly, they revealed that patients homozygous for the *4 allele had an inferior outcome compared with other patients receiving tamoxifen treatment. Whether expression of the *4 allele predicts a poor outcome has been examined in several recent reports, and whether it should be implemented for clinical testing to select patients for tamoxifen therapy remains an issue of debate [32]. Nevertheless, it illustrates an important principle – pharmacogenetics may be an important factor in selecting appropriate candidates for certain treatments in the future.
Phase IV studies
Phase IV studies include various kinds of research conducted after completion of the phase III studies that are pivotal for registration. Whether the treatments are used for metastatic disease or in the neoadjuvant (primary) setting, these studies may offer a suitable opportunity for translational research, with aims similar to those in phase II studies.
Conclusion
The extended need for translational research does not change the planning of phase I to III and even phase IV studies. However, the need for translational studies should be emphasized. Translational research should not be considered a 'secondary aim', proceeding in parallel with clinical studies in general. Rather, we should regard translational research as an integrated part of drug development during each phase of the programme.
Abbreviations
CYP: cytochrome P450; ER: oestrogen receptor; HER: human epidermal growth factor receptor; PM: poor metabolizing (allele).
Competing interests
The author declares that they have no competing interests.
Acknowledgments
Acknowledgements
This article has been published as part of Breast Cancer Research Volume 10 Supplement 4, 2008: Controversies in Breast Cancer 2008. The full contents of the supplement are available online at http://breast-cancer-research.com/supplements/10/S4
References
- Terman BI, Doughervermazen M. Biological properties of VEGF/VPF receptors. Cancer Metastasis Rev. 1996;15:159–163. doi: 10.1007/BF00437468. [DOI] [PubMed] [Google Scholar]
- Lønning PE, Knappskog S, Staalesen V, Chrisanthar R, Lillehaug JR. Breast cancer prognostication and prediction in the postgenomic era. Ann Oncol. 2007;18:1293–1306. doi: 10.1093/annonc/mdm013. [DOI] [PubMed] [Google Scholar]
- Ravdin PM. Docetaxel (Taxotere) for the treatment of anthra-cycline-resistant breast cancer. Semin Oncol. 1997;24:18–21. [PubMed] [Google Scholar]
- Mita AC, Olszanski AJ, Walovitch RC, Perez RP, MacKay K, Tuck DP, Simmons C, Hammond S, Mita MM, Beeram M, Stone AJ, Rowinsky EK, Lewis LD. Phase I and pharmacokinetic study of Al-850, a novel microparticle hydrophobic drug delivery system for paclitaxel. Clin Cancer Res. 2007;13:3293–3301. doi: 10.1158/1078-0432.CCR-06-2496. [DOI] [PubMed] [Google Scholar]
- Lønning PE. Dose response evaluation – use of plasma concentration confidence intervals as a tool to predict optimal drug dose ratio. Clin Pharmacokinet. 1993;25:1–5. doi: 10.2165/00003088-199325010-00001. [DOI] [PubMed] [Google Scholar]
- Silas JH, Lennard MS, Tucker GT, Smith AJ, Malcolm SL, Marten TR. Why hypertensive patients vary in their response to oral debrisoquine. Br Med J. 1977;1:422–425. doi: 10.1136/bmj.1.6058.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DA, Mahgoub A, Sloan TP, Idle JR, Smith RL. A family and population study of the genetic polymorphism of debrisoquine oxidation in a white British population. J Med Genet. 1980;17:102–105. doi: 10.1136/jmg.17.2.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JA. Clinical relevance of genetic polymorphisms in the human CYP2C subfamily. Br J Clin Pharmacol. 2001;52:349–355. doi: 10.1046/j.0306-5251.2001.01499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lønning PE, Kvinnsland S. Mechanisms of action of aminoglutethimide as endocrine therapy of breast cancer. Drugs. 1988;35:685–710. doi: 10.2165/00003495-198835060-00005. [DOI] [PubMed] [Google Scholar]
- Lønning PE, Schanche JS, Kvinnsland S, Ueland PM. Single-dose and steady-state pharmacokinetics of aminoglutethimide. Clin Pharmacokinet. 1985;10:353–364. doi: 10.2165/00003088-198510040-00005. [DOI] [PubMed] [Google Scholar]
- Lønning PE. Comparing cost/utility of giving an aromatase inhibitor as monotherapy for 5 years versus sequential administration following 2–3 or 5 years of tamoxifen as adjuvant treatment for postmenopausal breast cancer. Ann Oncol. 2006;17:217–225. doi: 10.1093/annonc/mdj048. [DOI] [PubMed] [Google Scholar]
- Norum J, Olsen JA, Wist EA, Lonning PE. Trastuzumab in adjuvant breast cancer therapy. A model based cost-effectiveness analysis. Acta Oncologica. 2007;46:153–164. doi: 10.1080/02841860601096841. [DOI] [PubMed] [Google Scholar]
- Lønning PE, Skulstad P, Sunde A, Thorsen T. Separation of urinary metabolites of radiolabelled estrogens in man by HPLC. J Steroid Biochem. 1989;32:91–97. doi: 10.1016/0022-4731(89)90019-8. [DOI] [PubMed] [Google Scholar]
- Jacobs S, Lønning PE, Haynes B, Griggs L, Dowsett M. Measurement of aromatisation by a urine technique suitable for the evaluation of aromatase inhibitors in vivo. J Enzyme Inhibition. 1991;4:315–325. doi: 10.3109/14756369109030396. [DOI] [PubMed] [Google Scholar]
- Lønning PE. Pharmacology of new aromatase inhibitors. Breast. 1996;5:202–208. doi: 10.1016/S0960-9776(96)90094-6. [DOI] [Google Scholar]
- Geisler J, King N, Dowsett M, Ottestad L, Lundgren S, Walton P, Kormeset PO, Lønning PE. Influence of anastrozole (Arimidex®), a selective, non-steroidal aromatase inhibitor, on in vivo aromatisation and plasma oestrogen levels in post-menopausal women with breast cancer. Br J Cancer. 1996;74:1286–1291. doi: 10.1038/bjc.1996.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzdar A, Hayes D, El-Khoudary A, Yan S, Lønning P, Lichinitser M, Gopal R, Falkson G, Pritchard K, Lipton A, Wolter K, Lee A, Fly K, Chew R, Alderdice M, Burke K, Eisenber P, Droloxifene 301 Study Group Phase III randomized trial of droloxifene and tamoxifen as first-line endocrine treatment of ER/PgR-positive advanced breast cancer. Br Cancer Res Treat. 2002;73:161–175. doi: 10.1023/A:1015229630260. [DOI] [PubMed] [Google Scholar]
- Bruning PF. Droloxifene, a new anti-oestrogen in post-menopausal advanced breast cancer: preliminary results of a double-blind dose-finding phase II trial. Eur J Cancer. 1992;28A:1404–1407. doi: 10.1016/0959-8049(92)90530-F. [DOI] [PubMed] [Google Scholar]
- Helle SI, Anker G, Tally M, Hall K, Lønning PE. Influence of droloxifene on plasma levels of insulin-like growth factor (IGF)-I, pro-IGF-IIE, insulin-like growth factor binding protein (IGFBP)-1 and IGFBP-3 in breast cancer patients. J Steroid Biochem Mol Biol. 1996;57:167–171. doi: 10.1016/0960-0760(95)00257-X. [DOI] [PubMed] [Google Scholar]
- Lønning PE, Hall K, Aakvaag A, Lien EA. Influence of tamoxifen on the plasma levels of insulin-like growth factor I and Insulin-like growth factor binding protein I in breast cancer patients. Cancer Res. 1992;52:4719–4723. [PubMed] [Google Scholar]
- Helle SI, Holly JMP, Tally M, Hall K, Stappen Jvd, Lønning PE. Influence of treatment with tamoxifen and change in tumor burden on the IGF-system in breast cancer patients. Int J Cancer (Pred Oncol) 1996;69:335–339. doi: 10.1002/(SICI)1097-0215(19960822)69:4<335::AID-IJC17>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Geisler J, Ekse D, Hösch S, Lønning PE. Influence of droloxifene (3-hydroxytamoxifen), 40 mg daily, on plasma gonadotrophins, sex hormone binding globulin and estrogen levels in postmenopausal breast cancer patients. J Steroid Biochem Mol Biol. 1995;55:193–195. doi: 10.1016/0960-0760(95)00163-T. [DOI] [PubMed] [Google Scholar]
- Geisler J, Haarstad H, Gundersen S, Raabe N, Kvinnsland S, Lønning PE. Influence of treatment with the anti-oestrogen 3-hydroxytamoxifen (droloxifene) on plasma sex hormone levels in postmenopausal patients with breast cancer. J Endocrinol. 1995;146:359–363. doi: 10.1677/joe.0.1460359. [DOI] [PubMed] [Google Scholar]
- Lønning PE. Genes causing inherited cancer as beacons identifying the mechanisms of chemoresistance. Trends Mol Med. 2004;10:113–118. doi: 10.1016/j.molmed.2004.01.005. [DOI] [PubMed] [Google Scholar]
- Geisler S, Lønning PE, Aas T, Johnsen H, Fluge O, Haugen DF, Lillehaug JR, Akslen LA, Børresen-Dale AL. Influence of TP53 gene alterations and c-erbB-2 expression on the response to treatment with doxorubicin in locally advanced breast cancer. Cancer Res. 2001;61:2505–2512. [PubMed] [Google Scholar]
- Lønning P. Study of suboptimum treatment response: lessons from breast cancer. Lancet Oncol. 2003;4:177–185. doi: 10.1016/S1470-2045(03)01022-2. [DOI] [PubMed] [Google Scholar]
- Staalesen V, Leirvaag B, Lillehaug JR, Lønning PE. Genetic and epigenetic changes in p21 and p21B do not correlate to resistance to doxorubicin or mitomycin and 5-fluorouracil in locally advanced breast cancer. Clin Cancer Res. 2004;10:3438–3443. doi: 10.1158/1078-0432.CCR-03-0796. [DOI] [PubMed] [Google Scholar]
- Knappskog S, Chrisanthar R, Staalesen V, Børresen-Dale AL, Gram IT, Lillehaug JR, Lønning PE. Mutations and polymorphisms of the p21B transcript in breast cancer. Int J Cancer. 2007;121:908–910. doi: 10.1002/ijc.22777. [DOI] [PubMed] [Google Scholar]
- Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006;6:909–923. doi: 10.1038/nrc2012. [DOI] [PubMed] [Google Scholar]
- Lien EA, Solheim E, Lea OA, Lundgren S, Kvinnsland S, Ueland PM. Distribution of 4-hydroxy-N-desmethyltamoxifen and other tamoxifen metabolites in human biological fluids during tamoxifen treatment. Cancer Res. 1989;49:2175–2183. [PubMed] [Google Scholar]
- Goetz MP, Rae JM, Suman VJ, Safgren SL, Ames MM, Visscher DW, Reynolds C, Couch FJ, Lingle WL, Flockhart DA, Desta Z, Perez EA, Ingle JN. Pharmacogenetics of tamoxifen biotrans-formation is associated with clinical outcomes of efficacy and hot flashes. J Clin Oncol. 2005;23:9312–9318. doi: 10.1200/JCO.2005.03.3266. [DOI] [PubMed] [Google Scholar]
- Hayes DF, Steams V, Rae J, Flockhart D. A model citizen? Is tamoxifen more effective than aromatase inhibitors if we pick the right patients? J Nat Cancer Instit. 2008;100:610–613. doi: 10.1093/jnci/djn127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrisanthar R, Knappskog S, Løkkevik E, Anker G, Østenstad B, Lundgren S, Berge EO, Risberg T, Mjaaland I, Maehle L, Engebretsen LF, Lillehaug JR, Lønning PE. CHEK2 mutations affecting kinase activity together with mutations in the TP53 indicate a functional pathway associated with resistance to epirubicin in primary breast cancer. PLoS One. 2008;3:e3062. doi: 10.1371/journal.pone.0003062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Børresen-Dale AL, Johnsen H, Aas T, Geisler J, Akslen LA, Anker G, Lønning PE. TP53 gene mutations predict the response to neoadjuvant treatment with FUMI in locally advanced breast cancer. Clin Cancer Res. 2003;9:5582–5588. [PubMed] [Google Scholar]
- Staalesen V, Falck J, Geisler S, Bartkova J, Børresen-Dale AL, Lukas J, Lillehaug JR, Bartek J, Lønning PE. Alternative splicing and mutation status of CHEK2 in stage III breast cancer. Oncogene. 2004;23:8535–8544. doi: 10.1038/sj.onc.1207928. [DOI] [PubMed] [Google Scholar]