

Abstract
Epigenetic mechanisms have been proposed to play a role in the etiology of autism. This hypothesis is supported by the discovery of increased MECP2 promoter methylation associated with decreased MeCP2 protein expression in autism male brain. To further understand the influence of female X chromosome inactivation (XCI) and neighboring methylation patterns on aberrant MECP2 promoter methylation in autism, multiple methylation analyses were peformed on brain and blood samples from individuals with autism. Bisulfite sequencing analyses of a region 0.6 kb upstream of MECP2 in brain DNA samples revealed an abrupt transition from a highly methylated region in both sexes to a region unmethylated in males and subject to XCI in females. Chromatin immunoprecipitation analysis demonstrated that the CCTC-binding factor (CTCF) bound to this transition region in neuronal cells, consistent with a chromatin boundary at the methylation transition. Male autism brain DNA samples displayed a slight increase in methylation in this transition region, suggesting a possible aberrant spreading of methylation into the MECP2 promoter in autism males across this boundary element. In addition, autistic female brain DNA samples showed evidence for aberrant MECP2 promoter methylation as an increase in the number of bisulfite sequenced clones with undefined XCI status for MECP2 but not androgen receptor (AR). To further investigate the specificity of MECP2 methylation alterations in autism, blood DNA samples from females and mothers of males with autism were also examined for XCI skewing at AR, but no significant increase in XCI skewing was observed compared to controls. These results suggest that the aberrant MECP2 methylation in autism brain DNA samples is due to locus-specific rather than global X chromosome methylation changes.
Keywords: epigenetics, X chromosome inactivation, MECP2, post-mortem brain
INTRODUCTION
Autism is a severe neurodevelopmental disorder affecting, according to most recent estimates, around 1 in 150 individuals (Rice et al., 2007). Autism is characterized by deficits in social interaction and communication and by gains in restricted and repetitive interests and behaviors. Although a large body of evidence indicates a major role for genetic variation, several observations point to an additional role for epigenetic variation in the etiology of autism. Decreased homologous 15q11-13 chromosome pairing, decreased H3K9 trimethylation, increased H3K9 acetylation, and aberrant UBE3A methylation have been observed in autism brain (Jiang et al., 2004; Thatcher et al., 2006; Thatcher et al., 2005). The higher incidence of autism in males (4:1 male to female) suggests the involvement of the sex chromosomes and possibly changes in epigenetic regulatory mechanisms that differ between the sexes.
X chromosome inactivation (XCI) is a random process by which the majority of genes on one of the two X chromosomes present in females are silenced by an epigenetic mechanism (Lyon, 1961). Increased XCI skewing has been observed in several human diseases with relevance to autism, such as Rett syndrome (Huppke et al., 2006; Knudsen et al., 2006) and X-linked mental retardation (MR) (Plenge et al., 2002). Increased skewing can result from multiple mechanisms including X-linked mutations and reduction in the pool of cells at the time of XCI in early embryogenesis (Brown et al., 2000). Skewed X inactivation was previously reported in autism female blood (Talebizadeh et al., 2005), but this report has yet to be independently replicated or examined in additional cell types. XCI skewing ratios in blood have been reported to significantly correlated with those from brain (Bittel et al., 2007), but XCI ratios in autism brain have not been previously determined.
MECP2 is a key epigenetic regulator, mutations of which cause the pervasive developmental disorder, Rett syndrome (Amir et al., 1999). Previously, we demonstrated a defect in MeCP2 expression in 79% of autism cerebral cortex samples (Nagarajan et al., 2006; Samaco et al., 2004). Decreased MeCP2 expression correlated with increased MECP2 promoter methylation in male brain samples. Here we examine possible explanations for aberrant MECP2 promoter methylation in males by defining the methylation boundary 0.6 kb upstream of MECP2 that corresponds to a binding site for the chromatin boundary factor CTCF. Furthermore, we examined X inactivation patterns in DNA samples isolated from autism female brain and blood cells from mothers of males with autism, hypothesizing that one manifestation of genetic and epigenetic dysregulation in autism might be altered X inactivation.
MATERIALS AND METHODS
Biological samples
Frozen frontal cerebral cortex (Brodmann Area 9) samples were obtained with assistance of the Autism Tissue Program from the University of Maryland Brain and Tissue Bank for Neurodevelopmental Disorders, the Harvard Brain Tissue Resource Center, and the University of Miami Brain and Tissue Bank for Neurodevelopmental Disorders. Red blood cell pellets containing polymorphonuclear cells were obtained from the UC Davis Childhood Autism Risks from Genetics and Environment (CHARGE) study, a large collaborative investigation of genetic, epigenetic, and environmental factors in autism (Hertz-Picciotto et al., 2006). Participants in the CHARGE study are a population-based sample of children 2-5 years of age from four groups: with autism, with developmental delays (DD), with autism spectrum disorders (ASD), and with typical development. Affected children and their families are recruited from a state-funded system that provides services for persons with developmental disabilities; controls with typical development are randomly sampled from state birth files, with frequency matching by age, gender and geographic area to autism cases. All autism diagnoses were confirmed by administration of the standardized instruments: Autism Diagnostic Interview-Revised (ADI-R) (Le Couteur et al., 2003) and Autism Diagnostic Observation Schedules (ADOS) (Lord et al., 2000).
DNA Purification
DNA was purified from all samples with the PureGene kit (Qiagen, Valencia, CA) per manufacturer’s instructions. RBC pellets were cleared of red blood cells by 2-3 washes in hypotonic RBC lysis solution. If necessary, samples were concentrated with microcon columns (Millipore, Temecula, CA).
Bisulfite sequencing
Frontal cortex (BA9) genomic DNA was isolated from frozen postmortem human samples using the PureGene Kit (Qiagen, Valencia, CA). Approximately 1 μg of frontal cortex genomic DNA was used for bisulfite conversion with the CpGenome kit (Millipore, Temecula, CA) following the manufacturer’s instructions. Briefly, DNA samples were treated overnight at 55° with sodium bisulfite, desalted, desulfonated, and eluted in 20 μl TE. One μl of converted DNA was used for bisulfite PCR.
Bisulfite primers were designed using the online tool Methprimer (http://www.urogene.org/methprimer/index.html)(Li et al., 2002). Primers for MECP2 Bisulfite Region I: F: GTTAGGTTTTAGGGTGGGTAATTTT, R:CCCCTCCAACTATTAATTAACTACTTTC; MECP2 Bisulfite Region II: F: GAGGTTTTGGTATGTATTTTTTTT, R: ATTACCCACCCTAAAACCTAAC; AR: F:GAGTTTTTTAGAATTTGTTTTAGAG, R: AACCAAATAACCTATAAAACCTCTAC. Primers were designed to regions without CpG sites to avoid amplification bias of methylated versus unmethylated. No polymorphisms were observed in the population analyzed for amplified regions from MECP2 bisulfite region I or II. A 50 μl volume PCR reaction was performed with 2.5 units Roche FastStart High Fidelity Taq, 1X Roche FastStart High Fidelity Reaction Buffer (with 1.8 mM MgCl2,) 0.2 mM of each dNTP, and 1X DMSO (Roche, Indianapolis, IN). PCR products were gel purified with the Qiaquick Gel Purification kit (Qiagen) and ligated overnight at 4°C with the pGEM-T-Easy kit (Promega, Madison, WI). Two to five μl of the ligation were used to transform JM109 competent cells (Promega) and bacteria were plated on LB ampicillin. White colonies were picked and grown up in overnight LB Amp cultures. Plasmids were purified with the Qiaprep kit (Qiagen) and tested for presence of the correct insert with a NotI digest (New England Biolabs, Ipswich, MA). Positive plasmids were sequenced using the T7 promoter sequencing primer. 10-20 colonies were sequenced for each brain sample in order to obtain a sufficient sampling of allele-specific methylation patterns.
X Chromosome Inactivation (XCI) Assay
XCI Assay was performed as described (Beever et al., 2003) with some modifications. Brain or RBC pellet genomic DNA was resuspended in TE to 100 ng/μl. 150 ng of this DNA were digested with 2 U RsaI or 2 U RsaI + 5 U HpaII in a final volume of 10 μl. Both enzymes were purchased from New England Biolabs (Ipswich, MA). Samples were incubated overnight at 37° C and the next day 5 U HpaII was added to RsaI + HpaII reactions. The reaction was again incubated for 3 hours at 37° C to insure complete digestion. Completeness of digestion was confirmed by PCR of 5′ UTR of MIC2, which escapes X inactivation in females and is therefore always unmethylated (Goodfellow et al., 1988). MIC2 PCR was performed at 33 cycles using Invitrogen taq (Carslbad, CA) with an annealing temperature of 62° C. The PCR products were visualized on a 1.5% agarose gel using Sybr-Gold (Invitrogen, Carslbad, CA). If any band was observed in an RsaI + HpaII reaction, the digest was repeated and tested again.
The human androgen receptor (AR; a.k.a. HUMARA) 5′ end was PCR amplified from the digested samples using 30 cycles with Invitrogen taq at an annealing temperature of 62° C. The forward primer was 5′ labeled with HEX. Amplicons were diluted 1:2 – 1:8, and capillary electrophoresis fragment analysis was performed on an ABI3100 (Applied Biosystems, Foster City, CA) with a ROX500 size standard. Allele sizes and peak areas were determined with the PeakScanner software package (Applied Biosystems). For any sample found to have >80% (or <20%) skewing, the assay was repeated, and results of both assays were averaged.
XCI skewing was calculated by measuring peak area of digested and undigested AR PCR products. Since PCR bias may result in preferential amplification of some AR alleles, the peak areas of digested samples were normalized to undigested samples. The degree of skewing was calculated with the formula: (d1/u1)/(d1/u1+d2/u2), where d1 and d2 are the peak areas of the smaller digested allele and the larger digested allele, respectively, and u1 and u2 are the undigested peak areas. Alleles close in size would result in shadow bands from one overlapping the main bands of the other. For such cases, peak areas were adjusted by assuming that the shadow band represented 30% of the peak area of the main allele. Statistical analysis was performed using a Fisher’s exact test.
Chromatin Immunoprecipitation
ChIP was performed using a protocol adapted from de Belle et al (de Belle et al., 1998). Briefly chromatin was prepared from formaldehyde crosslinked, PMA differentiated SH-SY5Y neurons and purified by urea gradient centrifugation. Purified chromatin was then digested with EarI and XcmI restriction enzymes (NEB, Beverly, MA) then immunoprecipitated (IP) with anti-CTCF (Upstate, Lake Placid NY) or control antibody. 10% of the total chromatin was reserved as input for PCR analysis. Chromatin/antibody complexes were washed extensively, digested with proteinase K and extracted with phenol/chloroform prior to precipitation of purified DNA from solution. DNA from CTCF IP, control IP and input was resuspended in 20 μl of 1XTE and amplified with MECP2 promoter primers (F: aggctcggatgaaataatgc and R:aggctcggatgaaataatgc) using 28, 30, 32, 34 and 36 cycles of 95°C for 30 s, 57°C for 30 s, 72°C for 30 s with a 10 min 95°C pre-incubation and a 10 min 72°C post-incubation. Amplicons were resolved by 1.5% agarose gel electrophoresis and visualized by SYBR Gold (Invitrogen, Carlsbad, CA) staining. Gel images were acquired using a Fluorchem (Alpha Innotech, Santa Leandro, CA) gel documentation system.
RESULTS
A boundary region for methylation differences observed in MECP2 upstream regulatory region
Our previous investigation demonstrated that the aberrant MECP2 methylation observed in autism male brain samples was most frequent in two 5′ sites (sites 1 and 3 of bisulfite region I, Fig. 1A) (Nagarajan et al., 2006). In addition, site 3 showed methylation specific to autism brain (filled circle with “A” in Fig. 1A). To determine if more 5′ regulatory CpG sites exhibited aberrant DNA methylation, bisulfite primers were designed to the adjacent region, spanning -700 to -509 bp and containing 6 CpG sites (bisulfite region II, Fig. 1A). Bisulfite sequencing of this region was performed on DNA isolated from cerebral cortex from autism and control samples. Figures 1B and 1C show the results for total percent methylation of bisulfite region II, and demonstrate that these regions are highly methylated but variable in both males and female brain DNA samples. The variability in methylation of bisulfite region II was independent of age or gender (Fig 1B). Autism and control brain DNA samples did not show significant differences in total percent methylation of bisulfite region II, but there was a trend towards higher methylation in male autism samples. Interestingly, in the analysis of percent methylation of each of the 6 CpG sites in this region (A-F, Fig. 1A), sites A-D showed significantly higher methylation than the 3′ sites E and F in male samples (P<0.0001, Figure 1D). This abrupt difference in methylation suggests the presence of an insulator element or chromatin boundary (represented as a hatched bar in Fig. 1A). Methylation of each CpG site was slightly increased in autism samples, although none reached statistical significance. Site F, at -533 bp, was nearly significantly increased in autism brain DNA samples (P=0.07).
Figure 1. Analysis of an upstream region of MECP2 reveals a methylation boundary element.
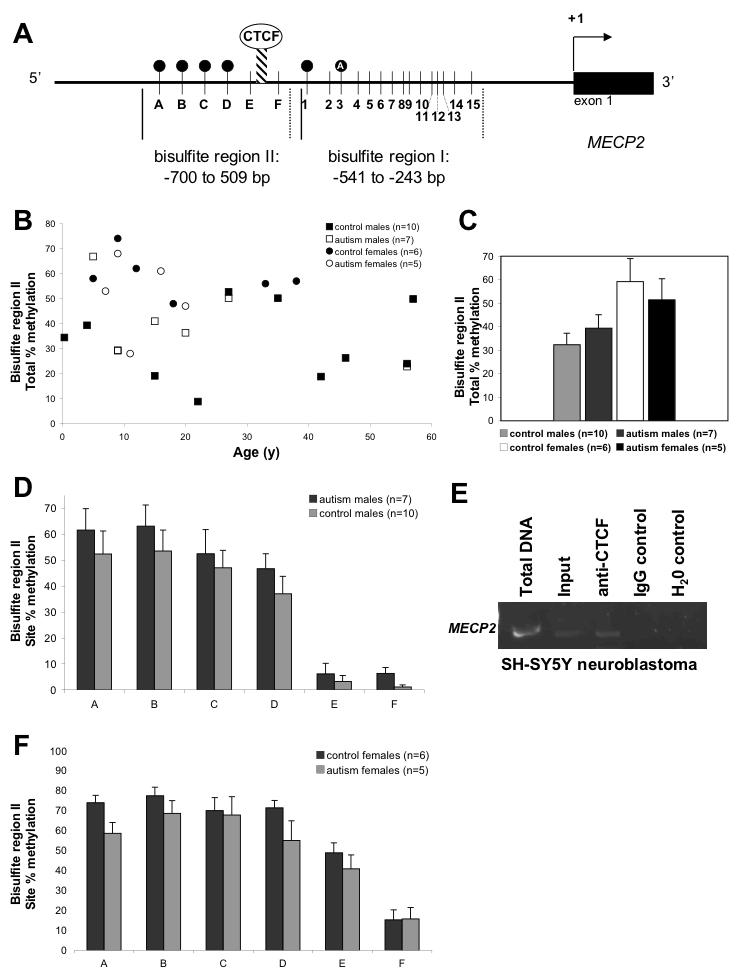
A) Schematic of MECP2 promoter regions analyzed by bisulfite sequencing. Vertical lines indicate CpG sites, designated as A-F for bisulfite region II and 1-15 for bisulfite region I. The hatched bar depicts the location of the boundary between methylated and unmethylated regions on the active X chromosome in males and females. Methylation sites are shown as filled circles and the filled circle with a “A” represents an autism-specific methylation site previously described (Nagarajan et al, 2006). B) The total percent methylation of all sites in bisulfite region II are graphed for each brain sample, designated as control males, autism males, control females and autism females. MECP2 region II shows a high degree of variable methylation independent of age, gender, or clinical status. C) The total percent methylation for MECP2 bisulfite region II is shown with each category grouped, showing an only modest nonsignificant increased methylation in females compared to males and no significant difference between autism and control. D) Male brain samples were compared for percent methylation of each CpG site within MECP2 bisulfite region II (A-F). While no significant differences were observed between autism and control males in the highly methylated sites A-D, there was an abrupt transition observed between sites D and E from a highly methylated region to a mostly unmethylated region at sites E and F. Control and autism brain samples showed significantly higher % DNA methylation of combined sites A-D compared to sites E-F by t-test (P<0.0001). Interestingly, autism males showed a trend of increased methylation (P=0.07) at site F which borders the sites in bisulfite region I previously shown to have increased methylation in autism males (A and Nagarajan et al, 2006). E) ChIP using chromatin isolated from SH-SY5Y and a specific anti-CTCF antibody or nonspecific antibody (IgG control). “Total DNA” represents non-manipulated genomic DNA from SH-SY5Y cells, while “input” is the DNA sample isolated from chromatin but without immunoprecipitation. The anti-CTCF (but not IgG control) showed specific enrichment of MECP2 (-595 to -389) over input. F) Female brain samples were compared for percent methylation of each CpG site within MECP2 bisulfite region II (A-F). Similar to male samples, no significant differences were observed between autism and control females, and sites A-D were methylated at higher than the expected 50% methylation (hatched line), demonstrating partial methylation of the active allele, as in males. Similar to male samples, sites A-D combined had significantly higher % methylation than sites E-F combined for female control and autism brain samples (P<0.0001).
Autism and control females showed very similar methylation patterns at individual sites within bisulfite region II (Figure 1E). Site E demonstrated approximately 50% methylation expected for X chromosome inactivation, while sites A-D showed 70-80% methylation, suggesting methylation of both active and inactive alleles for these four sites. Similar to male samples (Figure 1D), sites A-D exhibited significantly higher methylation than sites E-F for control female brain DNA samples (P<0.0001, Figure 1E). No significant differences were observed between autism and control females for any of the 6 CpG sites in bisulfite region II.
The DNA sequence between MECP2 sites D and F contained a putative binding site for the chromatin boundary factor CTCF (Filippova et al., 1996). In order to determine if CTCF binds to this region in neurons, chromatin immunoprecipitation (ChIP) was performed in the human neuroblastoma cell line SH-SY5Y. Figure 1F shows that ChIP with an anti-CTCF antibody specifically enriched MECP2 (-595 to -389) over input control, while a nonspecific antibody control (IgG) did not. These results demonstrate that CTCF binds to the methylation transition region in human neuronal cells and suggests that CTCF may mark the boundary of methylation on the active allele of MECP2.
Aberrant methylation status for MECP2 in female autism brain samples revealed by bisulfite sequencing
In order to separate active and inactive X chromosome alleles in female brain DNA samples, we analyzed individual clones from bisulfite sequencing for the number of CpG sites methylated. Both regions from the MECP2 promoter (Figure 1A) were analyzed as well as a highly characterized region of AR used for the XCI assay. At X-linked promoters subject to XCI, the inactivated X (Xi) tends to be methylated while the active X (Xa) is unmethylated in females, whereas the single X in males is unmethylated and active. Due to the presence of both inactive and active X chromosomes in female samples, we observed both hypo- and hypermethylated clones. For MECP2, however, control female brain DNA samples showed greater than the expected hypermethylated alleles (Supplementary Figure 1), likely due to a bias towards the methylated allele in the PCR reaction following bisulfite conversion for this highly CpG-rich promoter (Figure 2A, B, Supplementary Figure 1). The graphs in Figure 2 were used to divide clones as: Xa alleles, namely those with no CpG sites methylated; Xi alleles with >50% of maximum methylated sites; and a third infrequent category of clones that were not clearly Xa or Xi (X?). For both MECP2 promoter regions, autism females showed an increase in the percentage of alleles with undefined XCI status compared to controls, although for AR control the percentage of X? alleles was low in both autism and controls (Figure 2C, D). The presumed aberrant methylation in X? clones for autism females was not specific to one site but was spread throughout the different possible CpG sites for both MECP2 Regions I and II (Figure 2E, F). While these data are suggestive of aberrant methylation of the MECP2 promoter in autism females, problems with bias towards the methylated allele in control female DNA samples with the bisulfite sequencing assay complicate the analyses.
Figure 2. Determination of active and inactive X chromosome alleles of MECP2 and AR from bisulfite sequencing data.
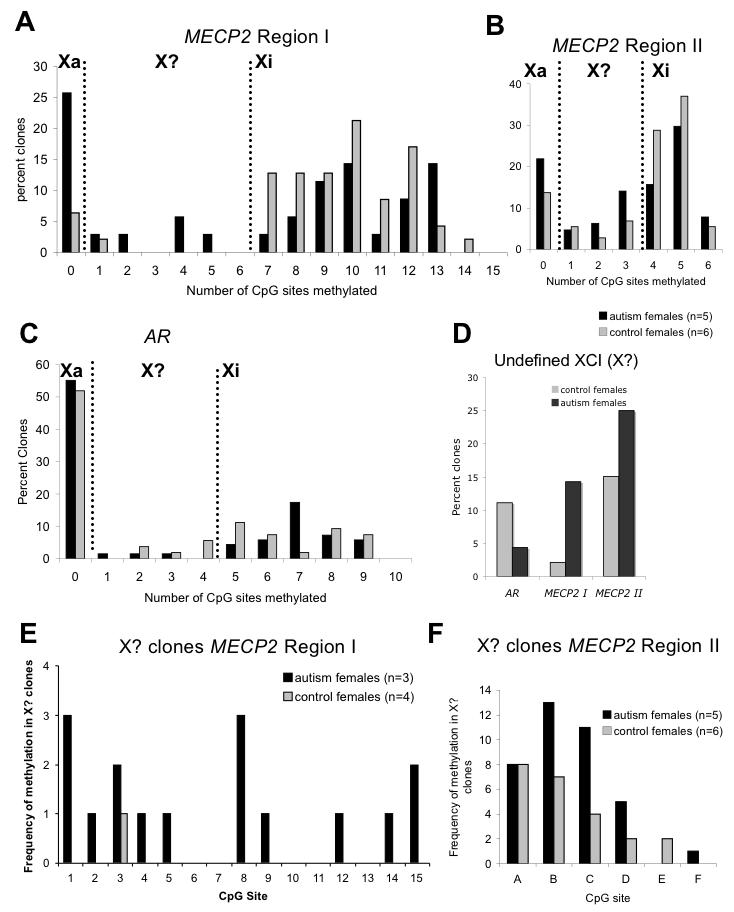
(A-D) Each clone from bisulfite sequencing data from autism or control females was analyzed for the number of CpG sites methylated in each clone (from data in Supplementary Figure 1). Histograms represent the distribution of percentage of clones based on degree of methylation. The bimodal peaks observed at all three loci are consistent with X chromosome inactivation. Clones with no sites methylated are designated as clear active alleles (Xa), clones with >50% of maximum number of methylated sites are designated as inactive (Xi), and the intervening clones as potentially undefined XCI status (X?). A) MECP2 region I has a maximum of 14 out of 15 possible methylation sites and control females show a clear bimodal peak between Xa and Xi, although there was an inherent bias towards Xi clones (7-14 sites methylated) for this highly CpG dense promoter. B) MECP2 region II has 6 possible methylation sites and more clones within the undefined XCI (X?) category (1-3 sites methylated). C) The control X linked gene AR has a maximum of 9 out of 10 possible methylation sites. Although most clones from AR fell clearly into either Xa or Xi categories, the number of methylated sites on the inactive X (Xi) was variable, ranging from 5-9 sites and no clones showing complete methylation of all 10 sites. D) The total percentage of undefined XCI (X?) clones for autism and control females is graphed for each of the three loci. Interestingly, both MECP2 loci (but not AR) show an increase in X? clones for autism compared to control females, suggesting aberrant methylation of MECP2 in females with autism. E-F) To examine the possibility of site specificity of methylation in clones showing undefined XCI status, the frequency of methylation of each possible CpG site is graphed for Region I (E) and Region II (F) only for X? clones.
X chromosome inactivation (XCI) in autism female frontal cortex
Since AR showed the expected patterns of XCI methylation in autism brain DNA samples, we decided to use the PCR-based XCI assay at AR that is commonly used to assess XCI ratios and determine skewed XCI in females. Skewed XCI was previously reported in small study of autism female blood cells (Talebizadeh et al., 2005) but not replicated in a much larger recent study (Gong et al., 2008). To date, XCI has not been described in DNA samples isolated from autism brain. We characterized XCI in autism brain DNA samples with the HpaII androgen receptor (AR) methylation assay. Table 1 shows XCI in DNA samples obtained from female postmortem frontal cortex and blood. DNA samples from female controls displayed a normal range of XCI values, ranging from 39.9% to 66.1%, with brain and blood derived DNA samples showing similar ranges. DNA isolated from females with autism had a comparable range of values, and no individual from either group displayed skewed X-inactivation. Although the small sample size precludes definitive conclusions, highly skewed X-inactivation does not appear to be frequent in female autism frontal cortex or blood.
Table 1.
XCI skewing determination in female autism brain and blood
| Sample # | Age (y) |
AR small allele (bp) |
AR large allele (bp) |
% XCI skew | |
|---|---|---|---|---|---|
| Control Brain | 1377 | 5 | 287 | 295 | 59.8 |
| 3835 | 9 | 262 | 283 | 42.0 | |
| 812 | 18 | 281 | 289 | 39.9 | |
| 1136 | 33 | 270 | 286 | 59.1 | |
| 1406 | 38 | 279 | 284 | 62.7 | |
|
Brain Control Average |
21 | 276 | 287 | 52.7 | |
| Autism Brain | 1174 | 7 | 276 | 287 | 54.1 |
| 1182 | 9 | 259 | 281 | 35.0 | |
| 5342 | 11 | 276 | 287 | 22.0 | |
| 3924 | 16 | 271 | 279 | 58.0 | |
| 1638 | 20 | 273 | 281 | 59.9 | |
|
Brain Autism Average |
13 | 271 | 284 | 42.7 | |
| Control Blood | Control 1 | 282 | 290 | 62.3 | |
| Control 2 | 276 | 293 | 66.1 | ||
| Control 3 | 270 | 287 | 61.2 | ||
| Control 4 | 284 | 298 | 58.5 | ||
|
Blood Control Average |
278 | 292 | 62.0 | ||
| Autism Blood | Autism 1 | 284 | 288 | 40.0 | |
| Autism 2 | 282 | 296 | 51.4 | ||
| Autism 3 | 276 | 282 | 78.4 | ||
| Autism 4 | 276 | 282 | 48.7 | ||
|
Blood Autism Average |
280 | 287 | 54.6 | ||
Blood X chromosome inactivation patterns in mothers of males with autism
As one possible explanation for aberrant methylation patterns observed in DNA samples isolated from autism males, we hypothesized that epigenetic dysregulation of the maternally inherited X chromosome could result in aberrant methylation in male offspring. This dysregulation might be observed as incomplete erasure of methylation patterns on the X chromosome in oocytes, which are the origin of the single X chromosome in males. X chromosome epigenetic dysregulation in mothers of male offspring might be reflected by skewed XCI in maternal somatic tissues, such as blood. Alternatively, skewed XCI in such mothers could result from protective inactivation of X-linked mutations, which can be passed on to affected males.
Using blood samples obtained from the UC Davis CHARGE study (Hertz-Picciotto et al., 2006), we extracted genomic DNA from red blood cell pellets from four groups of mothers, specifically those having: 1) sons with autistic disorder (AD); 2) sons with the broader phenotype of autism spectrum disorder (ASD); 3) sons with developmental delay (DD); and 4) typically developing sons (TD). Sample numbers and characteristics are shown in Table 2. The RBC pellets contain mostly enucleated erythrocytes and a smaller number of polymorphonuclear (PMN) cells, which we believe to be an ideal cell type for studying XCI because it does not undergo the cycling and clonal expansion common to lymphocytes. XCI patterns in DNA samples from control mothers (those with typically developing sons) are shown in Figure 3. A broad range of XCI values was observed, with some samples exhibiting skewed (< 15% or >85%) or highly skewed (<5% or >95%) XCI. AD, ASD, and DD mothers displayed similar overall XCI patterns compared to controls, without any significant differences between groups (Table 2). We conclude that XCI ratios are within the normal range in DNA samples from mothers of sons with autism or ASD, at least in the cell types under examination.
Table 2.
X inactivation in mothers of autism, ASD, developmental delay, and typically developing sons
| n= | number of uninformative samples (%) |
avg age (y)a |
avg small allele size (bp)a |
avg large allele size (bp)a |
avg % skewingab |
avg inactive allele size (bp)ac |
% mothers with < 5% or > 95% skewinga |
% mothers with < 15% or > 85% skewinga |
|
|---|---|---|---|---|---|---|---|---|---|
| Typical Development | 23 | 5 (22) | 33 | 274 | 286 | 53 | 280 | 11 | 17 |
| Delayed Development | 24 | 3 (13) | 32 | 275 | 287 | 45 | 282 | 0 | 10 |
| Autism | 27 | 2 (7) | 35 | 276 | 288 | 46 | 284 | 8 | 16 |
| ASD | 25 | 3 (12) | 36 | 277 | 286 | 60 | 280 | 9 | 18 |
Notes:
- for informative samples
- percent of cells with small allele inactive
- average of allele sizes (bp) of the alleles that are inactivated > 50%
Figure 3. X chromosome inactivation in blood of mothers with sons in four diagnostic categories.
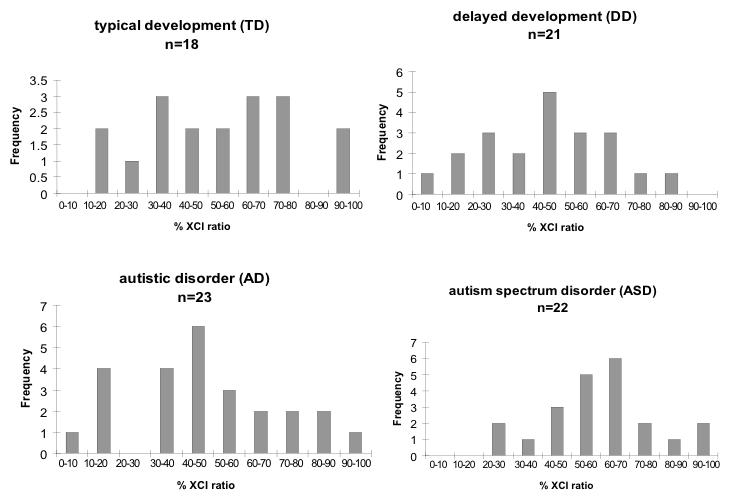
X-axes of histograms are binned into 10% intervals, showing the percent cells with the small allele inactivated. No significant differences were observed between groups (Fisher’s exact test).
We next hypothesized that mothers with skewed XCI would be observed to more frequently transmit the preferentially inactivated allele to sons with autism compared to typically developing sons, perhaps as a result of preferential inactivation of a deleterious X-linked mutation linked to AR. RBC pellet DNA was obtained for sons of mothers with skewed XCI >85:15 and <15:85. The size of the AR allele (which is inherited from one of the alleles present in the mother) was determined by capillary electrophoresis and shown in Table 3. Males with mothers displaying skewed XCI inherited both preferentially inactivated (skewed) and activated AR alleles, suggesting that protective skewing against X-linked mutations near AR is not specifically preferential in mothers of sons with autism, at least not in this small sample of primarily simplex cases.
Table 3.
Inheritance of AR alleles in sons of mothers with skewed X inactivation
| Child Diagnosis | Mom Age (Y) | Mom XCI % Skewing |
Mom small allele (bp) |
Mom large allele (bp) |
allele son inherited (bp) |
did son inherit skewed allele? |
|---|---|---|---|---|---|---|
| ASD | 36.3 | 92.0 | 279 | 282 | 282 | no |
| ASD | 36.3 | 85.0 | 286 | 292 | 286 | yes |
| ASD | 32.4 | 97.1 | 270 | 276 | ND | ND |
| Autism | 43.2 | 3.0 | 276 | 299 | 276 | no |
| Autism | 31.4 | 12.9 | 284 | 297 | 297 | yes |
| Autism | 37.5 | 12.1 | 277 | 291 | 291 | yes |
| Autism | 30.8 | 91.1 | 273 | 293 | 273 | yes |
| Delayed | 28.0 | 85.7 | 276 | 279 | ND | ND |
| Delayed | 39.5 | 12.7 | 271 | 282 | ND | ND |
| Delayed | 36.2 | 6.9 | 274 | 277 | 277 | yes |
| Typical | 35.1 | 93.4 | 267 | 276 | 276 | no |
| Typical | 25.6 | 96.6 | 270 | 276 | 270 | yes |
| Typical | 34.1 | 14.4 | 270 | 279 | 270 | no |
| Typical | 32.6 | 14.8 | 282 | 285 | 285 | yes |
DISCUSSION
Epigenetic defects, such as aberrant promoter methylation, can influence gene expression and result in pathological phenotypes, especially during tumorigenesis. As an example of an epigenetic defect observed in autism, we previously reported increased MECP2 promoter methylation in autism male frontal cortex that correlated with reduced MeCP2 expression in the frontal cortex (Nagarajan et al., 2006). In this study, we investigated several potential causes of aberrant MECP2 promoter methylation. First, we identified a novel boundary element 600 kb upstream of MECP2, suggesting the potential for methylation spreading from this boundary in males with autism exhibiting aberrant downstream promoter methylation. Second, we divided active and inactive alleles of MECP2 and AR in female brain samples and demonstrated evidence suggestive of aberrant MECP2 promoter methylation in females with autism compared to controls. Third, we provided evidence against skewed XCI as being a common epigenetic occurrence in females with autism or the mothers of males with autism. These results are novel and informative in understanding the extent of X-linked epigenetic alterations in the inheritance of autism.
Chromatin boundary elements are important epigenetic determinants in maintaining active promoters as separate from neighboring inactive DNA. The methylation boundary element we identified approximately 600 bp upstream of MECP2 contains several consensus binding sites for the known boundary element CTCF and here we directly demonstrate that CTCF binds to this region in human neuroblastoma cells. Since binding of CTCF is sensitive to DNA methylation, it is an excellent candidate for discriminating methylated from unmethylated sequences (Lewis et al., 2004). Furthermore, since CTCF binding also protects boundary elements from further methylation, CTCF binding could serve to maintain the active unmethylated MECP2 promoter free of the high degree of methylation observed in the immediately adjacent 5′ region. The trend for increased methylation of autism male DNA samples for two sites within the chromatin boundary region suggests that CTCF binding to these sites may be blocked, resulting in further spreading of methylation into the MECP2 promoter. CTCF has been shown to bind to boundaries of X chromosome inactivation in mouse (Filippova et al., 2005), consistent with this hypothesis. Future experiments could be designed to determine if CTCF binding or expression is deficient in autism brain samples based on these preliminary findings.
Aberrant methylation of MECP2 was more difficult to assess in females with autism because of the fewer number of brain samples currently available and the problem of primer bias toward the methylated allele of the highly CpG rich MECP2 promoter. Interestingly, female autism brain samples showed less primer bias than control female samples, presumably because of the aberrant methylation patterns reflected by the increased number of alleles not easily defined as active or inactive. Since female autism brain samples have also been observed to have aberrantly reduced MeCP2 expression in frontal cortex in addition to males, future studies with novel methods to determine aberrant MECP2 methylation with greater sensitivity in females are warranted.
Our study is novel in the examination of XCI patterns of individual alleles by bisulfite sequencing in addition to the commonly used AR methylation test that relies on restriction enzymes followed by PCR. While both methods revealed XCI in females, important insights into the number of individual methylation sites that are required for silencing AR and MECP2 on the inactive X chromosome were revealed exclusively by bisulfite sequencing, in which 50% methylation of CpG sites on a single allele appeared sufficient for inactivation. The AR PCR based method appears to be more accurate for determining XCI ratios as the number of cells sampled is much higher and PCR primers are not biased toward the methylated allele in CpG rich promoters. However, since the methyl sensitive restriction enzyme HpaII only targets two of the available CpG sites in AR exon 1 that are methylated an average of 90% of the time (data not shown), there is expected to be a roughly 10% false positive rate expected for Xa by the conventional PCR based assay of AR methylation.
Our data showing insignificant differences in XCI ratios between autism and control females or mothers of autism males are not consistent with a prior report that suggested frequent XCI skewing in autism females and their mothers (Talebizadeh, 2005). The differences in the two studies can likely be explained by differences in the small samples and different methodologies. Our small study is consistent with a recent very large study of 543 mothers and 163 females with ASD that also did not observe significant differences in XCI skewing in families with ASD (Gong et al., 2008). These studies make ASD mothers distinct from X linked mental retardation carrier mothers, who have a high frequency of skewed XCI (Plenge et al., 2002).
Recent evidence has implicated de novo genetic defects in autism (Marshall et al., 2008; Sebat et al., 2007). We propose that de novo mutations and specific epigenetic alterations, such as increased MECP2 promoter methylation in autism males, contribute substantially to the autism phenotype through dysregulation of genes important in brain development and function. One important next step is to use genomic approaches to assess both global and gene-specific epigenetic changes in multiple autism tissues. Identification of these molecular defects holds promise for the creation of diagnostic tests and eventually therapeutic targets.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Maria Peñaherrera and Courtney Hanna for technical assistance and Joe Schauer for human RBC samples, and Jane Pickett for identifying available brain samples. Human brain tissue samples were provided by the Autism Tissue Program, the NICHD Brain and Tissue Bank for Developmental Disorders, and the Harvard Brain Tissue Resource Center (supported in part by NIH R24MH-068855). This work was supported in part by NIH R01HD048799, R01ES015171, R01HD41462, and R01ES015359.
REFERENCES
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23(2):185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Beever CL, Stephenson MD, Penaherrera MS, Jiang RH, Kalousek DK, Hayden M, et al. Skewed X-chromosome inactivation is associated with trisomy in women ascertained on the basis of recurrent spontaneous abortion or chromosomally abnormal pregnancies. Am J Hum Genet. 2003;72(2):399–407. doi: 10.1086/346119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittel DC, Theodoro M, Kibiryeva N, Fischer W, Talebizadeh Z, Butler MG. Comparison of X chromosome inactivation patterns in multiple tissues from human females. J Med Genet. 2007 doi: 10.1136/jmg.2007.055244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CJ, Robinson WP. The causes and consequences of random and non-random X chromosome inactivation in humans. Clin Genet. 2000;58(5):353–363. doi: 10.1034/j.1399-0004.2000.580504.x. [DOI] [PubMed] [Google Scholar]
- de Belle I, Cai S, Kohwi-Shigematsu T. The genomic sequences bound to special AT-rich sequence-binding protein 1 (SATB1) in vivo in Jurkat T cells are tightly associated with the nuclear matrix at the bases of the chromatin loops. J Cell Biol. 1998;141(2):335–348. doi: 10.1083/jcb.141.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippova GN, Cheng MK, Moore JM, Truong JP, Hu YJ, Nguyen DK, et al. Boundaries between chromosomal domains of X inactivation and escape bind CTCF and lack CpG methylation during early development. Dev Cell. 2005;8(1):31–42. doi: 10.1016/j.devcel.2004.10.018. [DOI] [PubMed] [Google Scholar]
- Filippova GN, Fagerlie S, Klenova EM, Myers C, Dehner Y, Goodwin G, et al. An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol Cell Biol. 1996;16(6):2802–2813. doi: 10.1128/mcb.16.6.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong X, Bacchelli E, Blasi F, Toma C, Betancur C, Chaste P, et al. Analysis of X chromosome inactivation in autism spectrum disorders. Am J Med Genet B Neuropsychiatr Genet. 2008 doi: 10.1002/ajmg.b.30688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodfellow PJ, Mondello C, Darling SM, Pym B, Little P, Goodfellow PN. Absence of methylation of a CpG-rich region at the 5′ end of the MIC2 gene on the active X, the inactive X, and the Y chromosome. Proc Natl Acad Sci USA. 1988;85(15):5605–5609. doi: 10.1073/pnas.85.15.5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz-Picciotto I, Croen LA, Hansen R, Jones CR, van de Water J, Pessah IN. The CHARGE study: an epidemiologic investigation of genetic and environmental factors contributing to autism. Environ Health Perspect. 2006;114(7):1119–1125. doi: 10.1289/ehp.8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppke P, Maier EM, Warnke A, Brendel C, Laccone F, Gartner J. Very mild cases of Rett syndrome with skewed X inactivation. J Med Genet. 2006;43(10):814–816. doi: 10.1136/jmg.2006.042077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YH, Sahoo T, Michaelis RC, Bercovich D, Bressler J, Kashork CD, et al. A mixed epigenetic/genetic model for oligogenic inheritance of autism with a limited role for UBE3A. Am J Med Genet. 2004;131A(1):1–10. doi: 10.1002/ajmg.a.30297. [DOI] [PubMed] [Google Scholar]
- Knudsen GP, Neilson TC, Pedersen J, Kerr A, Schwartz M, Hulten M, et al. Increased skewing of X chromosome inactivation in Rett syndrome patients and their mothers. Eur J Hum Genet. 2006;14(11):1189–1194. doi: 10.1038/sj.ejhg.5201682. [DOI] [PubMed] [Google Scholar]
- Le Couteur A, Lord C, Rutter M. Western Psychological Sevices; Los Angeles: 2003. Autism Diagnostic Interview-Revised. [Google Scholar]
- Lewis A, Murrell A. Genomic imprinting: CTCF protects the boundaries. Curr Biol. 2004;14(7):R284–286. doi: 10.1016/j.cub.2004.03.026. [DOI] [PubMed] [Google Scholar]
- Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18(11):1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- Lord C, Rutter M, DiLavore P, Risi S. The Autism Diagnostic Obersrvation Schedule Manual. Western Psychological Sevices; Los Angeles: 2000. [Google Scholar]
- Lyon MF. Gene action in the X-chromosome of the mouse (Mus musculus L.) Nature. 1961;190:372–373. doi: 10.1038/190372a0. [DOI] [PubMed] [Google Scholar]
- Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82(2):477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan RP, Hogart AR, Gwye Y, Martin MR, Lasalle JM. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics. 2006;1(4):172–182. doi: 10.4161/epi.1.4.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plenge RM, Stevenson RA, Lubs HA, Schwartz CE, Willard HF. Skewed X-chromosome inactivation is a common feature of X-linked mental retardation disorders. Am J Hum Genet. 2002;71(1):168–173. doi: 10.1086/341123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice CE, Baio J, Van Naarden Braun K., Doernberg N, Meaney FJ, Kirby RS. A public health collaboration for the surveillance of autism spectrum disorders. Paediatr Perinat Epidemiol. 2007;21(2):179–190. doi: 10.1111/j.1365-3016.2007.00801.x. [DOI] [PubMed] [Google Scholar]
- Samaco RC, Nagarajan RP, Braunschweig D, LaSalle JM. Multiple pathways regulate MeCP2 expression in normal brain development and exhibit defects in autism-spectrum disorders. Hum Mol Genet. 2004;13(6):629–639. doi: 10.1093/hmg/ddh063. [DOI] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316(5823):445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talebizadeh Z, Bittel DC, Veatch OJ, Kibiryeva N, Butler MG. Brief report: non-random X chromosome inactivation in females with autism. J Autism Dev Disord. 2005;35(5):675–681. doi: 10.1007/s10803-005-0011-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thatcher KN, Lasalle JM. Dynamic changes in Histone H3 lysine 9 acetylation localization patterns during neuronal maturation require MeCP2. Epigenetics. 2006;1(1):24–31. doi: 10.4161/epi.1.1.2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thatcher KN, Peddada S, Yasui DH, Lasalle JM. Homologous pairing of 15q11-13 imprinted domains in brain is developmentally regulated but deficient in Rett and autism samples. Hum Mol Genet. 2005;14(6):785–797. doi: 10.1093/hmg/ddi073. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.