

Abstract
A common pathological finding in autism is a localized deficit in Purkinje cells (PCs). Cerebellar abnormalities have also been reported in schizophrenia. Using a mouse model that exploits a known risk factor for these disorders, maternal infection, we asked if the offspring of pregnant mice given a mid-gestation respiratory infection have cerebellar pathology resembling that seen in these disorders. We also tested the effects of maternal immune activation in the absence of virus by injection of the synthetic dsRNA, poly(I:C). We infected pregnant mice with influenza on embryonic day 9.5 (E9.5), or injected poly(I:C) i.p. on E12.5, and assessed the linear density of PCs in the cerebellum of adult or postnatal day 11 (P11) offspring. To study granule cell migration, we also injected BrdU on P11. Adult offspring of influenza- or poly(I:C)-exposed mice display a localized deficit in PCs in lobule VII of the cerebellum, as do P11 offspring. Coincident with this are heterotopic PCs, as well as delayed migration of granule cells in lobules VI and VII. The cerebellar pathology observed in the offspring of influenza- or poly(I:C)-exposed mice is strikingly similar to that observed in autism. The poly(I:C) findings indicate that deficits are likely caused by the activation of the maternal immune system. Finally, our data suggest that cerebellar abnormalities occur during embryonic development, and may be an early deficit in autism and schizophrenia.
Keywords: autism, schizophrenia, influenza, cerebellum, Purkinje cell, poly(I:C)
Introduction
Maternal respiratory infection can increase the risk in the offspring for mental disorders such as schizophrenia (reviewed by Brown, 2006; Penner and Brown, 2007). The most direct evidence for this comes from a prospective study of pregnant women with medically documented respiratory infections, where the risk for schizophrenia in the offspring is increased 3-fold by infection in the second trimester (Brown 2006). Moreover, the presence of anti-flu antibodies in maternal serum in the first half of pregnancy is associated with a 3–7-fold increase in risk, and elevated levels of the cytokine interleukin-8 in maternal serum is also associated with increased risk for schizophrenia in the offspring (Brown, et al., 2004a; Brown et al., 2004b). There is similar serological evidence linking rubella and toxoplasma maternal infections with schizophrenia (Penner and Brown, 2007). Although the epidemiology is much less extensive for autism, a >200-fold increase in autism incidence was found in the offspring of maternal rubella infection cases (Chess 1977). While rubella infections can also involve the fetus, smaller studies of other maternal viral infections support the idea that this can be a risk factor for autism (reviewed by Ciaranello 1995; Hyman et al., 2006; Moy and Nadler, 2007). These findings are remarkable given the strong genetic contribution to autism and schizophrenia, which, at present, cannot be controlled for in such epidemiological studies. If it were possible to confine epidemiological analysis to just those individuals with the appropriate susceptibility genotype, the figures for risk cited above could be much higher.
The offspring of mothers with infections display diverse neuropathology, depending in part on the nature and timing of the infection. Following late pregnancy intrauterine bacterial infections, cortical malformation and white matter damage is often seen in the offspring, leading to severe behavioral changes (Dammann et al., 2002; Hagberg et al., 2002). While pathology is more subtle and variable in mental illness, several reproducible changes have been identified. A common finding in autism is a localized loss of cerebellar PCs. In 8 studies involving 29 postmortem brains, 72% of the autism cases displayed such a deficit (Palmen et al., 2004). Another review recently concluded that 79% of the postmorten autism cases in which the cerebellum was studied displayed a deficit in PCs (Amaral et al., 2008). Although there are negative findings, this is remarkable consistency, particularly in light of the broad spectrum of ages, severity of symptoms and diversity of autism phenotypes included in these studies.
There is also a very strong inverse correlation between the magnitude of cerebellar lobule VI and VII hypoplasia and the degree of novel object exploration and stereotyped behavior in autistic children (Pierce and Couchesne, 2001; Akshoomoff et al., 2004). Moreover, functional MRI reveals abnormal cerebellar activation during motor and cognitive tasks in autistic subjects (Allen et al., 2003; Kates et al., 2004), and there are behavioral abnormalities in autism, such as abnormal eye blink conditioning and visual saccades, which are particularly relevant for the known functions of lobules VI and VII (Nowinski et al., 2005; Takarae et al., 2004). There are, however, reports of negative findings regarding the cerebellum and autism (reviewed by Kaufmann et al., 2003; Palmen et al., 2004).
There is also evidence for cerebellar pathology in schizophrenia, including PC deficits and reduced cerebellar volume, as well as behavioral evidence (saccades and eye blink conditioning) that points to pathology in lobules VI and VII (Bottmer and Schroder 2005; Brown and O’Donnell 2005; Ho et al., 2004). The identification of cerebellar pathology in these disorders supports an increasing recognition of a role for the cerebellum in higher functions such as cognition, language and learning (Allen 2006; Ramnani et al., 2001; Schmahmann 2001; Schutter et al., 2005).
In investigating the effects of maternal respiratory infection on fetal brain development, we found that adult offspring of pregnant mice given intranasal influenza on embryonic day 9.5 (E9.5) exhibit behavioral abnormalities reminiscent of autism and schizophrenia, some of which can be ameliorated by anti-psychotic drug treatment (Shi et al., 2003). Many of these behaviors can also be evoked in the absence of viral infection, by activating the maternal immune response with synthetic dsRNA (poly(I:C)) injection (Shi et al., 2003). We and Fatemi et al. found that the offspring of influenza-infected mice display histopathology consistent with that seen in schizophrenia, such as thinning of the neocortex and hippocampus, pyramidal cell atrophy, reduced levels of reelin immunoreactivity, and changes in the expression of neuronal nitric oxide synthase (NOS) and synaptosome associated protein of 25 kD (SNAP25) (Fatemi and Sidwell 2002; Fatemi and Sheikh 1999; Fatemi et al., 2005). Additional neuropathology and behavioral abnormalities have been subsequently reported in the poly (I:C) model of maternal immune activation (Meyer U et al 2005; Meyer 2006; Ozawa et al 2006; Zuckerman et al., 2003; Zuckerman et al., 2005), although no comments were made concerning cerebellar pathology. Using both the influenza-infection and poly(I:C) models, we here report a localized histopathology in the cerebellum that is very similar to that seen in autism and schizophrenia.
Materials and Methods
Viral infection
All procedures involving animals were approved by the Caltech Animal Care and Use Committee. The Balb/C mice were obtained from Simonson Laboratories (Gilroy, CA) and C57BL/6J mice were obtained from Jackson Laboratories (Bar Harbor, ME). Both strains were bred in the Caltech animal facility for several generations before use. Human influenza virus, strain A/NWS/33CHINI, was collected from the supernatant of infected MDCK cells, titered, and stored at −80 until use. Pregnant Balb/C mice were anesthetized intra-peritoneally (i.p.) with 10 mg/kg xylazine and 100 mg/kg ketamine on E9.5 (plugged day is day 0) and inoculated intra-nasally with either 6000 plaque-forming units (PFU) of human influenza virus in 90 μl phosphate buffered saline (PBS), or with PBS alone (sham-treatment), or received no treatment (naïve). When no differences were found between the sham and naive groups, the two groups were combined into a single control group. The number of pups born to control and infected mothers averaged 8 and 4, respectively. The first group of offspring (8 from infected, 8 from control; 4 males in each group; all from separate litters) were sacrificed at postnatal day 11 (P11). The second group of offspring (4 litters of infected and 3 litters of control) were injected with BrdU (dissolved at 10 mg/ml in saline and injected at 50 mg/kg) at P11. They were sacrificed either after 0.5 hour (6 from infected and 5 from control mothers), day 15 (P15) (6 from infected and 5 from control), or P17 (6 from infected and 7 from control). The third group of offspring was sacrificed as adults (9–14 months of age). These offspring were weaned at P21, and males and females were caged separately in groups of 2–4. Starting at 6 weeks, they were evaluated in open-field and novel object tests, and in a prepulse inhibition (PPI) assay, as described previously (Shi et al., 2003). We selected 9 mice from infected mothers with PPI and open field deficits (5 females and 4 male) and 6 from control mothers with normal behavior (3 females and 3 males) for histochemical analysis.
Poly(I:C) maternal immune activation
C57BL/6J mice were used for these experiments as they tended to give larger litter sizes than Balb/C mice following treatment. Four pregnant mice were injected i.p. with 20 mg/kg poly(I:C) (Sigma, Cat. P9582) freshly dissolved in 0.9% sterile PBS, in an injection volume of 8 ml/kg, on E12.5. Five control females were injected with the same volume of PBS. The offspring were undisturbed until weaning on P21 (32 control, 24 poly(I:C)). Offspring were behaviorally tested starting at 6 weeks and found to have significant deficits (p < 0.05) in latent inhibition, open field and novel object behavior and a trend toward significance in the PPI test (for descriptions of the behavior tests, see Smith et al., (2007) and Shi et al. (2003)). Four control and 4 poly (I:C)-exposed offspring (2 females in each group), each from different litters, were randomly selected for sacrifice at 4 months of age for histology.
Immunohistochemistry
All mice were perfused intracardially with 4% paraformaldehyde in PBS, and post-fixed in that solution at 4° overnight. Following post-fixation, the brains were transferred to a sucrose gradient (from 10 to 20%) at 4° until the tissue sank to the bottom of the container. The brains were then rapidly frozen using a methylbutane bath in dry ice and were stored at −80° until use. Cryostat sections of the cerebellum vermis were cut at 18 μm for adults and 14 μm for young mice. For calbindin immunostaining, sections were treated with 0.05% sodium citrate buffer and heated to boiling seven times over 30 min in a microwave oven. Sections were then cooled and treated with the M.O.M kit (Vector) to block endogenous mouse IgG, blocked with 10% normal goat or horse serum for 30 min, and incubated overnight at 4° with a 1:200 dilution of mouse monoclonal, anti-calbindin, ascites antibody (Sigma). For BrdU staining, slides were treated with 2 N HCl in Tris buffer (TBS) and 0.5% Tween for 60 min at 37°, followed by 10 minutes in 0.1 M sodium borate. They were incubated overnight with rat monoclonal, anti-BrdU antibody (Immunologicals.com) in TBS containing 10% normal goat serum, 0.5% Tween at 4°, washed in TBS (pH 8.0) with 0.5% Tween-20 for 20 min at room temperature. For GABA α6 receptor staining, sections were incubated in 10% normal goat serum for 30 min, followed by overnight treatment with rabbit anti-GABA α6 receptor antibody (1:500, Chemcon). For florescent images, AlexaFlour 488- or 568-conjugated, goat anti-mouse or anti-rat (1:200, Molecular Probes) antibodies were applied at RT for 2 hours, followed by several PBS washes and mounting with 50% glycerol. For immuohistochemistry, HRP-conjugated secondary antibody (goat anti-rat, Vector) was applied at RT for 2 hours, followed by several PBS washes. The DAB kit (Vector) was used for detection according to manufacturer’s instructions, followed by counterstaining with 0.45% Cresyl Violet, clearing through xylene, and mounting with Permount (Fisher).
Green NeuroTrace Fluorescent Nissl staining
Every fourth section of P11 and P17 cerebellum vermis in the BrdU-injected groups was stained for Green NeuroTrace Fluorescent Nissl Stain (Molecular Probes), which binds to Nissl substance and is present exclusively in the somata of neurons. Sections were treated with 0.1% Triton X-100 in PBS for 10 min, washed in PBS 3 times, incubated with 1:200 NeuroTrace in PBS for 20 min, and washed for 10 min in 0.1% Triton X-100, washed in PBS, and mounted.
Image capture
A Nikon fluorescence microscope was used to observe sections; sections of stained brains were first captured digitally using an RT Slider Spot digital camera (Nikon) at 2.5x, 4x, 10x and 20x magnification. Images were then imported into PhotoShop7.0 (Adobe) or ImageJ (NIH) to analyze. Images that were used for counting PCs were captured at 10x, and at this magnification we were able to capture the entire thickness of the section in the focal plane, thus ensuring complete counting of PCs without using confocal microscopy.
Quantitation
To ensure exhaustive sampling of the cerebellum, every tenth section in adult, and every fourth section in P11, were stained for calbindin. The linear density of PCs was determined by measuring the length of a line drawn along the PC layer from the middle of the cleft between lobules VI and VII to the cleft between lobules VII and VIII, and counting PCs along this line. In the case of lobule V, a line was drawn along the PC layer in the dorsal half of this very large lobule, and PC density determined along this line. The average linear density for each adult animal was calculated by averaging the linear density from between 7–12 sections covering the entire lobule VII. In P11 cerebella, there was a high variability in the length (size) of the lobules, while the PC number/lobule was relatively constant. Therefore, for this developmental stage we used the total PCs per lobule as the data for comparison between experimental groups.
Every fourth cerebellum vermis section in BrdU-injected animals was stained for BrdU. For the brains collected 0.5 hour after BrdU injection on P11, the linear density of granule cells (GCs) in lobule VII was determined by measuring the length of a line drawn along the outline of the lobule (defined as above), and counting all BrdU+ GCs. For the brains sacrificed at P15 and P17, BrdU+ cells in the molecular layer (ML) were counted.
Every fourth section in P17 brains was stained with Green NeuroTrace fluorescent dye. The thickness of the EGL and ML was measured (ImageJ) at three sites in each lobule.
Statistical analysis
Statistical analysis was performed with Prism software (Graphpad). In all experiments, we averaged data from sections from individual mice. The averages were then used to determine the mean ± SEM for each experimental group, so that the N reported reflects the number of mice used, not the number of sections analyzed. Statistical significance was assessed using Student’s T test.
Results
Purkinje cells in adult offspring
Given the common finding of PC deficits in autism, and the evidence for its localization to lobules VI and VII, we analyzed PCs in the adult offspring of influenza-infected Balb/c mothers. Using an anti-calbindin monoclonal antibody, which in the cerebellum selectively binds PCs, we find fewer of these neurons, specifically in lobule VII, compared to controls (Fig. 1). The section shown here is representative of the 33% difference in linear density between the experimental offspring and the offspring of control mothers (Fig. 2a). To illustrate the localized nature of the deficit, we counted PCs in lobule V and find no difference from the control (Fig. 2a). The deficit was equally pronounced in both male and female offspring; two-way ANOVA revealed a significant effect of prenatal treatment (p < 0.01), with no effect of gender. Qualitative examination of other lobules (I–IV and VIII–X) also indicates no difference between experimental and control cerebella (Fig. 1). The cross-sectional size of the lobules was also assessed, using the linear extent from the middle of the cleft between lobules to the middle of the next cleft. Every tenth section was measured through the entire vermis. Comparing the adult offspring of control and infected mothers, no difference was found in the size of lobule VII (1.655 ± 0.049 mm vs. 1.641 ± 0.048 mm, p = 0.84).
Figure 1.

Adult offspring of infected mothers display a PC deficit in lobule VII. Calbindin staining of adult cerebella from offspring of control (A, C) and infected mothers (B, D) reveals a deficit in lobule VII in the latter. Panels C and D (bar = 200 μm) are higher magnification views of panels A and B (bar =800 μm).
Figure 2.
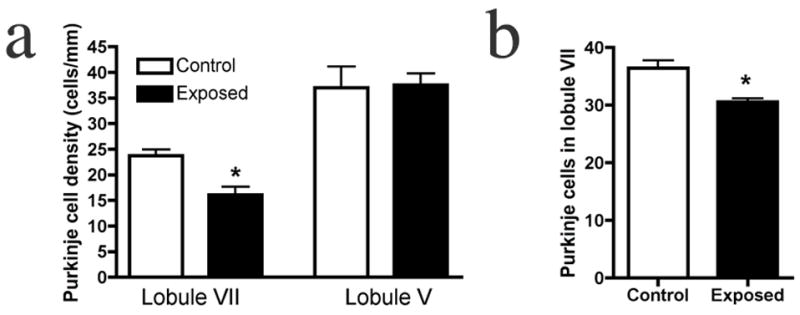
Young and adult offspring of infected mothers have a PC deficit specifically in lobule VII. (a) Quantification of PC linear density reveals a 33% deficit in lobule VII of the adult offspring of infected mothers, while no difference from controls is found in lobule V. (b) A similar, localized deficit is observed in the P11 offspring of infected mothers (* p < .01).
Purkinje cells in young offspring
To determine if the PC deficit occurs during development or as neurodegeneration in adulthood, we counted PCs at P11, the earliest stage at which these cells can be reliably stained and counted in a linear array. A statistically significant difference between the offspring of control and infected mothers, similar to that found in adults, is seen (Fig. 2b). As with adult animals, no within-group sex differences were found.
Heterotopic Purkinje cells
In further support of the deficit being a developmental phenomenon, we occasionally see heterotopic PCs in adult cerebella of mice born to infected mothers. These large, calbindin+ cells are found primarily in lobules VI and VII (Fig. 3), suggesting abnormal migration during development. We also find heterotopic PCs in the P11 offspring of infected mothers. In these younger animals, the heterotopy occurs at a higher frequency than in the adults, in some cases in every 2–3 sections. These findings, plus a lack of evidence of sick or degenerating PCs in the adult, lead us to conclude that the PC deficit largely emerges during early cerebellar development, prior to P11.
Figure 3.
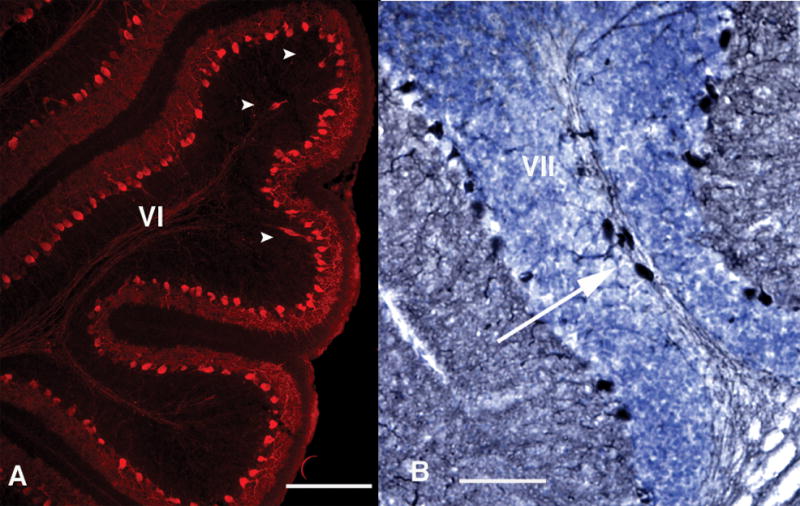
Heterotopic PCs are found in the offspring of infected mothers. Some P11 (A, bar = 200 μm) and adult (B, bar = 100 μm) offspring of infected mothers display large, calbindin+ cells (white arrowheads and arrow) in the white matter of lobules VI or VII. Such cells are rarely seen in other lobules, or in control cerebella.
Granule cells in young offspring
Considering the known developmental interactions between PCs and GCs, we charted the development of these cells as well. First, we measured the thickness of the molecular layer (ML), a layer in which GCs and PCs form synapses. In adult and P17 animals, there is no difference in the thickness of the ML (adult: control 95.0 ± 23.7 μm, exposed 99.5 ± 10.7 μm; P17: control 75.2 ± 3.2 μm, exposed 84.4 ± 4.8 μm). These data suggest that the PC reduction is not reflected in the size of the mature ML, perhaps due to the surviving PCs forming more synapses per cell to compensate.
During development, GCs are born in the external granular layer (EGL) and migrate through the ML to their final position in the internal granular layer (IGL). On E17, a time when the EGL in controls is disappearing, the EGL is significantly thicker in offspring of infected mothers (Fig. 4a–d). This effect is most pronounced in lobules VI and VII (control EGL 9.5 ± 0.4 μm, exposed EGL 15.8 ± 1.2 μm, p < 0.001), consistent with the localized deficit in PCs. The abnormally persistent EGL is eventually lost, however, as Nissl staining in adult animals reveals the normal absence of an EGL in both control and exposed offspring (Fig. 4e,f). To determine if the thicker EGL at P17 is due to a migrational delay, BrdU was injected at P11 to label newly generated GCs, and the mice sacrificed at 0.5 hours, P15 or P17. At 0.5 hours after BrdU injection, we find no difference in BrdU+ GCs in the EGL (cells per section: control, 166 ± 1.7; exposed, 174 ± 26.5), indicating normal levels of proliferation. On P15, there are similar numbers of GCs migrating through the ML towards their final position in the IGL (cells per section: control 74 ± 16, exposed 86 ± 21). However, in mice sacrificed at P17, we find significantly more BrdU+ GCs in the ML of lobule VII of exposed mice (cells per section: control 23 ± 3.5, exposed 51 ± 4.3, p < 0.001), suggesting a spatially localized migrational delay in exposed animals. In adult animals, however, no GCs are found in the ML (Fig. 4g,h).
Figure 4.

Granule cell development is abnormal in the offspring of infected mothers. At P17, control mice lack an EGL (A, C), while a persistent EGL is observed in the offspring of infected mothers (B, D), particularly around lobules VI and VII (arrowhead). In the adult, Nissl staining reveals the normal absence of an EGL in both control (E) and experimental (F) offspring. Moreover, no GABAR α6 staining is found in the ML of the adult control (G) or experimental (H) offspring, indicating that the GCs have completed their migration into the IGL. Scale bars A, B = 200μm; C–H = 100μm.
Purkinje cells in adult offspring of poly(I:C)-immune activated mothers
Many of the behavioral abnormalities seen in the offspring of infected mothers, such as deficits in prepulse inhibition of the acoustic startle (PPI), open field exploration and social interaction, can also be found in the offspring of mothers whose immune systems were activated by injection of poly(I:C) rather than by virus (Meyer U et al 2005; Meyer 2006; Ozawa et al 2006; Shi 2003; Smith 2007). Thus, it was of interest to ask if the PC deficit is also shared between these two animal models. When the cerebella of adult offspring of poly(I:C)-injected C57Bl/6 mothers are compared to controls, a difference in PC linear density is seen in lobule VII (Fig. 5). This 20% deficit is similar to that observed in the offspring of infected mothers, indicating that the mother’s inflammatory response is likely mediating the deficit seen in the maternal infection model. No differences in PC density from controls are found in lobules V or VIII (Fig. 5). No difference between control and experimental groups is found in the length of lobule VII (1.89 ± 0.061 vs. 1.92 ± 0.041 mm, p < 0.66).
Figure 5.

Purkinje cell loss in the adult offspring of immune-activated mothers is localized to lobule VII. A single poly(I:C) injection in pregnant mice causes a deficit in PC density in the adult offspring, specifically in lobule VII. (* p < 0.02)
Discussion
Although there is a strong genetic component to schizophrenia and autism, it is clear that certain environmental factors can strongly raise the risk for these disorders. One of the best studied of these environmental risk factors in schizophrenia is maternal infection, particularly respiratory infection (reviewed by Penner and Brown, 2007; Brown 2006). In autism, a highly significant effect was seen with maternal rubella infection, although increased risk has also been reported for other maternal viral infections (reviewed by Patterson 2002; Hyman et al., 2006; Moy and Nadler, 2007). In modeling this risk factor in rodents, major abnormalities have been reported in the offspring of infected or immune-activated mothers using behavioral assays that are relevant for schizophrenia and autism. These assays include PPI of the acoustic startle, social interaction, anxiety behavior in the open field and with a novel object, amphetamine-induced locomotion, and latent inhibition (Borrell et al., 2002; Fortier 2004; Meyer U et al 2005; Meyer 2006; Ozawa et al., 2006; Shi 2003; Zuckerman 2003).
Given the construct and face validity of this model for schizophrenia and autism, it was of interest to examine the cerebellum to determine if there is pathology that resembles that seen in these disorders. This is particularly important in the context of autism, where cerebellar pathology is a commonly reported histological and imaging abnormality. Our finding of a localized deficit in PCs strikingly resembles that seen in autism both in location and magnitude, with a 25% reduction in PC linear density reported in autism (Ritvo 1986) and 33% (influenza) and 20% (poly(I:C)) reductions reported here. While significant changes in the cerebellum have also been found in schizophrenia, there has been little evidence reported as to whether such changes may be more prominent in particular lobules. A preliminary report did indicate, however, that the volume of lobules VI and VII inversely correlate with positive symptoms and hallucinations in schizophrenia (Pierson et al., 2003).
What is the basis for the discrete, localized deficit in PCs? At 50–80 μm in diameter, PCs are very large neurons. Each cell has over 200,000 synapses, giving the cell an exceptionally high metabolic demand, which predisposes PCs to both excitotoxicity and ischemic death (Kern 2003). While histologically uniform at a superficial level, the cerebellum can be compartmentalized into a variety of patterns based on expression of particular molecules, the spatial effects of mutations, and its connectivity. The PCs in lobules VI and VII are distinctive, and perhaps more vulnerable to developmental insults, for several reasons. (i) These cells express a unique combination of molecular markers, both in the neonate and the adult (Ozol et al., 1999; Rogers et al., 1999). (ii) They receive a distinct set of afferent inputs (olivocerebellar, pontocerebellar and cuneocerebellar mossy fibers), and they project to specific regions of the fastigial (pursuit eye movements) and interposed nuclei (Armstrong et al., 2001). (iii) They receive unique migrational cues; in the weaver mutant mouse (girk2), PC progenitors specifically in lobules VI and VII appear not to migrate outward to form a monolayer (Armstrong et al., 2001). (iv) PCs in lobules VI and VII (and in IX and X) preferentially survive following 3-acetylpyridine ablation of the inferior olive in Shaker mutant rats (Tolbert et al., 2000). Numerous mutations and toxic insults have been associated with distinct patterns of PC loss and survival (Sarna et al., 2003), some of which are inversely correlated with expression of the neuroprotective protein HSP25/27. Since the pattern of cell loss in each case is specific to the type of insult, the selective loss of PCs in lobule VII in the influenza model is a particularly important parallel with autism.
When we administer influenza virus at E9.5, the peak of the inflammatory response occurs around E12 (Fritz et al., 1999; Swiergiel, et al., 1999; Swiergiel et al., 1997a; Swiergiel et al., 1997b), which is also the time at which we administer poly(I:C). This time window of maximal cytokine production corresponds with the timing of PC neurogenesis. Precursor PCs are born during embryonic days E11–E13 in the mouse. Postmitotic PCs migrate radially from the neuroepithelium of the ventricle towards the cortical surface between E13–E17 along radial glial fibers (Hatten 1999; Hatten et al., 1997; Hatten et al., 1995; Miale and Sidmen, 1961; Uzman 1960; Yuasa et al., 1991). By the time of birth, all PCs occupy their position between the EGL and IGL. The activated immune system produces many molecules, such as cytokines and chemokines, which have the potential to alter the neurogenesis and migration of PCs. Our data suggest that the primary deficit occurs in this early stage, with maternal immune activation resulting in abnormal migration of PC precursors. It is also possible that PC precursor proliferation is affected.
The abnormal GC development that we observe may be secondary to the PC deficit, as granule cell development is dependent on signals from PCs. Sonic hedgehog is produced by PCs and is required for proliferation of GC precursors, and it induces increased migration of GCs from cortical explants in vitro (Dahmane et al., 1999). Our data show that at P17, a time when most BrdU+ GCs have migrated to the IGL in controls, many GCs still remain in the ML in exposed animals. Furthermore, these animals have a persistent EGL that is most prominent in lobules VI and VII, suggesting that a pool of GCs have yet to migrate. PC deficits have the potential to slow GC migration due to the lack of Shh or other factors normally produced by PCs. However, it seems that GCs eventually do receive the proper migration cues and form the IGL, because we do not find GCs in the ML of adult animals.
There are several possible functional consequences of the PC deficit observed here. Lobules VI and VII are also called the oculomotor vermis, since their function is linked to eye movements. Our finding that offspring of poly(I:C)-treated mice display abnormalities in classical eye blink conditioning (Lee 2007) could be related to the PC deficits reported here. The fact that abnormalities in eye blink conditioning are also found in autism (Sears et al., 1994; Steinmetz et al., 2001) and schizophrenia (Brown and O’Donnell 2005; Marenco et al., 2003; Sears 2000) is a further link between these disorders and the maternal immune activation model. Recent evidence suggests that abnormalities in eye tracking are present in infants with high risk for autism, suggesting that impaired eye tracking or eye contact may play a role in later deficits in social interaction (Merin et al., 2007). Due to the importance of the oculomotor vermis in these behaviors, understanding the mechanisms that lead to PC deficits could be crucial in understanding the pathology of autism and schizophrenia.
In retrospect, it is perhaps not surprising that this model of maternal immune activation yields offspring with behaviors and pathologies in common with both autism and schizophrenia. Both disorders share the epidemiological risk factor of maternal infection, and in his original description of autism, (Kanner 1943) noted the similarities with the negative symptoms of schizophrenia. A new challenge is to study those abnormalities in the mouse model that may be specific for autism or schizophrenia.
Acknowledgments
We thank L. Chahal, K. Christenson and A. Mihalas for help with the experiments, B. Deverman and J. Jankowsky for useful comments on the manuscript, and D. McDowell and K. Hamilton for administrative support. This work was supported by gifts from Ginger and Ted Jenkins and Ruben Mettler, a McKnight Foundation Neuroscience of Brain Disorder Award, and grants from the Cure Autism Now Foundation, the National Institute of Mental Health (RO1 MH067978), and the Stanley Medical Research Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akshoomoff N, Lord C, Lincoln A, Courchesne R, Carper R, Townsend J, Courchesne E. Outcome classification of preschool children with autism spectrum disorders using MRI brain measures. Journal of the American Academy Child and Adolescent Psychiatry. 2004;43:349–357. doi: 10.1097/00004583-200403000-00018. [DOI] [PubMed] [Google Scholar]
- Allen G. Cerebellar contributions to autism spectrum disorders. Clinical Neuroscience Research. 2006;6:195–207. [Google Scholar]
- Allen G, Courchesne E. Differential effects of developmental cerebellar abnormality on cognitive and motor functions in the cerebellum: an fMRI study of autism. American Journal of Psychiatry. 2003;160:262–273. doi: 10.1176/appi.ajp.160.2.262. [DOI] [PubMed] [Google Scholar]
- Armstrong C, Hawkes R. Selective Purkinje cell ectopia in the cerebellum of the weaver mouse. Journal of Comparative Neurology. 2001;439:151–161. doi: 10.1002/cne.1339. [DOI] [PubMed] [Google Scholar]
- Borrell J, Vela JM, Arevalo-Martin A, Molina-Holgado ECG. Prenatal immune challenge disrupts sensorimotor gating in adult rats. Implications for the etiopathogenesis of schizophrenia. Neuropsychopharmacology. 2002;26:204–215. doi: 10.1016/S0893-133X(01)00360-8. [DOI] [PubMed] [Google Scholar]
- Bottmer C, Bachmann S, Pantel J, Essig M, Amann M, Schad LR, Magnotta V, Schroder J. Reduced cerebellar volume and neurological soft signs in first-episode schizophrenia. Psychiatry Research. 2005;140:239–250. doi: 10.1016/j.pscychresns.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Brown AS. Prenatal infection as a risk factor for schizophrenia. Schizophrenia Bulletin. 2006;32:200–202. doi: 10.1093/schbul/sbj052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AS, Begg M, Gravenstein S, Schaefer C, Wyatt R, Bresnahan M, Babulas V, Susser E. Serologic evidence of prenatal influenza in the etiology of schizophrenia. Archives of General Psychiatry. 2004a;161:774–480. doi: 10.1001/archpsyc.61.8.774. [DOI] [PubMed] [Google Scholar]
- Brown A, Hooton J, Schaefer C, Zhang H, Petkova E, Babulas V, Perrin M, Gorman J, Susser E. Elevated maternal interleukin-8 levels and risk of schizophrenia in adult offspring. American Journal of Psychiatry. 2004b;161:889–895. doi: 10.1176/appi.ajp.161.5.889. [DOI] [PubMed] [Google Scholar]
- Brown A, Susser ES. In utero infection and adult schizophrenia. Mental Retardation and Developmental Disabilities Research Reviews. 2002;8:51–57. doi: 10.1002/mrdd.10004. [DOI] [PubMed] [Google Scholar]
- Brown S, Kieffaber PD, Carroll CA, Vohs JL, Tracy JA, Shekhar A, O’Donnell BF, Steinmetz JE, Hetrick WP. Eyeblink conditioning deficits indicate timing and cerebellar abnormalities in schizophrenia. Brain and Cognition. 2005;58:94–108. doi: 10.1016/j.bandc.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Chess S. Follow-up report on autism in congenital rubella. Journal of Autism and Childhood Schizophrenia. 1977;7:69–81. doi: 10.1007/BF01531116. [DOI] [PubMed] [Google Scholar]
- Ciaranello A, Ciaranello RD. The neurobiology of infantile autism. Annual Review of Neuroscience. 1995;18:101–128. doi: 10.1146/annurev.ne.18.030195.000533. [DOI] [PubMed] [Google Scholar]
- Dahmane N, Ruiz I, Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development. 1999;126:3089–3100. doi: 10.1242/dev.126.14.3089. [DOI] [PubMed] [Google Scholar]
- Dammann O, Kuban KC, Leviton A. Perinatal infection, fetal inflammatory response, white matter damage, and cognitive limitations in children born preterm. Mental Retardation and Developmental Disabilities Research Reviews. 2002;8:46–50. doi: 10.1002/mrdd.10005. [DOI] [PubMed] [Google Scholar]
- Fatemi S, Earle J, Kanodia R, Kist D, Emamian ES, Patterson PH, Shi L, Sidwell R. Prenatal viral infection leads to pyramidal cell atrophy and macrocephaly in adulthood: implications for genesis of autism and schizophrenia. Cellular and Molecular Neurobiology. 2002;22:25–33. doi: 10.1023/a:1015337611258. [DOI] [PubMed] [Google Scholar]
- Fatemi S, Emamian ES, Kist D, Sidwell RW, Nakajima K, Akhter P, Shier A, Sheikh S, Bailey K. Defective corticogenesis and reduction in Reelin immunoreactivity in cortex and hippocampus of prenatally infected neonatal mice. Molecular Psychiatry. 1999;4:145–154. doi: 10.1038/sj.mp.4000520. [DOI] [PubMed] [Google Scholar]
- Fatemi S, Pearce DA, Brooks AI, Sidwell RW. Prenatal viral infection in mouse causes differential expression of genes in brains of mouse progeny: a potential animal model for schizophrenia and autism. Synapse. 2005;57:91–99. doi: 10.1002/syn.20162. [DOI] [PubMed] [Google Scholar]
- Fortier M, Joober R, Luheshi GN, Boksa P. Maternal exposure to bacterial endotoxin during pregnancy enhances amphetamine-induced locomotion and startle responses in adult rat offspring. J Psychiatr Res. 2004;38:335–345. doi: 10.1016/j.jpsychires.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Fritz R, Hayden FG, Calfee DP, Cass LM, Peng AW, Alvord WG, Strober W, Straus SE. Nasal cytokine and chemokine responses in experimental influenza A virus infection: results of a placebo-controlled trial of intravenous zanamivir treatment. Journal of Infectious Diseases. 1999;180:586–593. doi: 10.1086/314938. [DOI] [PubMed] [Google Scholar]
- Hagberg H, Peebles D, Mallard C. Models of white matter injury: comparison of infectious, hypoxic-ischemic, and excitotoxic insults. Mental Retardation and Developmental Disability Research Reviews. 2002;8:30–38. doi: 10.1002/mrdd.10007. [DOI] [PubMed] [Google Scholar]
- Hatten ME. Central nervous system neuronal migration. Annual Review in Neuroscience. 1999;22:511–539. doi: 10.1146/annurev.neuro.22.1.511. [DOI] [PubMed] [Google Scholar]
- Hatten M, Alder J, Zimmerman K, Heintz N. Genes involved in cerebellar cell specification and differentiation. Current Opinion in Neurobiology. 1997;7:40–47. doi: 10.1016/s0959-4388(97)80118-3. [DOI] [PubMed] [Google Scholar]
- Hatten M, Heintz N. Mechanisms of neural patterning and specification in the developing cerebellum. Annual Review of Neuroscience. 1995;18:385–408. doi: 10.1146/annurev.ne.18.030195.002125. [DOI] [PubMed] [Google Scholar]
- Ho B, Mola C, Andreasen NC. Cerebellar dysfunction in neuroleptic naive schizophrenia patients: clinical, cognitive, and neuroanatomic correlates of cerebellar neurologic signs. Biological Psychiatry. 2004;55:1146–1153. doi: 10.1016/j.biopsych.2004.02.020. [DOI] [PubMed] [Google Scholar]
- Hyman S, Arndt TL, Rodier PM. Environmental agents and autism: Once and future associations. International Review of Research in Mental Retardation. 2006;30:171–194. [Google Scholar]
- Kates W, Burnette CP, Eliez S, Strunge LA, Kaplan D, Landa R, Reiss AL, Pearlson GD. Neuroanatomic variation in monozygotic twin pairs discordant for the narrow phenotype for autism. American Journal of Psychiatry. 2004;161:539–546. doi: 10.1176/appi.ajp.161.3.539. [DOI] [PubMed] [Google Scholar]
- Kaufmann W, Cooper KL, Mostofsky SH, Capone GT, Kates WR, Newschaffer CJ, Bukelis I, Stump MH, Jann AE, Lanham DC. Specificity of cerebellar vermian abnormalities in autism: a quantitative magnetic resonance imaging study. Journal of Child Neurology. 2003;18:463–470. doi: 10.1177/08830738030180070501. [DOI] [PubMed] [Google Scholar]
- Kern JK. Purkinje cell vulnerability and autism: a possible etiological connection. Brain Development. 2003;6:377–382. doi: 10.1016/s0387-7604(03)00056-1. [DOI] [PubMed] [Google Scholar]
- Marenco S, Weinberger DR, Schreurs BG. Single-cue delay and trace classical conditioning in schizophrenia. Biological Psychiatry. 2003;53:390–402. doi: 10.1016/s0006-3223(02)01506-8. [DOI] [PubMed] [Google Scholar]
- Mednick S, Machon RA, Huttunen MO, Bonett D. Adult schizophrenia following prenatal exposure to an influenza epidemic. Archives of General Psychiatry. 1988;45:189–192. doi: 10.1001/archpsyc.1988.01800260109013. [DOI] [PubMed] [Google Scholar]
- Merin N, Young GS, Ozonoff S, Rogers SJ. Visual Fixation Patterns during Reciprocal Social Interaction Distinguish a Subgroup of 6-Month-Old Infants At-Risk for Autism from Comparison Infants. Journal of Autism and Developmental Desorders. 2007;37:108–121. doi: 10.1007/s10803-006-0342-4. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Schedlowski M, BK Y. Towards an immuno-precipitated neurodevelopmental animal model of schizophrenia. Neurosci Biobehav Rev. 2005;29:913–947. doi: 10.1016/j.neubiorev.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Schedlowski M, Yee BK. Immunological stress at the maternal-fetal interface: a link between neurodevelopment and adult psychopathology. Brain, Behavior and Immunity. 2006;20:378–388. doi: 10.1016/j.bbi.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Miale I, Sidman RL. An autoradiographic analysis of histogenesis in the mouse cerebellum. Experimental Neurology. 1961;4:277–296. doi: 10.1016/0014-4886(61)90055-3. [DOI] [PubMed] [Google Scholar]
- Nowinski C, Minshew NJ, Luna B, Takarae Y, Sweeney JA. Oculomotor studies of cerebellar function in autism. Psychiatry Research. 2005;137:11–19. doi: 10.1016/j.psychres.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Ozawa K, Hashimoto K, Kishimoto T, Shimizu E, Ishikura HMI. Immune activation during pregnancy in mice leads to dopaminergic hyperfunction and cognitive impairment in the offspring: a neurodevelopmental animal model of schizophrenia. Biol Psychiatry. 2006;59:546–554. doi: 10.1016/j.biopsych.2005.07.031. [DOI] [PubMed] [Google Scholar]
- Ozol K, Hayden JM, Oberdick J, Hawkes R. Transverse zones in the vermis of the mouse cerebellum. J Comp Neurol. 1999;412:95–111. [PubMed] [Google Scholar]
- Palmen S, van Engeland H, Hof PR, Schmitz C. Neuropathological findings in autism. Brain. 2004;127:2572–2583. doi: 10.1093/brain/awh287. [DOI] [PubMed] [Google Scholar]
- Patterson PH. Maternal infection: window on neuroimmune interactions in fetal brain development and mental illness. Current Opinion in Neurobiology. 2002;12:115–118. doi: 10.1016/s0959-4388(02)00299-4. [DOI] [PubMed] [Google Scholar]
- Pierce K, Courchesne E. Evidence for a cerebellar role in reduced exploration and stereotyped behavior in autism. Biological Psychiatry. 2001;49:655–664. doi: 10.1016/s0006-3223(00)01008-8. [DOI] [PubMed] [Google Scholar]
- Pierson R, Alicata D, Nopoulos P, O’Leary D, Andreasen NC. Cerebellar lobe morphology and its relation to symptoms in schizophrenia. Schizophrenia Research. 2003;60:205. [Google Scholar]
- Ramnani N, Miall C. Expanding cerebellar horizons. Trends in Cognitive Science. 2001;5:135–136. doi: 10.1016/s1364-6613(00)01635-1. [DOI] [PubMed] [Google Scholar]
- Ritvo E, Freeman BJ, Scheibel AB, Duong T, Robinson H, Guthrie D, Ritvo A. Lower Purkinje cell counts in the cerebella of four autistic subjected: Initial findings of the UCLA-NSAC autopsy research center. American Journal of Psychiatry. 1986;143:7. doi: 10.1176/ajp.143.7.862. [DOI] [PubMed] [Google Scholar]
- Rogers J, Ciossek T, Menzel P, Pasquale EB. Eph receptors and ephrins demarcate cerebellar lobules before and during their formation. Mechanisms of Development. 1999;87:119–128. doi: 10.1016/s0925-4773(99)00154-9. [DOI] [PubMed] [Google Scholar]
- Sarna J, Hawkes R. Patterned Purkinje cell death in the cerebellum. Progress in Neurobiology. 2003;70:473–507. doi: 10.1016/s0301-0082(03)00114-x. [DOI] [PubMed] [Google Scholar]
- Schmahmann JD. The cerebrocerebellar system: Anatomic substrates of the cerebellar contribution to cognition and emotion. International Review of Psychiatry. 2001;13:247–260. [Google Scholar]
- Schutter D, van Honk J. The cerebellum on the rise in human emotion. Cerebellum. 2005;4:290–294. doi: 10.1080/14734220500348584. [DOI] [PubMed] [Google Scholar]
- Sears L, Andreasen NC, O’Leary DS. Cerebellar functional abnormalities in schizophrenia are suggested by classical eye blink conditioning. Biological Psychiatry. 2000;48:204–209. doi: 10.1016/s0006-3223(00)00247-x. [DOI] [PubMed] [Google Scholar]
- Sears LL, Finn PR, Steinmetz JE. Abnormal classical eye-blink conditioning in autism. Journal of Autism and Developmental Disorders. 1994:24. doi: 10.1007/BF02172283. [DOI] [PubMed] [Google Scholar]
- Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. Journal of Neuroscience. 2003;23:297–302. doi: 10.1523/JNEUROSCI.23-01-00297.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SEP, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. Journal of Neuroscience. 2007;27:10695–10702. doi: 10.1523/JNEUROSCI.2178-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz J, Tracy JA, Green JT. Classical eyeblink conditioning: clinical models and applications. Integrative Physiological and Behavioral Science. 2001;36:220–238. doi: 10.1007/BF02734095. [DOI] [PubMed] [Google Scholar]
- Swiergiel A, Dunn AJ. The roles of IL-1, IL-6, and TNFalpha in the feeding responses to endotoxin and influenza virus infection in mice. Brain, Behavior and Immunity. 1999;13:252–265. doi: 10.1006/brbi.1999.0565. [DOI] [PubMed] [Google Scholar]
- Swiergiel A, Smagin GN, Dunn AJ. Influenza virus infection of mice induces anorexia: comparison with endotoxin and interleukin-1 and the effects of indomethacin. Pharmacology, Biochemistry and Behavior. 1997a;57:389–396. doi: 10.1016/s0091-3057(96)00335-8. [DOI] [PubMed] [Google Scholar]
- Swiergiel A, Smagin GN, Johnson LJ, Dunn AJ. The role of cytokines in the behavioral responses to endotoxin and influenza virus infection in mice: effects of acute and chronic administration of the interleukin-1-receptor antagonist (IL-1ra) Brain Research. 1997b;776:96–104. doi: 10.1016/s0006-8993(97)01009-3. [DOI] [PubMed] [Google Scholar]
- Takarae Y, Minshew NJ, Luna B, Sweeney JA. Oculomotor abnormalities parallel cerebellar histopathology in autism. Journal of Neurology, Neurosurgery and Psychiatry. 2004;75:1359–1361. doi: 10.1136/jnnp.2003.022491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolbert DL, Clark BR. Olivocerebellar projections modify hereditary Purkinje cell degeneration. Neuroscience. 2000:101. doi: 10.1016/s0306-4522(00)00362-6. [DOI] [PubMed] [Google Scholar]
- Uzman LL. The histogenesis of the mouse cerebellum as studied by its tritiated thymidine uptake. Journal of Comparative Neurology. 1960;114:137–159. doi: 10.1002/cne.901140204. [DOI] [PubMed] [Google Scholar]
- Yuasa S, Kawamura K, Ono K, Yamakuni T, Takahashi Y. Development and migration of Purkinje cells in the mouse cerebellar primordium. Anatomy and Embryology (Berl) 1991;184:195–212. doi: 10.1007/BF01673256. [DOI] [PubMed] [Google Scholar]
- Zuckerman L, Rehavi M, Nachman R, Weiner I. Immune activation during pregnancy in rats leads to a postpubertal emergence of disrupted latent inhibition, dopaminergic hyperfunction, and altered limbic morphology in the offspring: a novel neurodevelopmental model of schizophrenia. Neuropsychopharmacology. 2003;28:1778–1789. doi: 10.1038/sj.npp.1300248. [DOI] [PubMed] [Google Scholar]
- Zuckerman L, Weiner I. Maternal immune activation leads to behavioral and pharmacological changes in the adult offspring. Journal of Psychiatric Research. 2005;39:311–323. doi: 10.1016/j.jpsychires.2004.08.008. [DOI] [PubMed] [Google Scholar]