

Abstract
S-Nitrosothiols have generated considerable interest due to their ability to act as nitric oxide (NO) donors and due to their possible involvement in bioregulatory systems—e.g., NO transfer reactions. Elucidation of the reaction pathways involved in the modification of the thiol group by S-nitrosothiols is important for understanding the role of S-nitroso compounds in vivo. The modification of glutathione (GSH) in the presence of S-nitrosoglutathione (GSNO) was examined as a model reaction. Incubation of GSNO (1 mM) with GSH at various concentrations (1–10 mM) in phosphate buffer (pH 7.4) yielded oxidized glutathione, nitrite, nitrous oxide, and ammonia as end products. The product yields were dependent on the concentrations of GSH and oxygen. Transient signals corresponding to GSH conjugates, which increased by one mass unit when the reaction was carried out with 15N-labeled GSNO, were identified by electrospray ionization mass spectrometry. When morpholine was present in the reaction system, N-nitrosomorpholine was formed. Increasing concentrations of either phosphate or GSH led to lower yields of N-nitrosomorpholine. The inhibitory effect of phosphate may be due to reaction with the nitrosating agent, nitrous anhydride (N2O3), formed by oxidation of NO. This supports the release of NO during the reaction of GSNO with GSH. The products noted above account quantitatively for virtually all of the GSNO nitrogen consumed during the reaction, and it is now possible to construct a complete set of pathways for the complex transformations arising from GSNO + GSH.
Keywords: nitric oxide, glutathionyl radical, sulfenamide, transnitrosation, N-nitrosomorpholine
S-Nitrosothiols, RSNO, with certain exceptions, are unstable in aqueous solution. For example, S-nitrosoglutathione (GSNO) undergoes decomposition over hours, whereas S-nitrosocysteine has a half-life of less than 2 min. The initial step in the decomposition of RSNO is believed to be homolytic cleavage of the S—N bond to give nitric oxide (NO) and a thiyl radical (1, 2). These compounds are involved in many bioregulatory functions, including vasodilation and inhibition of platelet aggregation. The existence of more stable transport forms of NO has been postulated in view of the short half-life of authentic NO in vivo (3). Low molecular weight thiols such as cysteine, glutathione (GSH), and penicillamine are prime candidates for such carrier molecules, and they can form S-nitrosothiols on reaction with oxides of nitrogen (4). It has been assumed that the biological effects of these compounds are due to the spontaneous release of NO; however, this hypothesis is not supported by currently available data (5–7).
Although a few studies have been carried out in an attempt to determine the reaction products and to deduce the mechanism of the modification of the thiol group by S-nitrosothiols, the experiments were purely qualitative and no clear mechanistic picture has emerged (8, 9). In this report, we describe the reaction of GSNO with GSH, a tripeptide with intracelluclar concentrations as high as 10 mM (10). It is involved in the cell’s antioxidant defense, xenobiotic detoxification, and amino acid transport (11). GSNO is also believed to be involved in many physiologic and pathophysiologic processes. It has recently been shown that exposure of human neutrophils to NO results in the conversion of intracellular GSH to GSNO and that intracellular GSNO activates the hexose monophosphate shunt of the cells (12). Since GSH concentration is millimolar in cells and NO concentration is less than micromolar, it follows that the concentration of GSNO will always be small compared that of GSH.
We report here that, in addition to release of NO from GSNO in the presence of GSH, an important pathway for GSNO decomposition is reaction with GSH to form glutathione conjugates similar to those reported for the reaction of GSH with other electrophilic metabolites (13–16). Our data further show that the major end product from this transformation is oxidized GSH, GSSG. The amount formed is dependent on the availability of oxygen in the system, suggesting the possible involvement of radicals.
MATERIALS AND METHODS
Materials.
Thiazolidine-4-carboxylic acid, GSH, N-nitrosomorpholine (NMOR), and GSSG were from Sigma. Sodium nitrite (15N-labeled) was from MSD isotopes. Morpholine (MOR) was from Aldrich. GSNO and GS15NO were prepared according to Hart (17) by acid-catalyzed nitrosation of GSH; these compounds were quantitated by measuring the absorbance at 334 nm (A334 = 800 M−1·cm−1). N-Nitroso-N′,N′,N-trimethylurea (NTMU) was synthesized by nitrosating trimethylurea (Alfa) at pH 2.5 in the presence of acetic acid and was quantitated by measuring the absorbance at 374 nm (A374 = 88 M−1·cm−1) (18). N-Acetylthiazolidine-4-carboxylic acid was prepared by addition of acetic anhydride to a hot solution of thiazolidine-4-carboxylic acid in acetic acid (19). Nitrous oxide (N2O) and NO were from Matheson, and isobutane was from Med Tech (Medford, MA).
Experiments.
The reactions were conducted in 0.01 M or 0.05 M potassium phosphate buffer (pH 7.4) at room temperature in vials equipped with rubber septa. The pH of GSH and GSNO solutions in the appropriate buffers was adjusted to 7.4 with diluted KOH. When needed, MOR/trimethylurea (1 mM each) solutions were prepared similarly and added to the GSH/GSNO solution. A fresh solution of GSNO was prepared immediately before each experiment, and the concentration was confirmed spectrophotometrically at 334 nm. Typically, the GSNO concentration was fixed at 1 mM and the GSH concentration was varied between 1 and 10 mM. For reactions under argon or nitrogen, the vials, containing the appropriate buffer, were deaerated for 30 min prior to use. Concentrated solutions of GSNO that were not deaerated were added in small amounts (≈50 μl) along with deaerated GSH solutions by using a Hamilton syringe. For experiments in which NMOR was measured, the MOR solution was added to the buffer solution prior to deaerating. The vials were then stored in argon- or nitrogen-filled balloons for 24 or 40 h. Internal standard was added to the vial at the end of the reaction and the solutions were then analyzed for the compounds of interest.
Electrospray Ionization Mass Spectrometry (ESI-MS).
These experiments were done on a Hewlett Packard model 5989B mass spectrophotometer in the negative ion mode. The reactions were conducted in ammonium acetate (1 mM; pH ≈7.5) buffer at room temperature or at 4°C, and the reaction mixture was analyzed at the end of 2 h. Typically, 50 μl of reaction solution was added to 100 μl of the electrospray solvent [50:50 water/methanol containing 1% acetic acid or 50:40:10 (vol/vol) methanol/water/acetonitrile containing 1% ammonium hydroxide]. A Harvard syringe pump was used to introduce the electrospray solvent into the mass spectrometer at 20 μl/min and the samples were flow-injected into the solvent stream by means of a Valco injector with a 10-μl sample loop. Under these conditions, the residence time of the samples in the mass spectrometer was about 0.6 min. Spectra were averaged over the maximum central portion ≈0.3 min) of the resulting total-ion plot.
Determination of Nitrite and Nitrate.
Nitrate and nitrite were measured in an automated system that has been described in detail elsewhere (20).
GC-MS Analysis.
These analyses were on a Hewlett–Packard 5971 mass-selective detector (MSD) or a Hewlett–Packard 5989A GC-MS. For NMOR and NTMU analysis, a fused-silica capillary column coated with Supelcowax 10 (Supelco) (15 m × 0.25 mm) was used. The mass spectrometer was operated in the selective ion mode using ions at m/z 116 and m/z 86 for NMOR, m/z 131 and m/z 72 for NTMU, and m/z 123 for the internal standard nitrobenzene. Due to the instability of NTMU, the analysis was done by lowering the temperatures of the injector (150°C) and column (initial value 60°C). For the analysis of gaseous products, reactions were conducted in septum-capped vials. The column was fused-silica (25 m × 0.32 mm) packed with Poraplot Q (Hewlett–Packard). A fixed amount (50 μl) of internal standard (isobutane) from a 1000-ml flask was added to the reaction vial at the end of the reaction and allowed to equilibrate for 15 min. Head-space samples were withdrawn with a 25-μl Hamilton gas-tight syringe and injected into the column at 45°C; the temperature was then increased at the rate of 40°C/min to 220°C after 3 min. The mass spectrometer was set to detect ions at m/z 44 and 30 for N2O, m/z 30 for NO, m/z 58 for isobutane, and m/z 30 for 15N2. The peak areas at the appropriate retention times (as determined by authentic standards) were measured by the integration routine included in the data system. The areas of NMOR and N2O were normalized to the areas of the internal standards nitrobenzene and isobutane, respectively.
Generation of N2O.
N2O standard was generated by the addition of a 5-fold molar excess of sodium nitrite solution to a solution of sodium azide, in 0.01 M potassium phosphate buffer (pH 3) (21). The concentration of N2O was determined from the concentration of sodium azide used in the reaction. The reaction was repeated with various concentrations of azide to obtain a standard calibration curve. The detector was calibrated on each experimental day by duplicate injections of 25 μl of head-space after addition of internal standard as discussed above.
Ammonia Assay.
The concentrations of NH3 were measured with a commercial ammonia diagnostic kit from Sigma. The assay involved the reductive amination of 2-oxoglutarate by glutamate dehydrogenase and NADPH.
HPLC Analysis.
HPLC studies, for quantification of GSSG, were conducted using a reversed-phase column (Supelcosil LC-18-DB, 25 cm × 4.6 mm). The gradient was obtained with eluent A (0.1% phosphoric acid) and eluent B (acetonitrile). After injecting the sample (20 μl in 1 ml of 0.1% phosphoric acid) along with a fixed amount of an internal standard (N-acetylthiazolidine-4-carboxylic acid), a linear change from 100% A to 80% A over 20 min was carried out. The GSSG area was normalized to the area of the internal standard. The flow rate was 1 ml/min, and the detector absorbance was at 215 nm.
RESULTS
Nitrite Formation.
The half-life of GSNO was dependent on the concentrations of GSNO and GSH. As measured by the decrease in absorbance at 334 nm, GSNO (1 mM) was stable for at least 90 min in the absence of GSH. Decomposition of GSNO was observed upon addition of GSH (0.5–10 mM). The effect was dependent on the GSH concentration (Fig. 1). This is in contrast to previously reported chemical stabilization of S-nitrosothiols in the presence of excess thiol (22). In the absence of oxygen, the rate of GSNO decomposition in the presence of GSH decreased by a factor of 2 (data not shown).
Figure 1.
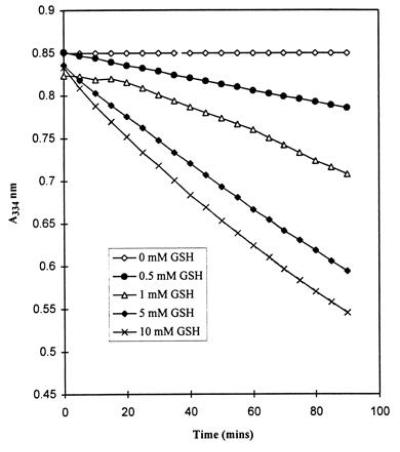
Absorbance vs. time plots for the decomposition of GSNO (1 mM) in 50 mM potassium phosphate buffer solutions (pH 7.4) with increasing GSH concentration.
GSNO (1 mM) was incubated with various amounts of GSH (1–10 mM) in aerated or deaerated phosphate buffer, and the reaction mixtures were analyzed for nitrite and nitrate. Nitrite was the only product detected, and the amounts formed varied with the GSNO-to-GSH ratio and whether the solution was aerated or deaerated (Table 1). The maximum amount of nitrite from GSNO was obtained in the presence of equimolar or less than equimolar concentrations of GSH. When GSH was present in large excess, nitrite formation decreased significantly. A similar observation was made by Pietraforte et al. (23) for the reaction of GSNO with l-cysteine, but the question of missing nitrite was not addressed and no new products were reported.
Table 1.
Relative amounts of nitrogen-containing products from the reaction of GSNO (1 mM) with GSH under aerobic and anaerobic conditions
| GSH, mM | Product obtained, μM
|
|||
|---|---|---|---|---|
| Nitrite | N2O* | NH3 | Total N† | |
| Aerobic conditions | ||||
| 1 | 826 | 5 ± 3 | 63 | 899 |
| 5 | 636 | 29 ± 11 | 226 | 920 |
| 10 | 420 | 11 ± 1 | 454 | 896 |
| Anaerobic conditions | ||||
| 1 | 410 | 213 ± 22 | 65 | 901 |
| 5 | 314 | 168 ± 26 | 323 | 973 |
| 10 | 157 | 78 ± 19 | 586 | 899 |
Unless otherwise stated, all values reported are an average of two experiments. The reactions were conducted in 0.05 M potassium phosphate buffer (pH 7.4) and analyzed at the end of 40 h. No reaction products were obtained in the presence of GSNO alone.
Each value represents the mean ± SD of four experiments. Every mole of N2O accounts for 2 moles of GSNO.
Nitrogen arising from GSNO (1000 μM).
To determine whether the nitrite formed was a product of NO oxidation, the reaction was performed under a nitrogen atmosphere. At the end of the reaction (GSNO/GSH = 1), one vial was purged with nitrogen before adding oxygen. The amount of nitrite present in the nitrogen-purged reaction vials was significantly lower compared with the unpurged reaction vials (190 μM and 410 μM, respectively) indicating that NO was being released from GSNO in the presence of GSH. This was confirmed by GC-MS analysis of the head-space; NO was detected under anaerobic conditions with various GSH concentrations. No NO was detected in the absence of GSH over the same time period, suggesting that the release of NO became possible only in the presence of GSH. The addition of EDTA (0.1 mM) did not alter the yield of nitrite, thus ruling out the involvement of metal ions in nitrite formation.
Effect of Phosphate and GSH on N-Nitrosation.
N-Nitrosation can occur during the decomposition of S-nitroso compounds in the presence of secondary amines and requires the presence of oxygen (24). The results are rationalized in terms of release of NO, which, in the presence of oxygen, produces N2O3, which is a good electrophilic nitrosating agent. N2O3 can undergo hydrolysis to nitrite, nitrosate secondary amines (MOR in this study) to form N-nitrosamines, or react with thiols to form S-nitroso compounds.
Reaction of GSNO with MOR in the presence and absence of GSH, in 0.01 and 0.05 M phosphate buffer, yielded NMOR. As shown in Fig. 2, the yield of NMOR is maximal with equimolar or less-than-equimolar concentrations of GSH and negligible with 5- to 10-fold molar excess of GSH. In the presence of excess GSH, N2O3 reacts preferentially with GSH to form GSNO, thereby resulting in decreased NMOR formation. Further, the rate of NMOR formation is faster (by a factor of at least 2) in the presence of equimolar and less-than-equimolar GSH concentrations than in the complete absence of GSH, consistent with the enhanced release of NO in the presence of GSH.
Figure 2.
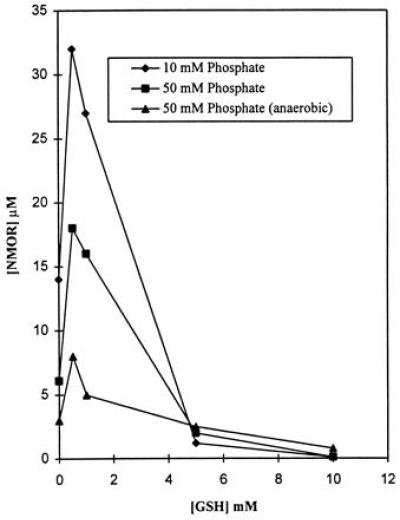
NMOR formation at the end of 24 h during the reaction of GSNO with MOR. The reactions were carried out in 10 and 50 mM aerated and 50 mM deaerated potassium phosphate buffer solutions (pH 7.4), in the presence and absence of GSH.
NMOR formation was significantly inhibited by 0.05 M phosphate buffer (Fig. 2), consistent with the observation that phosphate reacts competitively with N2O3 to form nitrite, thereby inhibiting N-nitrosation (25). The inhibition observed with phosphate suggests that NMOR formation occurs by release of NO followed by formation of N2O3. In deaerated solutions (Fig. 2), NMOR probably arises from trace amounts of oxygen still present in solution as well as by a possible direct transnitrosation reaction between GSNO and MOR.
Nitrosonium Ion (NO+).
The decomposition of GSNO (1 mM) in the presence of both GSH (1–10 mM) and trimethylurea (1 mM) (a substrate that can be nitrosated only with a strong nitrosating agent—e.g., NO+) (26) was studied to determine whether NTMU and, by inference, NO+ were produced. No NTMU was detected (data not shown), thus ruling out NO+ as an intermediate.
N2O and N2 Formation.
The gaseous species involved in the reaction were examined in a closed system by GC-MS. Although N2O, arising by HNO elimination (27), has been suggested as a major reaction product in the reaction of thiols with S-nitrosothiols, the reports to date are purely qualitative (8, 9). We measured the amount of N2O generated at various GSH concentrations and a fixed concentration of GSNO. N2O was detected in the head-space in insignificant amounts under aerobic conditions and in significant amounts under anaerobic conditions (Table 1). The yield of N2O under anaerobic conditions is inversely dependent on the GSH concentration, a trend similar to that noted for nitrite. These results suggest the existence of other reaction pathways in the presence of excess GSH. The gas phase was also analyzed for N2 formation by conducting the reaction in an atmosphere of argon and using 15N-labeled GSNO. Trace amounts of 15N2 were detected with various GSH concentrations.
ESI-MS studies.
The reaction of GSNO (1 mM) with GSH (5 mM) was studied by ESI-MS. Transient signals at m/z 626 and m/z 642, which increased by 1 mass unit when the reaction was conducted with 15N-labeled GSNO, were detected. The signals were assigned to the sulfenamide GS—NH—SG and the N-hydroxysulfenamide GS—N(OH)—SG or the sulfinamide GS—NH—S(O)G, respectively. Similar conjugates have been detected in the reaction of nitrosoarenes with GSH. Klehr et al. (14), for example, reported that nitrosobenzene reacts with GSH to form phenylhydroxylamine, GSSG, and a GSH conjugate identified as GSH sulfinanilide [GS(O)—NH—Ph]. In acidic solutions, the GSH sulfinanilide decomposes to form aniline and GSH sulfinic acid (GSO2H). Mulder et al. (15) reported that p-nitrosophenetole reacts with GSH to form the sulfenamide N-(glutathionyl-S-yl)-p-phenetidine, which is unstable in the presence of excess GSH and decomposes to p-phenetidine. No corresponding sulfinanilide, which could arise from nitrosobenzene, was reported. The signal at m/z 642 was much more intense when the reaction was carried out at 4°C. However, the signals at m/z 642 and m/z 626 did not accumulate over a period of time, suggesting that both species were labile. Neither GSO2H nor GSNH2 was detected under our reaction conditions. This suggests that if GSNH2 does form it reacts rapidly with GSH to form NH3.
NH3 Formation.
Table 1 shows the amount of NH3 formed at various GSH concentrations. Although this is again a function of the GSH concentration, it follows an opposite trend to nitrite and N2O. The sum of nitrite, N2O, and NH3 accounted for 90% or more of the nitrogen introduced as GSNO (Table 1).
GSSG.
When GSNO was allowed to react with GSH the only product detected by HPLC was GSSG. GSNO, prepared according to Hart (17), contained 5% contamination of GSSG. The amount of GSSG generated (after subtraction of values obtained in control incubations with only GSH) increased with increasing concentrations of GSH. To test if this was related to the presence of oxygen in the system, the reaction was performed while rigorously excluding oxygen. The amounts of GSSG obtained under aerobic and anaerobic condition appear to be the same (Table 2).
Table 2.
Formation of GSSG during reaction of GSNO (1 mM) in the presence of GSH under aerobic and anaerobic conditions
| GSH, mM | GSSG formation, mM
|
|||
|---|---|---|---|---|
| Total* | NH3 basis† | N2O basis‡ | Other§ | |
| Aerobic conditions | ||||
| 1 | 0.7 | 0.2 | 0.01 | 0.5 |
| 5 | 2.0 | 0.7 | 0.06 | 1.2 |
| 10 | 2.5 | 1.4 | 0.02 | 1.1 |
| Anaerobic conditions | ||||
| 1 | 0.9 | 0.2 | 0.4 | 0.3 |
| 5 | 1.6 | 1.0 | 0.3 | 0.3 |
| 10 | 2.1 | 1.8 | 0.2 | 0.1 |
After subtraction of control values obtained in incubation minus GSNO. The reactions were conducted in 0.05 M phosphate buffer (pH 7.4) and analyzed at the end of 40 h. All values reported are average of two experiments.
GSSG values calculated on the basis of the experimental values of NH3 obtained in Table 1, assuming a GSSG-to-NH3 molar ratio of 3:1.
GSSG values calculated on the basis of the experimental values of N2O obtained in Table 1, assuming a GSSG-to-N2O molar ratio of 2:1.
GSSG unaccounted for by the NH3 and N2O pathway.
DISCUSSION
It has recently been suggested that the bioactivity of S-nitrosothiols may not be due to the spontaneous liberation of NO (5–7). Kowaluk and Fung (5) studied the effect of added thiol, N-acetylpenicillamine (NAP), on the decomposition of GSNO and S-nitroso-N-acetylpenicillamine (SNAP). They found that the rate of decomposition and the initial rate of NO formation, from both GSNO and SNAP, were increased in the presence of NAP. We have shown here that the decomposition of GSNO is also accelerated in the presence of an added thiol—i.e., GSH. Release of NO, however, decreases in the presence of excess GSH. Instead, the reaction of excess GSH with GSNO appears to be similar to the reaction of GSH with electrophilic metabolites to form glutathione conjugates. GSH conjugation is believed to be one of the primary detoxification mechanisms in vivo.
The scheme presented in Fig. 3 summarizes the reactions of GSNO with GSH and illustrates that the reaction is more complex than might be inferred from the few final reaction products. Under our experimental conditions, GSH reacts with GSNO to form the GSH conjugate N-hydroxysulfenamide, GS—N(OH)—SG. In the case of nitrosoarenes, the N-hydroxysulfenamides are known to rearrange to sulfinamides (S-oxides) that are sufficiently stable for characterization (14). The labile nature of the conjugate and lack of formation of GSO2H under acidic conditions suggest that the rearranged sulfinamide is not obtained under our reaction conditions.
Figure 3.
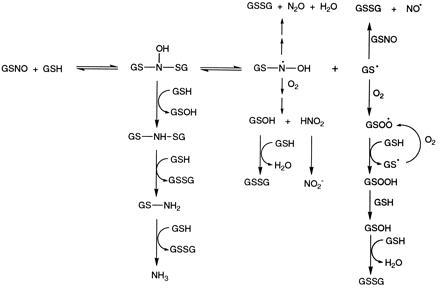
Reactions of GSNO with GSH.
The N-hydroxysulfenamide can react via different pathways depending on the availability of GSH and oxygen. Reaction of N-hydroxysulfenamide with GSH (which predominates when excess GSH is present) results in the sulfenamide GS—NH—SG. Subsequent reductions with GSH result in the formation of NH3. GSSG and NH3, the end products of this transformation, are obtained in a 3:1 ratio as depicted in Fig. 3.
Stamler (28) has suggested that nucleophilic attack of the thiolate anion (RS−) on the S-nitrosothiol (R′SNO) sulfur results in the nominal release of NO−. The end product of this transformation is N2O and the disulfide RSSR′. Stamler’s hypothesis (28) cannot explain the rapid transnitrosation (exchange reaction) (29), characteristic of any system containing thiol (RSH) and S-nitrosothiol (R′SNO). We propose that transnitrosation (Eq. 1) and N2O formation occur through the N-hydroxysulfenamide that arises from nucleophilic attack of the thiol on the R′SNO nitrogen rather than sulfur.
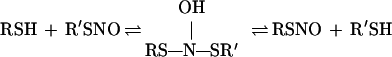 |
1 |
Homolytic decomposition of tribenzenesulfenamide [(PhS)3N] is an example of thermal cleavage of the S—N bond (30). Similarly, homolytic cleavage of the S—N bond of GSH N-hydroxysulfenamide would generate the glutathionyl radical (GS·) and the GSH N-hydroxyl radical (Eq. 2). The comparison is limited to the cleavage of the S—N bond and is not intended to imply anything about the stability of the resulting radicals. The fate of the glutathionyl radical and the GSH N-hydroxyl radical in the presence and absence of oxygen is discussed below.
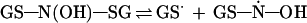 |
2 |
The GSH N-hydroxyl radical can dimerize to the unstable dihydroxyhydrazine and subsequently form GSSG and hyponitrous acid (Eq. 3) (31). This latter compound is known to eliminate N2O, the gaseous product that we observe (27). Similarly, the GS· can dimerize to form GSSG (Eq. 4)
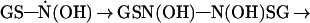 |
3 |
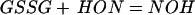 |
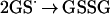 |
4 |
Combining Eqs. 2–4 results in a 2:1 ratio for the end products, GSSG and N2O.
The two pathways discussed so far, which result in the formation of GSSG, are those involving formation of NH3 (NH3:GSSG, 1:3) and N2O (N2O:GSSG, 1:2). The GSSG formed (Table 2) from these two pathways accounts for virtually all of the GSSG formed under anaerobic conditions. Under aerobic conditions, however, a significant proportion of GSSG is formed by other mechanisms. Oxygen therefore appears essential for maximal levels of thiol oxidation.
The reaction of glutathionyl radicals with oxygen, and subsequently with excess GSH to give extensive oxidation of thiol, has been studied by Ross et al. (32, 33). We believe that a similar pathway may be responsible for the significantly greater GSSG formation under aerobic vs. anaerobic conditions (Fig. 3). The glutathionyl radical (GS·) may dimerize (Eq. 4), interact with oxygen, or react with GS− to form GSSG⨪ (34, 35). GSSG⨪ can reduce oxygen to superoxide (O2⨪), which on reaction with NO forms peroxynitrite (reactions not included in Fig. 3). The GSH peroxysulfenyl radical (GSOO·), arising from the addition of oxygen to the glutathionyl radical, can abstract a hydrogen atom from GSH to form GSH sulfenyl hydroperoxide (GSOOH) and another GS·. This could lead to a cyclic reaction sequence, depending upon the availability of oxygen and GSH. A small amount of GS· radical could thus lead to extensive thiol oxidation. The GSOOH could interact with GSH to form the hypothetical intermediate GSH sulfenic acid (GSOH), which could then interact with GSH to produce GSSG (36). In the absence of oxygen, the ultimate fate of the glutathionyl radical would be formation of GSSG.
The glutathionyl radical could also react with GSNO to form NO and GSSG (24, 37). This would explain the faster rate of formation of NO from GSNO in the presence of GSH. The ultimate fate of NO in aerated phosphate buffer at pH 7.4 is formation of nitrite but little or no nitrate, consistent with our observations (38, 39).
The formation of significantly greater amounts of N2O under anaerobic vs. aerobic conditions can be explained as follows. In the presence of oxygen, the GSH N-hydroxyl radical can undergo the reaction sequence outlined in Eq. 5. The radical [GSN(OH)OO·] obtained by addition of oxygen to the GSH N-hydroxyl radical can abstract a H atom from GSH to form GSN(OH)OOH and GS·. Rearrangement of the GSN(OH)OOH could yield HNO2 and GSOH (Eq. 6). In the presence of sufficient GSH, the GSOH can react with GSH to form GSSG (36). The GSSG produced by this pathway and the NO pathway will be proportional to the amount of nitrite formed. This accounts for all the GSSG obtained under anaerobic conditions but not accounted for by the NH3 and N2O pathways (Table 2). However, it cannot account for all the GSSG obtained under aerobic conditions, thus strengthening the hypothesis that glutathionyl radicals are involved in this transformation.
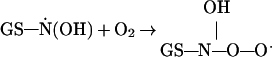 |
5 |
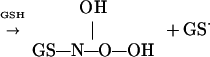 |
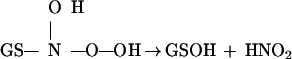 |
6 |
Eq. 6 provides a route for nitrite formation without the involvement of NO. This pathway would result in a decrease in N2O formation and an increase in nitrite formation in the presence of oxygen, in agreement with our observations (Table 1). Further—in the presence of oxygen—the equilibrium described by Eq. 2 shifts to the right due to interactions of the radical with oxygen, thereby increasing the rate of disappearance of GSNO. The decrease in N2O and nitrite formation, and increase in NH3 formation, with increasing GSH concentrations, reflect the increased reduction of the N-hydroxysulfenamide in the presence of excess GSH leading to increased formation of NH3.
In summary, we can account quantitatively for virtually all of the GSNO consumed in this reaction. In the presence of oxygen, nitrite forms from NO as well as from an NO-independent pathway. The major product in the presence of excess GSH is not NO as had been assumed but, instead, NH3. The formation of N2O, nitrite, and NH3—via GSH conjugates—reveals new chemistry of S-nitrosothiols. These results strengthen the idea that the biological activity of S-nitrosothiols may be associated with both heterolytic and homolytic mechanisms of decomposition.
Acknowledgments
We thank Dr. Paul Skipper for helpful discussions and Joe Glogowski for analysis of nitrite/nitrate. This work was supported by National Institutes of Health grants CA 26731 and ES 07020.
Footnotes
Abbreviations: GSH, glutathione; GSSG, oxidized GSH; GSNO, S-nitrosoglutathione; MOR, morpholine; NMOR, N-nitrosomorpholine; NTMU, N-nitroso-N′,N′,N-trimethylurea; ESI-MS, electrospray ionization mass spectrometry.
References
- 1.Josephy P D, Rehorek D, Janzen E G. Tetrahedron Lett. 1984;25:1685–1688. [Google Scholar]
- 2.Williams D L H. Chem Soc Rev. 1985;14:171–196. [Google Scholar]
- 3.Kelm M, Schrader J. Circ Res. 1990;66:1561–1575. doi: 10.1161/01.res.66.6.1561. [DOI] [PubMed] [Google Scholar]
- 4.Oae S, Shinhama K. Org Prep Proced Int. 1983;15:165–198. [Google Scholar]
- 5.Kowaluk E A, Fung H L. J Pharmacol Exp Ther. 1990;255:1256–1264. [PubMed] [Google Scholar]
- 6.Rockett K A, Auburn M M, Cowden W B, Clark I A. Infect Immun. 1991;59:3280–3283. doi: 10.1128/iai.59.9.3280-3283.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gaston B, Drazen J M, Jansen A, Sugarbaker D A, Loscalzo J, Richards W, Stamler J S. J Pharmacol Exp Ther. 1994;268:978–984. [PubMed] [Google Scholar]
- 8.Park J W. Biochem Biophys Res Commun. 1988;152:916–920. doi: 10.1016/s0006-291x(88)80127-x. [DOI] [PubMed] [Google Scholar]
- 9.Park J W, Billman G E, Means G E. Biochem Mol Biol Int. 1993;30:885–891. [PubMed] [Google Scholar]
- 10.Sies H, Brigelius R, Akerboom T P M. In: Functions of Glutathione: Biochemical, Physiological, and Clinical Aspects. Larsson A L, Orrenius S, Holmgren A, Monnerik B, editors. New York: Raven; 1983. pp. 51–64. [Google Scholar]
- 11.Beutler E, Dale G L. In: Coenzymes and Cofactors. Dolphin D, Avramovic O, Poulson R, editors. New York: Wiley; 1989. pp. 291–317. [Google Scholar]
- 12.Clancy R M, Levartovsky D, Leszczynska-Piziak J, Yegudin J, Abramson S B. Proc Natl Acad Sci USA. 1994;91:3680–3684. doi: 10.1073/pnas.91.9.3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eyer P. Chem Biol Interact. 1979;24:227–239. doi: 10.1016/0009-2797(79)90011-5. [DOI] [PubMed] [Google Scholar]
- 14.Klehr H, Eyer P, Schafer W. Biol Chem Hoppe-Seyler. 1985;366:755–760. doi: 10.1515/bchm3.1985.366.2.755. [DOI] [PubMed] [Google Scholar]
- 15.Mulder G J, Kadlubar F F, Mays J B, Hinson J A. Mol Pharmacol. 1984;26:342–347. [PubMed] [Google Scholar]
- 16.Mulder G J, Unruh L E, Evans F E, Ketterer B, Kadlubar F F. Chem Biol Interact. 1982;39:111–127. doi: 10.1016/0009-2797(82)90010-2. [DOI] [PubMed] [Google Scholar]
- 17.Hart T W. Tetrahedron Lett. 1985;26:2013–2016. [Google Scholar]
- 18.Elespuru R K, Lijinsky W. Food Cosmet Toxicol. 1973;11:807–811. doi: 10.1016/0015-6264(73)90139-9. [DOI] [PubMed] [Google Scholar]
- 19.Herbst R M, Shemin D. In: Organic Syntheses. Blatt A H, editor. New York: Wiley; 1946. pp. 11–12. [Google Scholar]
- 20.Green L C, Wagner D A, Glogowski J, Skipper P L, Wishnok J S, Tannenbaum S R. Anal Biochem. 1982;126:131–138. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- 21.Block H. In: Supplement to Mellor’s Comprehensive Treatise on Inorganic Chemistry. Eldridge A A, Dyson G M, Welch A J E, Pantony D A, editors. New York: Wiley; 1967. pp. 352–364. [Google Scholar]
- 22.Feelisch M, Poel M, Zamora Z, Deussen A, Moncada S. Nature (London) 1991;368:62–65. doi: 10.1038/368062a0. [DOI] [PubMed] [Google Scholar]
- 23.Pietraforte D, Mallozzi C, Scorza G, Minetti M. Biochemistry. 1995;34:7177–7185. doi: 10.1021/bi00021a032. [DOI] [PubMed] [Google Scholar]
- 24.Askew S C, Barnett D J, McAninly J, Williams D L H. J Chem Soc, Perkin Trans. 1995;2:741–745. [Google Scholar]
- 25.Lewis R S, Tannenbaum S R, Deen W M. J Am Chem Soc. 1995;117:3933–3939. [Google Scholar]
- 26.Kosaka H, Wishnok J S, Miwa M, Leaf C D, Tannenbaum S R. Carcinogenesis. 1989;10:563–566. doi: 10.1093/carcin/10.3.563. [DOI] [PubMed] [Google Scholar]
- 27.Addison C C, Lewis J. Q Rev Chem Soc. 1955;9:115–149. [Google Scholar]
- 28.Stamler J S. In: Current Topics in Microbiology and Immunology. Koprowski H, Maeda H, editors. Berlin: Springer; 1995. pp. 19–36. [Google Scholar]
- 29.Meyer D J, Kramer H, Ozer N, Coles B, Ketterer B. FEBS Lett. 1994;345:177–180. doi: 10.1016/0014-5793(94)00429-3. [DOI] [PubMed] [Google Scholar]
- 30.Barton D H R, Blair I A, Magnus P D, Norris R K. J Chem Soc, Perkin Trans. 1973;2:1031–1037. [Google Scholar]
- 31.Pryor W A, Church D F, Govindan C K, Crank G. J Org Chem. 1982;47:156–159. [Google Scholar]
- 32.Ross D, Norbeck K, Moldeus P. J Biol Chem. 1985;260:15028–15032. [PubMed] [Google Scholar]
- 33.Ross D, Cotgreave I, Moldeus P. Biochim Biophys Acta. 1985;841:278–282. doi: 10.1016/0304-4165(85)90069-8. [DOI] [PubMed] [Google Scholar]
- 34.Karoui H, Hogg N, Frejaville C, Tordo P, Kalyanaraman B. J Biol Chem. 1996;271:6000–6009. doi: 10.1074/jbc.271.11.6000. [DOI] [PubMed] [Google Scholar]
- 35.Winterbourn C C, Metodiewa D. Arch Biochem Biophys. 1994;314:284–290. doi: 10.1006/abbi.1994.1444. [DOI] [PubMed] [Google Scholar]
- 36.Allison W S. Acc Chem Res. 1976;9:293–299. [Google Scholar]
- 37.Oae S, Kim Y H, Fukushima D, Shinhama K. J Chem Soc, Perkin Trans. 1978;1:913–917. [Google Scholar]
- 38.Lewis R S, Deen W M. Chem Res Toxicol. 1994;7:568–574. doi: 10.1021/tx00040a013. [DOI] [PubMed] [Google Scholar]
- 39.Pires M, Rossi M J, Ross D S. Int J Chem Kinet. 1994;26:1207–1227. [Google Scholar]