

Abstract
Isolated from Hypericum species H. chinese L. var. salicifolium, biyouyanagin A was assigned structure 1a or 1b on the basis of NMR spectroscopic anaylsis. This novel natural product exhibited significant anti-HIV properties and inhibition of lipopolysaccharide-induced cytokine production. Described herein are the total syntheses of biyouyanagin A and several analogs (3-11), structural revision of biyouyanagin A to 2b, and the biological properties of all synthesized compounds. The total synthesis proceeded through cascade sequences that efficiently produced enantiomerically pure or enriched key building blocks 15b (ent-zingiberene) and 18 (hyperolactone C), and featured a novel [2+2] photoinduced cycloaddition reaction which occurred with complete regio- and stereoselectivity. Biological investigations with the synthesized biyouyangagins A (2-11) and hyperolactones C (12-16) revealed that the activity of biyouyanagin A most likely resides in its hyperolactone C structural domain.
Introduction
The Hypericum species H. chinese L. var. salicifolium has been used in Japan as a folk medicine for the treatment of female disorders for centuries.1 Investigations with this plant led to the discovery of biyouyanagin A, a substance which was originally assigned structure 1a or 1b (Figure 1) on the basis of NMR spectroscopic analysis.2 Biyouyanagin A exhibited significant and selective inhibitory activity against HIV replication in H9 lymphocytes (EC50 = 0.798 μg mL-1) as compared to noninfected H9 lymphocytes (EC50 >25 μg mL-1), thus demonstrating a therapeutic index (TI) of greater than 31.3.2 In addition, this substance inhibited strongly lipopolysaccharide (LPs)-induced cytokine production at 10 μg mL-1 [IL-10 = 0.03; IL-12 = 0.02; tumor necrosis factor-α (TNFα) = 0.48].2 In a recent communication we reported the total synthesis and structural revision of biyouyanagin A (2b) and its 24-epimer, 24-epi-biyouyanagin A (2a, Figure 2).3 In this article we report our detailed investigations in the biyouyanagin field, including an X-ray crystallographic analysis of biyouyanagin A (2b) and the synthesis and biological evaluation of several analogs (3-16, Figure 3) of this novel natural product.
Figure 1.

Originally proposed structures for biyouyanagin A.
Figure 2.

Revised structures of biyouyanagin A (2b) and 24-epi-biyouyanagin A (2a).
Figure 3.

Synthesized biyouyanagin A (3-11) and hyperolactone C (12-16) analogs.
Results and Discussion
By virtue of its novel molecular architecture, the remaining ambiguity shadowing its structure, and its significant biological properties, biyouyanagin A presented itself as an attractive synthetic target. Of particular interest was the verification of the all-cis stereochemistry of the substituents around the cyclobutane ring, an arrangement that looked rather odd at the outset,2 given the steric congestion associated with it. Another impetus for undertaking the total synthesis of biyouyanagin A was to advance further the advent of cascade reactions4 and exploit recent developments in organocatalysis5 for total synthesis purposes.
Retrosynthetic Analysis
While there are myriad ways to disassemble the biyouyanagin A molecule retrosynthetically, the one made possible by a retro [2+2] cycloaddition reaction (Figure 4) is both aesthetically and practically most appealing. In the synthetic direction such a reaction can, in principle, be realized by irradiation with UV light, although no precedent existed at the outset of this work for the photoinduced [2+2] cycloaddition of substrates such as the two components defined by the proposed cyclobutane disconnection (i.e. triene 17b or its 7-epimer 17a and enone 18, Figure 4). If successful, however, this approach would consititute a highly convergent strategy for the total synthesis of the natural product and might also have implications in its biosynthesis. To be sure, however, this rather obvious hypothesis had been proposed as a plausible biosynthetic pathway towards biyouyanagin A by its discoverers.2 In considering such a scenario, inspection of the two transition states that could lead to the biyouyanagin A molecule (I: exo and II: endo, Figure 4) was instructive. Thus, formation of the cis, cis, cis, cis structure (1a or 1b) originally proposed for biyouyanagin A2 requires the endo transition state II, an arrangement that suffers from severe steric congestion between the γ-lactone moiety of the enone and the side chain of the triene component as demonstrated by manual molecular models, and shown in Figure 4. On the other hand, the alternative arrangement of the reacting components as shown in the exo transition state I is free of such unfavorable interactions. This realization created a suspicion in our minds with regards to the structure of biyouyanagin A as proposed in the isolation paper.2 Specifically, we began to favor the cis, cis, trans, trans stereochemistry as shown in structure 2b, although the cloud of ambiguity over the configuration of the C-24 stereocenter (see structure 2a) remained. In addition, the NOE interactions reported for biyouyanagin A,2 in conjunction with manual molecular models, did not exclude the cis, cis, trans, trans structure 2b (or 2a), a fact that fueled further our skepticism about the true structure of the natural product. It was with this background that we embarked on the synthetic journey to biyouyanagin A, whose true structure became our immediate puzzle to solve.
Figure 4.

Retrosynthetic analysis of biyouyanagin A and transition states of the proposed photoinduced [2+2] cycloaddition reaction. For simplicity, only the 24(R) isomers (1b and 2b) are shown.
Construction of Building Blocks for Biyouyanagin A
Having defined the required building blocks for the devised [2+2] photocycloaddition strategy towards biyouyanagin A, their construction evolved as shown in Scheme 1 (17a: ent-7-epi-zingiberene and 17b: ent-zingiberene) and Schemes 2 and 3 (18: hyperolactone C). Interestingly, hyperolactone C (18) is already known in the literature as a naturally occurring substance,6 while 17a and 17b are enantiomers of natural terpenes (7-epi-zingiberene and zingiberene, respectively).7 The initial choice of these particular enantiomeric forms of 17a and 17b was based on the assumption that the hyperolactone C structural motif of biyouyanagin A would be of the same absolute configuration as the naturally occurring hyperolactone C,6 a postulate that, in turn, suggested the absolute configuration of the terpenoid fragments as shown.
Scheme 1.

Synthesis of Terpenoids 17a and 17b.a
aReagents and conditions: (a) 19a or 19b (1.0 equiv), MVK (1.5 equiv), 20 (5 mol%), ethyl 3,4-dihydroxybenzoate (20 mol%), 0 °C, 24 h; then KOH (0.1 N aq., 1.0 equiv), n-Bu4NOH (40% aq., cat.), Et2O:THF:H2O (3:1:3), reflux, 6 h, 72% yield, 93% de for 22a; 68% yield, 86% de for 22b; (b) KHMDS (1.5 equiv), THF, -78 °C, 3 h; then Comins’ reagent (1.5 equiv), THF, -78 °C, 1 h; c) MeMgI (3.0 M in Et2O, 1.5 equiv), CuI (2 mol%), THF, 0 °C, 15 min, 80% over the two steps. MVK = methyl vinyl ketone; THF = tetrahydrofuran; KHMDS = potassium hexamethyldisilazanide; Tf = trifluoromethanesulfonyl
Scheme 2.

Synthesis of Propargyl Alcohol 26.a
aReagents and conditions: (a) DMP (2.0 equiv), CH2Cl2, 25 °C, 5 h, 92%; (b) acetylene, n-BuLi, THF, - 78 °C, 1 h, 92%, 3:1 d.r.; (c) 3,5-dinitrobenzoyl chloride (1.2 equiv), NEt3 (1.2 equiv), 4-DMAP (0.1 equiv), CH2Cl2, 25 °C, 3 h, 98%. Abbreviations: DMP = Dess-Martin periodinane; 4-DMAP = 4-dimethylaminopyridine.
Scheme 3.

Separation of Propargyl Alcohols 26 and 4-epi-26 (biyouyanagin numbering).a
aReagents and conditions: (a) 3,5-dinitrobenzoyl chloride (1.2 equiv), Et3N (1.2 equiv), 4-DMAP (0.1 equiv), CH2Cl2, 25 °C, 3 h; (b) K2CO3 (5.0 equiv), MeOH, 0 → 25 °C, 30 min, 98%.
The synthesis of the terpenoid fragments 17a (ent-7-epi-zingiberene) and 17b (ent-zingiberene) begun from (S)-citronellal (19a) and (R)-citronellal (19b), respectively, and proceeded according to the sequence depicted in Scheme 1. Thus, enamine-mediated 1,4-addition of (S)-citronellal (19a) to methyl vinyl ketone (MVK) facilitated by the proline-derived catalyst 20 (5 mol%)8 and ethyl 3,4-dihydroxybenzoate (21) as a co-catalyst (20 mol%)9 furnished, after an intramolecular aldol condensation (KOH, n-Bu4NOH cat.) of the resulting ketoaldehyde product, 24(S) enone 22a in 72% overall yield and 93% de. The conversion of the latter compound to the desired building block 17a (ent-7-epi-zingiberene) proceeded smoothly, and in 80% overall yield, through a Kumada coupling of the corresponding vinyl triflate (KHMDS, Comins’ reagent)10 with MeMgI (CuI cat.).11 A similar route starting from (R)-citronellal (19b) and MVK, and proceeding through intermediate enone 22b (68% yield, and 86% de), furnished ent-zingiberene (17b), the other targeted terpenoid fragment (80% overall yield for the last two steps).
The synthesis of hyperolactone C (18, Scheme 2)6e begun with (S)-malic acid (23), and proceeded, sequentially, through lactone 26 (Scheme 2) and spirolactone intermediate 27 (Scheme 4). As shown in Scheme 2, (S)-malic acid was converted in five steps, and by literature procedures,6e to hydroxy lactone 24, which was oxidized with DMP to ketolactone 25 in 92% yield. The latter compound reacted with lithium acetylide (generated from acetylene and n-BuLi in THF at -78 °C) to afford, stereoselectively, propargylic alcohol 26 (92% yield, ca 3:1 isomeric ratio). Although this mixture could not be conveniently resolved chromatographically, the desired stereoisomer could be isolated easily by fractional crystallization from CH2Cl2/hexanes (62% yield). Alternatively, the two isomers could be separated by flash column chromatography of their 4-nitrobenzoates (4-nitrobenzoyl chloride, Et3N, 4-DMAP, 95% combined yield), and then two free alcohols (26 and 4-epi-26) were released after hydrolysis (K2CO3, MeOH, quantitative yields, Scheme 3). The stereoselectivity observed in this reaction is attributed to the presumed chelation control exerted by the benzyloxy group. The stereochemistry of 26 was initially confirmed by an X-ray crystallographic analysis (see ORTEP drawing, Figure 5) of its 2,4-dinitrobenzoate (26a, mp 159-160 °C, toluene; obtained by reaction of 26 with 2,4-dinitrobenzoyl chloride in the presence of Et3N and 4-DMAP, 98% yield), and subsequently by an X-ray crystallographic analysis performed on the compound itself (26), which crystallized upon prolonged standing in a mixture of CH2Cl2 and hexanes (mp 84-86 °C, CH2Cl2/hexanes, see ORTEP drawing, Figure 6).
Scheme 4.

Synthesis of Hyperolactone C (18) and 4-epi-Hyperolactone C (16) Through Palladium-catalyzed Cascade Sequences (biyouyanagin numbering).a
aReagents and conditions: (a) Pd(PPh3)4 (5 mol%), PhI (1.2 equiv), CO (200 psi), CO2 (200 psi), Et3N, 100 °C, 5 h, 79%; (b) BBr3 (1.5 equiv), CH2Cl2, -78 °C, 30 min, 95%; (c) o-NO2C6H4SeCN (1.2 equiv), P(n-Bu)3 (1.2 equiv), THF, 25 °C, 4 h, 90%; (d) H2O2 (30% aqueous, 10 equiv), THF, 0 → 25 °C, 1 h, 85%, (e) same as (a), 71%; (f) same conditions as for (b); (g) same conditions as for (c); (h) same conditions as for (d), 71% over the three steps.
Figure 5.

ORTEP drawing of compound 26a with the thermal ellipsoids at 30% probability level.
Figure 6.

ORTEP drawing of compound 26 with the thermal ellipsoids at 30% probability level.
What was developed next was a highly efficient and pleasing cascade sequence that produced spirolactone 27 from propargylic alcohol 26 and phenyl iodide in one pot. This remarkable palladium-catalyzed cascade was brought about by Pd(PPh3)4 (5 mol%) and Et3N under an atmosphere of a mixture of CO and CO2 (200 psi) as depicted in Scheme 4 (79% yield, single diastereomer).12 An X-ray crystallographic analysis of 27 (mp 176-177 °C, toluene, see ORTEP drawing, Figure 7) allowed its unambiguous stereochemical assignment. The retention of stereochemistry at the spirocenter of product 27 as compared to the starting substrate (26) of the cascade sequence is consistent with the proposed mechanism involving intermediates I-VI shown in Scheme 4. Specifically, it is postulated12 that a Sonogashira-type process under the reaction conditions allows the initial formation of acetylenic ketone I, which absorbs a molecule of CO2 to furnish species II, an intermediate that serves as the precursor to cyclic carbonate III, whose palladium-catalyzed extrusion of CO2 leads to the π-palladium complex IV↔V, stereospecifically. Finally, rearrangement of V, as shown, leads to palladacycle VI, which readily loses Pd0 to afford the observed product (27) with retention of stereochemistry. The completion of the synthesis of hyperolactone C (18) required debenzylation (BBr3), selenenylation [o-(NO2)C6H4SeCN, n-Bu3P], and oxidation/syn-elimination (H2O2), a sequence that proceeded in 79% overall yield {[α]D25 = -272.8 (c = 0.69, CHCl3); lit.,6b [α]D25 = -270.7 (c = 0.11, CHCl3)} as shown in Scheme 4. Similarly, 4-epi-hyperolactone C (16) was synthesized from 4-epi-26 without loss of integrity of stereochemistry (Scheme 4).
Figure 7.

ORTEP drawing of compound 27 with the thermal ellipsoids at 30% probability level.
Total Synthesis of Biyouyanagin A and 24-epi-Biyouyanagin
Having established enantioselective and practical routes to fragments 17a and 17b, and 18, the next task became their photoinduced fusion into the biyouyanagin A framework (Scheme 5). To this end, each of the two terpenoid fragments 17a and 17b (4.0 equiv) were separately mixed with hyperolactone C (18, 1.0 equiv) and irradiated with UV light in the presence of 2′-acetonaphthone13 (1.0 equiv) as a triplet sensitizer in concentrated CH2Cl2 solution in a quartz cell (320 nm filter) at 5 °C for 8 h. Under these conditions, we were pleased to observe regio- and stereoselective formation of the desired cyclobutane product in each case, leading to 2a (48% yield, based on 18) and 2b (54% yield, based on 18). The two compounds exhibited NMR spectroscopic data consistent with their structures, with 2b revealing identical signals to those reported for the natural product, biyouyanagin A.2 Crucial NOE data (see Figure 8) for this compound, however, supported 2b, rather than the originally proposed structure 1b. Particularly diagnostic for the cis, cis, trans, trans stereochemistry were the NOEs between H-6 and H-17, H-6 and H-22, and H-17 and H-22. Note that adjacent protons of the cyclobutane ring may exhibit an NOE, even if they are trans to each other, as it is the case here. In addition, the indicated NOEs between the aromatic and C-23 methyl protons (see Figure 8) revealed the syn orientation of these substituents. The absolute structures of 2a (mp 94-95 °C, hexanes) and 2b (mp 75-76 °C, hexanes) were ultimately solved by X-ray crystallographic analysis (see ORTEP drawings, Figure 9 and 10, respectively), which unambiguously established the complete structure of biyouyanagin A as 2b. Its absolute stereochemistry was deduced from those of the starting materials [(R)-citronellal and (S)-malic acid] employed for the synthesis of the natural enantiomer {[α]D25 = -256.6 (c = 0.80, CHCl3); lit.,2 [α]D25 = -240.0 (c = 0.50, CHCl3)}.
Scheme 5.

Completion of the Total Synthesis of 24-epi-Biyouyanagin A (2a) and Biyouyanagin A (2b).a
aReagents and conditions: (a) 17a or 17b (4.0 equiv), 2′-acetonaphthone (1.0 equiv), CH2Cl2 (0.5 M for 18), 320 nm filter, 5 °C, 8 h, 48% for 2a, 54% for 2b.
Figure 8.

Selected NOEs exhibited by biyouyanagin A (2b).
Figure 9.

ORTEP drawing of compound 2a with the thermal ellipsoids at 30% probability level.
Figure 10.

ORTEP drawing of compound 2b with the thermal ellipsoids at 30% probability level.
Design and Synthesis of Biyouyanagin A Analogs
With an efficient route to the biyouyanagin A molecule and the establishment of its relative and absolute stereochemistry, we then turned our attention to the application of the developed synthetic technology to the construction of designed analogs for biological investigations. The biyouyanagin A analogs 3-11 (Figure 3) were designed in order to take advantage of the flexibility and scope of the developed synthetic strategy and to test the effect of substituents at C-3, C-7, C-19 and C-22, while the truncated structures 12-16 (hyperolactone C analogs) were designed in order to explore structure activity relationships within the hyperolactone C (18) structural motif, which was suspected to be the active pharmacophore of the biyouyanagin A molecule.
The required building blocks were constructed by procedures either as described above (30, 12, 13) for biyouyanagin A, or as reported previously in the literature (31,14 3215), except for enone 14 and ketone 15. Enone 14 was obtained from hydrogenation of hyperolactone C (18) with H2 (1 atm) and 10 mol% Pd/C cat. in quantitative yield while a stereoselective 1,4-reduction reaction of 18 with L-selectride gave ketone 15 in 70% yield (Scheme 6). Analogs 3-9 (see Table 1 and Figure 2) were synthesized from the corresponding terpenoid dienes and enone components through [2+2] photocycloaddition reactions as indicated in Table 1. The structures of these products were consistent with their spectroscopic data; that of 8 was further confirmed by X-ray crystallographic analysis (mp 170-171 °C, hexanes, see Figure 11 for ORTEP drawing).
Scheme 6.

Synthesis of Enone 14 and Ketone 15.a
aReagents and conditions: (a) H2 (1 atm), 10 wt% Pd/C (5 mol%), MeOH, 25 °C, 3 h, 99%; (b) L-Selectride (3.0 equiv), THF, -78 → -10 °C, 1 h, 70%.
Table 1.
Syntheses of Biyouyanagin A Analogs by [2+2] Photocycloadditiona
| entry | terpenoid component | enone component | Product (%yield) |
|---|---|---|---|
| 1 |  |
 |
3 (49) |
| 2 |  |
 |
4 (15) |
| 3 | 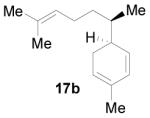 |
 |
5 (43) |
| 4 |  |
 |
6 (53) |
| 5 |  |
 |
7 (49) |
| 6 |  |
 |
8 (46) |
| 7 |  |
 |
9 (50) |
Reactions were carried out by irradiating (quartz cell, 320 nm filter) a concentrated solution (0.5 M) of the terpenoid (4.0 equiv) and the enone (1.0 equiv) components in CH2Cl2 at 5 °C for 8 h.
Figure 11.

ORTEP drawing of compound 8 with the thermal ellipsoids at 30% probability level.
The hexacyclic compound 10 was unexpectedly obtained (40% yield) upon irradiation of a mixture of terpenoid component 17b and enone 28a in the presence of 2′-acetonaphthone through the presumed intermediacy of the initially formed adduct 33 as shown in Scheme 7. The spontaneous ring closure of the hydroxyl moiety upon the carbonyl function and the stability of its lactol form (10) are clearly favored within the latter molecule, as opposed to the architecture of the starting enone, which apparently (1H and 13C NMR spectroscopic analysis) remains open as shown. The structure of compound 10 was initially based on its spectroscopic data and its facile conversion to biyouyanagin A (2b) through iodination (I2, PPh3, imid.), followed by sequential exposure of the resulting iodide to o-NO2C6H4SeNa and H2O2 (51% overall yield for the three steps). Ultimately, an X-ray crystallographic analysis of 10 (mp 127-128 °C, CH2Cl2/hexanes) unambiguously established its assigned structure (see ORTEP drawing, Figure 12).
Scheme 7.

Synthesis of Biyouyanagin A Analog 10.a
aReagents and conditions: (a) 17b (4.0 equiv), 2′-acetonaphthone (1.0 equiv), CH2Cl2 (0.5 M for 28a), 320 nm filter, 5 °C, 8 h, 40%; (b) I2 (3.0 equiv), PPh3 (3.0 equiv), imidazole (4.0 equiv), CH2Cl2; (c) o-NO2C6H4SeNa (2.0 equiv), THF; then H2O2 (30% aqueous, 10 equiv), THF, 25 °C, 5 h, 51% over the three steps.
Figure 12.

ORTEP drawing of compound 10 with the thermal ellipsoids at 30% probability level.
Finally, compound 11 was constructed from biyouyanagin A (2b) by a cross metathesis with (Z)-2-butene-1,4-diol diacetate (34, Scheme 8). This remarkably chemoselective reaction was brought about, in 73% yield, by exposure of a mixture of 2b and 34 (5.0 equiv) to Grubbs catalyst II (10 mol%) in CH2Cl2 solution at 40 °C.16 Interestingly, no products arising from cross metathesis with any of the other olefinic bonds of biyouyanagin A (2b) were observed.
Scheme 8.

Synthesis of Analog 11 Via Olefin Cross Metathesis.a
aReagents and conditions: Grubbs II cat. (10 mol%), 34 (5.0 equiv), CH2Cl2, 40 °C, 24 h, 74%.
Biological Evaluation of Biyouyanagin A and Analogs
The synthesized compounds were tested for their inhibitory activity against HIV-1 replication in MT-2 lymphocytes and their cytotoxicity against non-infected MT-2 lymphocytes using the TZM-bl/luciferase assay and AZT as a control. As seen in Table 2, synthetic biyouyanagin A (2b) exhibited significant activity against HIV-1 replication in this assay (IC50 = 26 μM) and lower cytotoxicity against the MT-2 lymphocytes, scoring a therapeutic index (TI) of 7.5. The lower inhibitory potency observed in these experiments compared to those reported by Tanaka et al. (IC50 = 1.7 μM)2 may be due to sensitivity differences in the two assays employed. 24-epi-Biyouyanagin A (2a) and the other biyouyanagin A analogs listed in Table 2 (and shown in Figures 2 - 4) displayed similar activities against HIV-1 replication and cytotoxicities against the MT-2 lymphocytes (IC50 = 19 - 31 μM; CC50 = 143 - 512 μM) and TIs (7.5 - 18.9), with analogs 7 and 10 being the most potent against HIV-1 (IC50 = 19 μM), and analog 11 possessing the most favorable TI (18.9). Interestingly, however, we discovered that hyperolactone C (18, Figure 4) and its analogs (12-16, Figure 3) showed comparable, and in some cases more potent, anti-HIV activities compared to biyouyanagin A and its analogs. Thus, hyperolactone C (18, entry 17), itself, exhibited an IC50 value of 29 μM against HIV-1 infected cells, and a CC50 value of 925 μM (TI = 32.0). The most potent member of the series against HIV-1 replication was found to be hyperolactone C analog 13 (IC50 = 12 μM), possessing a para-methoxy group on its phenyl moiety. The most impressive compound of the class, however, turned out to be hyperolactone C analog 12 which exhibited a TI of 80.0 (IC50 = 16 μM; CC50 = 1301 μM). With a fluoride residue in its structure, this relatively small molecule may serve as a new lead for a drug discovery program in the anti-HIV area of pharmaceutical research.
Table 2.
Anti-HIV-1 Activities and Cytotoxicities of Biyouyanagin A and Analogsa
| Entry | compound | HIV-1 IC50 (μM)b | MT-2 CC50 (μM)c | TId |
|---|---|---|---|---|
| 1 | AZT | 0.078 | >50 | >641 |
| 2 | 2a | 20 | 285 | 14.4 |
| 3 | 2b | 26 | 198 | 7.5 |
| 4 | 3 | 25 | 444 | 17.5 |
| 5 | 4 | 25 | 268 | 10.8 |
| 6 | 5 | 20 | 197 | 10.0 |
| 7 | 6 | 19 | 213 | 11.1 |
| 8 | 7 | 23 | 333 | 14.4 |
| 9 | 9 | 31 | 232 | 7.5 |
| 10 | 10 | 19 | 143 | 7.5 |
| 11 | 11 | 27 | 512 | 18.9 |
| 12 | 12 | 16 | 1301 | 80.0 |
| 13 | 13 | 12 | 417 | 35.6 |
| 14 | 14 | 34 | 1377 | 40.0 |
| 15 | 15 | 23 | 459 | 20.0 |
| 16 | 16 | 35 | 462 | 13.3 |
| 17 | 18 | 29 | 925 | 32.0 |
Average value from quintuplicate experiments.
IC50 is the concentration at which 50% inhibition of HIV-1 replication is observed as measured by a TZM-bl/luciferase assay.
CC50 is the concentration at which 50% inhibition of cell metabolism is observed as measured by a colorimetric XTT assay.
Therapeutic index (CC50/IC50).
Conclusion
Relying on a highly convergent strategy, the described total synthesis of biyouyanagin A and its 24-epimer highlights the importance of cascade reactions in total synthesis and the continuing role the later discipline plays in natural products chemistry and biology. Thus, based on both organocatalysis and metallocatalysis, the developed synthetic technology culminated in the revision of the originally assigned structure of biyouyanagin A and determination of its absolute stereochemistry. It also delivered a series of analogs of the natural product, whose biological evaluation established the first structure activity relationships in the field, and led to the discovery that hyperolactone C, a plausible biogenetic precursor of biyouyanagin A, possesses higher and significant potency against HIV-1 replication. Based on these biological results and considering the efficiency of the established synthetic strategy, further investigations into chemical biology and medicinal chemistry may be warranted.
Supplementary Material
Acknowledgments
Dedicated to Professor Duilio Arigoni on the occasion of his 80th birthday. We thank Dr. D. H. Huang and Dr. L. Pasterneck for NMR spectroscopic assistance, and Dr. Siuzak and Dr. R. Chadha for mass spectrometric and X-ray crystallographic assistance, respectively. Financial support for this work was provided by the National Institute of Health (USA) and the Skaggs Institute for Chemical Biology and A*STAR (postdoctoral fellowship to T.R.W.).
References
- (1).Murakami K. Tokushima-ken Yakusouzukan. 1984:102–103. [Google Scholar]
- (2).Tanaka N, Okasaka M, Ishimaru Y, Yakaishi Y, Sato M, Okamoto M, Oshikawa T, Ahmed SU, Consentino LM, Lee K-H. Org. Lett. 2005;7:2997–2999. doi: 10.1021/ol050960w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Nicolaou KC, Sarlah D, Shaw DM. Angew. Chem. Int. Ed. 2007;46:4708–4711. doi: 10.1002/anie.200701552. [DOI] [PubMed] [Google Scholar]
- (4).Nicolaou KC, Edmonds DJ, Bulger PG. Angew. Chem. Int. Ed. 2006;45:7134–7186. doi: 10.1002/anie.200601872. For a recent review article on cascade reactions in total synthesis, see. [DOI] [PubMed] [Google Scholar]
- (5).Berkessel A, Gröger H. Asymmetric Organocatalysis: From Biomimetic Concepts to Applications in Asymmetric Synthesis. Wiley-VCH, Weinheim; 2006. p. 454. [Google Scholar]
- (6)(a).Aramaki Y, Chiba K, Tada M. Phytochemistry. 1995;38:1419–1421. For selected papers on hyperolactone C, see: isolation and structure. [Google Scholar]; (b) Crockett SL, Schuhly W, Belaj F, Khan IA. Acta Crystallogr. Sect. E. 2004;60:o2174–o2176. X-ray structure. [Google Scholar]; (c) Ichinari D, Ueki T, Yoshihara K, Kinoshita T. Chem. Commun. 1997:1743–1743. Synthesis. [Google Scholar]; (d) Ueki T, Ichinari D, Yoshihara K, Morimoto Y, Kinoshita T. Tetrahedron Lett. 1998;39:667–668. [Google Scholar]; (e) Ueki T, Doe M, Tanaka R, Morimoto Y, Yoshihara K, Kinoshita T. J. Heterocycl. Chem. 2001;38:165–172. [Google Scholar]; (f) Kraus GA, Wei JJ. Nat. Prod. 2004;67:1039–1040. doi: 10.1021/np0498962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7)(a).Eschenmoser A, Schinz H. Helv. Chim. Acta. 1950;33:171–177. For selected papers on zingiberenes, see. [Google Scholar]; (b) Arigoni D, Jeger O. Helv. Chim. Acta. 1954;37:881–883. [Google Scholar]; (c) Joshi GD, Kulkarni SN. Indian J. Chem. 1965;3:91–92. [Google Scholar]; (d) Uhde G, Ohloff G. Helv. Chim. Acta. 1972;55:2621–2625. doi: 10.1002/hlca.19720550712. [DOI] [PubMed] [Google Scholar]; (e) Breeden DC, Coates RM. Tetrahedron. 1994;50:11123–11132. [Google Scholar]; (f) Bhonsle JB, Deshpande VH, Ravindranathan T. Indian J. Chem. Sect. B. 1994;33:313–316. [Google Scholar]
- (8).Enders D, Kipphardt H, Gerdes P, Brena-Valle LJ, Bhushan V. Bull. Soc. Chim. Belg. 1988;97:691–704. [Google Scholar]
- (9).Chi Y, Gellman SH. Org. Lett. 2005;7:4253–4256. doi: 10.1021/ol0517729. [DOI] [PubMed] [Google Scholar]
- (10).Comins DL, Dehghani A. Tetrahedron Lett. 1992;33:6299–6302. [Google Scholar]
- (11).Karlström ASE, Rönn M, Thorarensen A, Bäckvall J-E. J. Org. Chem. 1998;63:2517–2522. doi: 10.1021/jo971737i. [DOI] [PubMed] [Google Scholar]
- (12).Inoue Y, Ohuchi K, Yen I-F, Imaizumi S. Bull. Chem. Soc. Jpn. 1989;62:3518–3522. [Google Scholar]
- (13)(a).Cantrell TS. J. Org. Chem. 1974;39:3063–3070. [Google Scholar]; (b) Schenck GO, Kuhls J, Krauch CH. Justus Liebigs Ann. Chem. 1966;693:20–43. [Google Scholar]
- (14).Sen A, Grosch W. Flavour Frag. J. 1990;5:233–234. [Google Scholar]
- (15).Buttrus NH, Cornforth J, Hitchcock PB, Kumar A, Stuart AS. J. Chem. Soc., Perkin Trans. I. 1987:851–857. [Google Scholar]
- (16).Chatterjee AK, Choi TL, Sanders DP, Grubbs RH. J. Am. Chem. Soc. 2003;125:11360–11370. doi: 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.