

Abstract
A fundamental issue in cell biology is how signals are transmitted across membranes. A variety of transmembrane receptors, including multichain immune recognition receptors, lack catalytic activity and require Src family kinases (SFKs) for signal transduction. However, many receptors only bind and activate SFKs after ligand-induced receptor dimerization. This presents a conundrum: How do SFKs sense the dimerization of receptors to which they are not already bound? Most proposals to resolve this enigma invoke additional players, such as lipid rafts or receptor conformational changes. Here we used simple thermodynamics to show that SFK activation is a natural outcome of clustering of receptors with SFK phosphorylation sites, provided that there is phosphorylation-dependent receptor-SFK association and an SFK bound to one receptor can phosphorylate the second receptor or its associated SFK in a dimer. A simple system of receptor, SFK and an unregulated protein tyrosine phosphatase (PTP) can account for ligand-induced changes in phosphorylation observed in cells. We suggest that a core signaling system comprising a receptor with SFK phosphorylation sites, an SFK and an unregulated PTP provides a robust mechanism for transmembrane signal transduction. Other events that regulate signaling in specific cases may have evolved for fine-tuning of this basic mechanism.
Keywords: Ephrin, C-type lectin, Nephrin, CDCP1, Reelin, Dab1, Multichain immune recognition receptors, T cell receptor, B cell receptor, FcεRI
Cells detect their environment through a wide variety of receptors. While signaling via receptors with intracellular catalytic activities or channel functions is well understood, signaling by a variety of receptors that are linked to Src family kinases (SFKs) remains unresolved (1–11). SFKs are a subgroup of the non-receptor tyrosine kinases. They are allosteric enzymes with at least two conformational states that are differentially stabilized by protein-protein interactions and by phosphorylation-dephosphorylation at two different sites (12–14). Active SFKs can bind to other proteins through a phosphorylation-independent SH3 domain and a phosphorylation-dependent SH2 domain. This allows for stabilization of the active state.
Some receptors, such as CD4, are constitutively bound to an SFK (15, 16). Clustering of CD4 by its ligand brings the bound SFKs together and allows for a stimulating intermolecular phosphorylation event (17). This mechanism is comparable to that established for receptor tyrosine kinases (RTKs)(Fig. 1b) (18, 19). RTK kinase domains have a low basal activity, which is effectively opposed by cellular protein tyrosine phosphatases (PTPs). When the kinase domains are brought together they undergo intermolecular phosphorylation, which stabilizes a new conformation with increased activity.
FIGURE 1. Models for regulation of receptor tyrosine kinases and SFK-dependent non-catalytic receptors.
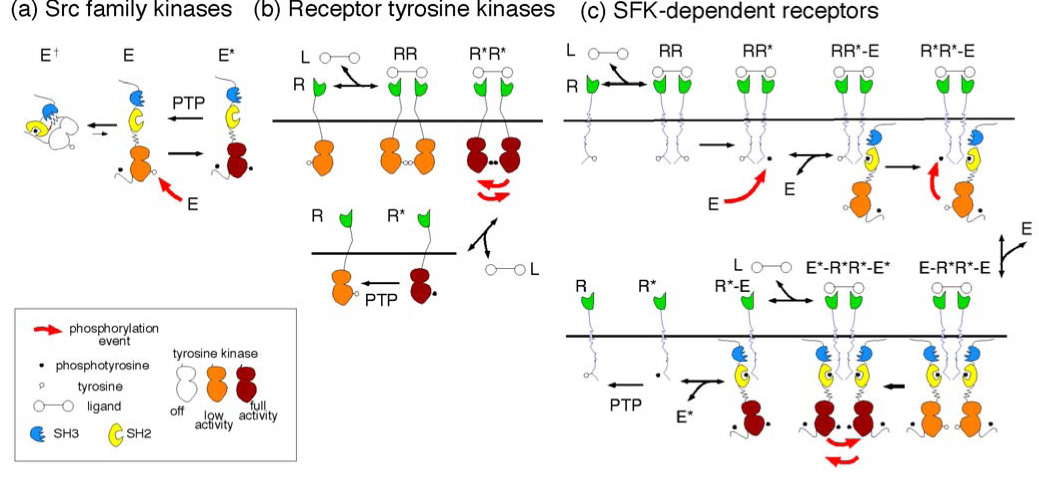
(a) Structure and regulation of SFKs. SFKs have 3 domains: SH3 (blue, phosphorylation-independent binding, not considered here), SH2 (yellow, phosphorylation-dependent binding) and kinase (white, orange or crimson according to increasing activity). The SFK can adopt two main conformation states: "closed" inactive (E†) and "open" low (E) and high (E*) activity forms. E is converted to E* by intermolecular phosphorylation in the activation loop. In addition, phosphorylation at the C terminus alters the balance between the closed and open conformations by stabilizing the closed form. This phosphorylation is not regulated in the model, so is not considered further.
(b) Regulation of RTKs. The kinase domains of monomeric RTKs, R, are inhibited by intramolecular interactions, and have low basal activity (orange) which is readily reversed by PTPs. Following dimerization by ligand, L, intermolecular phosphorylation of the activation loop occurs, and the kinase is activated (crimson). If ligand dissociates, monomeric receptors will remain in the phosphorylated, active state until dephosphorylated by PTPs. This allows for hysteresis in signaling.
(c) Proposed model for regulation of SDRs. Only a small fraction of monomeric SDRs (R) are phosphorylated. After dimerization by ligand, this phosphorylation may allow binding of an SFK (RR*-E complex). This may lead to an "intramolecular" phosphorylation to form a R*R*-E complex. This stimulates receptor phosphorylation. If a second E binds, to form an E-R*R*-E complex, then intermolecular phosphorylation of E will be stimulated, and the E*-R*R*-E* complex will have high kinase activity. Phosphorylated E* may be released into the cytosol, to phosphorylate more receptors. This would provide a mechanism for hysteresis in signaling.
Unlike CD4, however, many other SFK-dependent receptors show little SFK binding prior to clustering, so the RTK model does not apply. Some such receptors are listed in Table1. They include immune regulatory receptors and several cell-matrix and cell-cell signaling receptors (1–11). Here we call these receptors SDRs (SFK-dependent receptors). SDR signaling is initiated by clustering (dimerization or oligomerization) of the receptors by ligand (1–11). This induces SDR tyrosine phosphorylation, SFK activation and formation of phosphorylation-dependent SFK-SDR complexes via the SFK SH2 domain.
Table 1.
SFK-Dependent Receptors (SDRs)
| SDR | SFK phosphorylation site(s) a | References |
|---|---|---|
| Multichain immune recognition receptors, e.g., T cell receptor ξ chain, B cell receptor Igα and β chains, FcεRIγ | YXXL/I(6–8 b)YXXL/I | (1–3) |
| Inhibitory immune receptors, e.g., FcεRIIB, SHPS, Siglecs | (I/V/L/S)xYxx(L/V) | (4) |
| Lectins CLEC-2 and Dectin-1 | DEDGYXXL | (5) |
| Ephrin B family | YXXV(3)YXXPXYXXQ(10)YYKV | (6) |
| Nephrin c | Five repeats of YDXV | (7) |
| CDCP1 | YXXI(5)YXXL | (8) |
| Myelin-associated glycoprotein | YAEI | (9) |
| Reelin receptor-Dab1 complexes | YQXI(9)YQXI | (10, 11) |
Y, confirmed SFK phosphorylation site; Y, phosphorylation not shown.
Numbers in parentheses indicate spacing between sites.
Nephrin also binds to SFK SH3 domains.
Various mechanisms have been proposed to explain how dimerization of an SDR is communicated to an SFK that is not previously bound. For example, dimerization of the T cell receptor (TCR) may alter the conformation of its cytoplasmic domain so that it partitions into lipid rafts, where it is exposed to raft-associated SFKs (20–23). Alternatively, antigen-presenting cells may displace transmembrane PTPs with bulky extracellular domains from the vicinity of TCRs (24). Or, an SFK inhibitor, Csk, may be lost from the vicinity of the TCR due to rapid dephosphorylation of its membrane anchor, Cbp/PAG (25). However, none of these mechanisms is straightforward. Lipid rafts provide an environment that may inhibit SFKs (26), they are more important for maintaining than for initiating TCR signaling (27) and they do not coincide with initial SFK-dependent clusters of TCRs (28). Physical displacement of transmembrane PTPs seems unlikely for receptors with small ligands. And in myeloid cells Cbp/PAG phosphorylation increases during the initial activation of FcεRI, which would seem to oppose SFK activation (25). Furthermore, none of these mechanisms satisfactorily explains a recent observation (29). The Dab1 component of the Reelin receptor-Dab1 complex contains SFK sites that are phosphorylated when the Reelin receptors are clustered. This phosphorylation is also induced when the Dab1 component is artificially dimerized in the cytosol (29). This suggests that activation can occur by clustering phosphorylation sites in the absence of membranes or apparent mechanisms for regulating kinases or phosphatases.
Here we have explored the properties of a minimal signaling system. We asked what happens when a hypothetical SDR changes from monomer to dimer state. We assumed that the only association between SDR and SFK requires phosphorylated SDR, and there are no conformation changes or compartmentalization of the receptor and no external influences on SFK or PTP activity. Using first principles, we found that that mass action can explain significant increases in SDR phosphorylation and SFK activation following SDR dimerization.
RESULTS
The model is shown in Fig 1c. In the absence of ligand, basal activity of the SFK (E) causes low-level phosphorylation of the monomeric receptor (R) to create R* (where asterisk represents phosphate). The SH2 domain of E can associate with R* to form R*-E, protecting R* from dephosphorylation (30). However, association-dissociation is rapid (31), and constant PTP activity keeps the phosphorylation level low. In the presence of ligand, the receptor is a dimer (RR). Now, phosphorylation of one receptor molecule creates RR* and allows binding of E to create RR*-E. The second receptor in the dimer can now be phosphorylated in an essentially "intra-molecular" reaction (32, 33). This is a key step, and creates R*R*-E to which a second E may bind, creating E-R*R*-E. In the second key step, the two SFKs in this tetramer can undergo intermolecular phosphorylation and become more active (14, 34–36). This creates the highly active E*-R*R*-E*. Subsequent dissociation of ligand will allow release of E*, raising the activity of E in the cell and stimulating further phosphorylation of RR. With time, the system returns to baseline by PTP action.
Even though the model contains only SDR, SFK and PTP, it is more complex than the RTK model, and a priori seemed unlikely to explain the experimentally-observed increases in SDR phosphorylation or SFK activation after ligand-induced receptor dimerization. We therefore tested the model by analyzing the component reactions and applying the laws of mass action.
We first analyzed the effect of SDR-SFK association on SDR phosphorylation (Appendices 1 and 2; all Appendices are available as Supporting Information). We then analyzed the effect of SDR-SFK association on SFK phosphorylation (Appendix 3). This breakdown allowed us to solve the simplest kinetic model representing each effect algebraically, without resort to complex simulation or numerical methods. The Supporting Information includes Excel spreadsheets that will calculate results for any chosen values of input parameters, and selected results are plotted in Fig. 2– Fig. 4.
1. Positive and negative feedback effects due to SFK binding to monomeric receptors
We analyzed the effects of SFK (E) binding to phosphorylated monomeric receptors (R*) (Fig. 2a). SFKs are allosteric enzymes with at least two conformational states (12–14): closed, inactive E† and open, active E (Fig. 1a; E can be further activated by phosphorylation in the activation loop, but for the present we consider only E in its low activity state). The relative amount of E† (the ratio Q) is regulated by C-terminal phosphorylation at a residue that stabilizes the E† state. We are not proposing any regulation of phosphorylation of the C-terminal site, so we assume that Q does not change when receptors are dimerized. The active E phosphorylates R with a bimolecular rate constant k1, and binds R* with association constant K3. The PTP is in excess and not regulated, so appears as a pseudo first order rate constant, k2, for dephosphorylating R*.
FIGURE 2. Positive and negative feedback effects due to SFK binding to monomeric receptors.
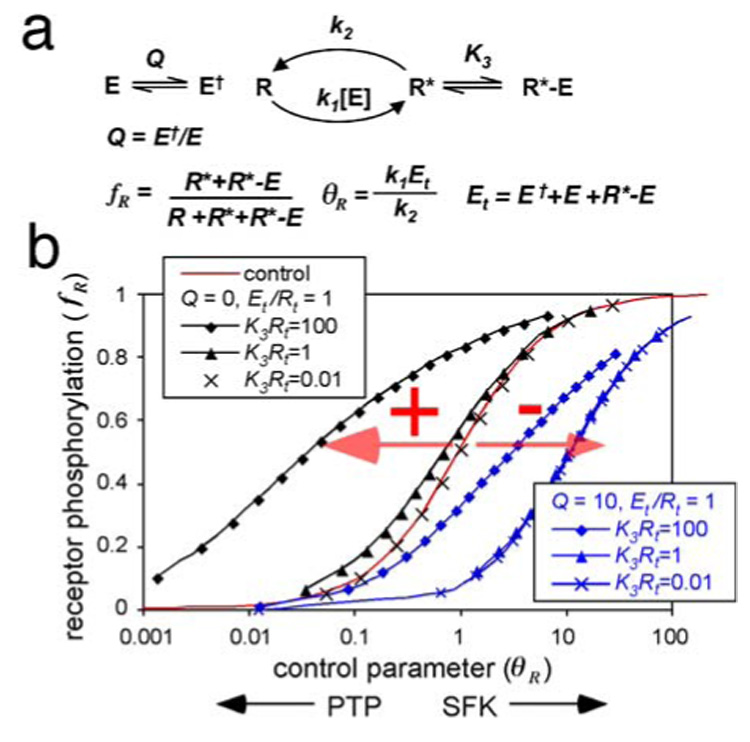
(a) Reactions and equations describing the conformation states of the SFK and monomeric receptor phosphorylation. These reactions are solved in Appendix 1.
(b) Fractional receptor phosphorylation (fR) as a function of the ratio of SFK and PTP activities (the control parameter for receptor phosphorylation, θR). Results are shown for different values of the equilibrium between closed and open SFK (Q), the ratio of total SFK to receptor (Et/Rt), and the product of receptor concentration and binding affinity (K3Rt). The control (red line) is for no binding of E to R*. Black lines show results where positive feedback (+) predominates. Blue lines show where negative feedback predominates (−).
We calculated the fractional phosphorylation of receptors, fR, under different conditions of SFK and PTP activity (Appendix 1). fR is commonly measured experimentally, for example, by Western blotting, and represents the amount of R* molecules relative to the total (Fig. 2a). The cellular environment was represented by the ratio of SFK and PTP activities, θR (Fig. 2a). Note that the cellular environment does not change when receptors dimerize, but will differ according to cell type and conditions.
The best way to understand the effect of binding of E to R* is to compare it with the simplest situation of no binding. In this case, we have the standard “hyperbolic response curve” with 50% phosphorylation (fR = 0.5) when the rates of phosphorylation and dephosphorylation are balanced (θR = 1). Increasing or decreasing the ratio of kinase to phosphatase activities increases or decreases fR accordingly (Fig. 2b, red line). Binding of E to R* has two effects: First, it reduces the amount of free E able to phosphorylate more R. This is a negative feedback or sequestration effect (37). In addition, the R*-E complexes are protected from dephosphorylation (30), so this stabilizes R* and causes a positive feedback. These two counteracting influences mean that more or less E activity may be needed for 50% phosphorylation - the response curve is shifted left by positive feedback and right by negative feedback (Fig. 2b, + and - red arrows, respectively). Fig. 2b (black lines) shows the effect of changing the association constant of E for R* (K3, Fig. 2a). At very low binding (Fig. 2b, x's), the curve overlays the control (red line), but with high binding (diamonds), the response curve is shifted far to the left. The positive feedback effect predominates. On the other hand, reducing the total concentration of E relative to R, or raising Q, which allows for a reservoir of closed-conformation, inactive E†, causes negative feedback to predominate and shifts the curve to the right (Fig 2b, blue lines).
2. Effect of trans-phosphorylation in dimeric receptor complexes
The essence of our model for SDR mediated signaling is an additional positive feedback pathway for a dimeric receptor: an “intramolecular” phosphorylation of the RR*-E complex to form R*R*-E (32, 33).. RR of course can still be phosphorylated on the two subunits independently by the bimolecular reaction, and phosphorylated subunit(s) can bind to E (Fig.3a). This receptor trans-phosphorylation effect is quantified by parameter σ, which includes the association constant K3 and the uni-molecular trans-phosphorylation constant, k4 (Fig. 3a). When R is a monomer, σ = 0 (Fig. 3b, red line). However, when R is a dimer, σ has some positive value and the value of fR is increased (Fig 3b, black lines; Appendix 2). Thus, for given θR, receptor dimerization increases fR, moving vertically on Fig 3b from the red line to the appropriate black line depending on the value of σ for the dimer (red arrow). In the example shown, dimerization of receptors with σ = 1000 causes a ~15-fold increase in phosphorylation at θR = 0.05 (Fig. 3c, red arrow), but a smaller change at either higher or lower values of θR. The importance of θR fits with biological expectations: increasing the PTP activity too high will prevent receptor activation even at high levels of extracellular stimulus, and decreasing the PTP too low will result in activation even in the absence of stimulus. The concentration and activity of PTP set a threshold and appropriate range for receptor activation.
FIGURE 3. Trans-phosphorylation of dimeric receptors.
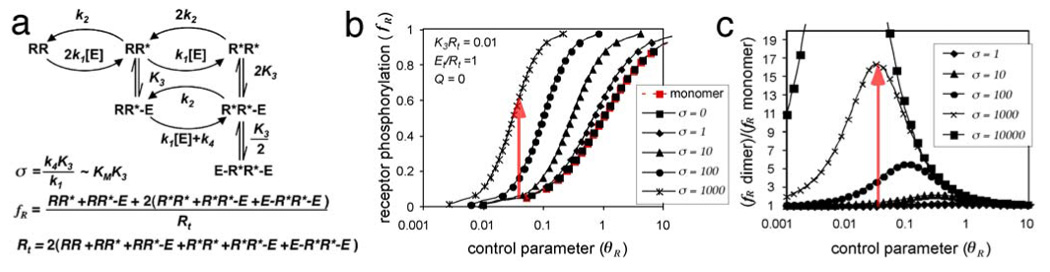
(a) Reactions and equations describing the interactions of open-conformation E with dimeric receptor RR. The phosphorylation/dephosphorylation and association constants are as in Fig. 2, except k4 represents receptor trans-phosphorylation within a RR*-E complex. These reactions are solved in Appendix 2.
(b) Fractional receptor phosphorylation (fR) as a function of the control parameter for receptor phosphorylation (θR), for particular values of Q, Et/Rt and K3Rt. The control (red) is for monomeric receptors. Black lines show results for dimeric receptors with various values of the receptor trans-phosphorylation parameter (σ). Dimerization does not change any parameter except σ, which increases fR (red arrow).
(c) Relative increase in receptor phosphorylation due to dimerization.
Surprisingly, we found that receptor monomers and dimers are similarly (but not identically) affected by negative and positive feedback in Sec. 1 (Appendix 2, Fig S1). We calculated receptor phosphorylation for a variety of parameter values that shift the monomer response to left or right (Fig. 2b), and found that they similarly affect the dimer, so the set of curves shown in Fig 3b shifts left or right along the abscissa (Fig. S1). Therefore, the effect of trans-phosphorylation in the RR*-E complex (Fig. 3b) is similar over a wide range of starting concentrations and other constants, such as Q (the fraction of E in the inactive conformation). A surprising result is that even if all the SFK is in the active conformation (Q = 0, e.g., in cells expressing a mutationally-activated allele, or in cells where the C-terminal tyrosine of the SFK is not phosphorylated), dimerization of receptors can still cause a considerable increase in phosphorylation (Fig. S1). The effects of feedback on receptor monomers and dimers are not identical however, so changes in K3, Rt, Et or Q do have small effects on the fold increase in phosphorylation induced by receptor dimerization.
3. Effect of SFK trans-phosphorylation
We next analyzed the effect of SFK phosphorylation in its activation loop. The open, low activity form of SFK (E) can be activated ϕ-fold by intermolecular phosphorylation (14, 34–36), with bimolecular rate constant q1 (Fig. 4a). E bound to various forms of R* can also be phosphorylated by E. All forms of E*, whether free or bound, are dephosphorylated with pseudo-first order rate constant q2. All other binding and phosphorylation reactions are as before.
FIGURE 4. Trans-phosphorylation of SFK.
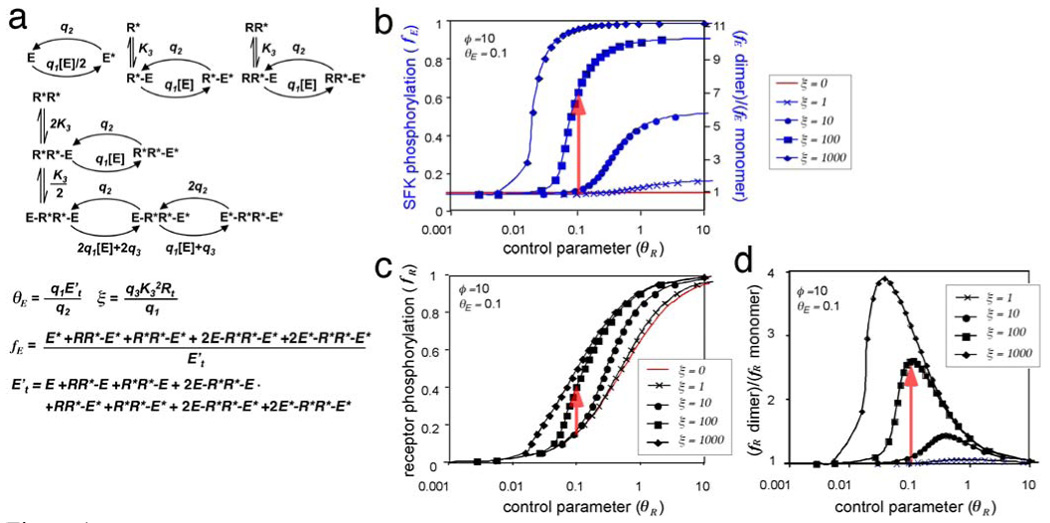
(a) Reactions and equations describing the intermolecular phosphorylation of E. Phosphorylation of E, alone or complexed with various forms of R*, occurs with bi-molecular rate constant q1, and dephosphorylation occurs with pseudo-first order rate constant q2. q4 represents SFK trans-phosphorylation within a E-R*R*-E complex. These reactions are solved in Appendix 3.
(b) Fractional SFK phosphorylation (fE) as a function of the control parameter for receptor phosphorylation (θR), for particular values of the phosphorylation-induced increase in SFK activity (ϕ) and the control parameter for SFK phosphorylation (θE). The control (red) is for monomeric receptors. Blue lines show results for dimeric receptors with various values of the SFK trans-phosphorylation parameter (ξ). Dimerization does not change any parameter except σ, which increases fE (red arrow).
(c) Fractional receptor phosphorylation. Same parameters as in (b).
(d) Relative increase in receptor phosphorylation due to dimerization. These graphs were constructed by interpolation of the data in (c).
We introduce a control parameter for SFK phosphorylation (θE), which represents the balance between bi-molecular phosphorylation of E and its dephosphorylation by PTP. Like θR, θE is the same for monomer and dimer receptors. It sets the tone for the system, and can vary in different cell types. For receptor monomers, where E influences R phosphorylation but not vice versa, the fraction of E that is phosphorylated, fE, depends only on θE. The relationship between θE and fE is the same as between θR and fR (eg. see red line in Fig. 2b). Note that the only role of Q, the equilibrium between inactive and active conformations, is to change θE (Appendix 3, Eqns. 1 and Eqn. 4). This means that the following argument applies equally to cells in which the SFK is or is not phosphorylated at its C terminal tyrosine residue.
With receptor dimers, doubly-phosphorylated R*R* can act as a scaffold for forming E-R*R*-E complexes, and phosphorylation and activation of E in these complexes becomes uni-molecular (rate constant q3), and is greatly increased. This SFK trans-phosphorylation effect is represented by ξ. When receptors are monomers, there is no trans-phosphorylation, ξ = 0, and fE depends on θE but not θR (Fig. 4b, red line). However, with receptor dimers, ξ is a positive number, and fE now depends on θE, ξ, and on the concentration of R*R*. This latter depends on fR, which in turn depends on θR, θE, ξ and ϕ (E* activity relative to E). This circular relationship between phosphorylation of E (fE) and R (fR) represents positive feedback, in which increased R phosphorylation leads to increased R*R*, increased scaffolding effect, increased E* and increased E activity to phosphorylate R.
The fraction of E phosphorylation (fE) is graphed relative to θR for given θE and ϕ and various ξ in Fig. 4b (blue lines). Receptor dimerization at given θR increases E phosphorylation vertically from the horizontal red line to the appropriate blue line, dependent on ξ(red arrow). The fraction of phosphorylated receptors, fR, also increases, moving vertically on Fig 4c from the red line to the appropriate black line depending on the value of ξ for the dimer (red arrow). In the example shown, with ξ = 100, ϕ = 10, θE = 0.1 and θR = 0.1, we find that fE increases ~7-fold and fR increases ~2.7-fold (Fig. 4b and d, squares). Depending on starting conditions, the increases in SFK and receptor phosphorylation due to SFK trans-phosphorylation can be quite large (Fig. S2). Note that we ignored the receptor trans-phosphorylation effect (Fig. 3) in these calculations. Receptor trans-phosphorylation would further increase fR, which would further increase fE, which would further increase fR.
4. Values for the parameters from experimental measurements
Values for receptor trans-phosphorylation (σ), SFK trans-phosphorylation (ξ), SFK activation by phosphorylation (ϕ) and total cellular concentrations of SFK (Et) and receptor (Rt) were obtained from the literature (Table 2, available in Supporting Information). Each of these values may range over several orders of magnitude: σ from 20 to 5000, ξ from 2 to 800, and ϕ from 4 to 20. As explained above, the parameter Q, the relative amount of SFK in the inactive conformation, has approximately the same effect on phosphorylation of receptor monomers as it does on receptor dimers. To estimate the control parameters for receptor and SFK phosphorylation, θR and θE, we made use of the observation that low levels of phosphorylated receptors and SFKs are detectable in unstimulated cells (38–40), suggesting that these quantities are in the range of ~0.05–0.1. The preceding values were used for Fig. 2–Fig. 4. The results show that receptor and SFK trans-phosphorylation effects both contribute to ligand-induced phosphorylation of SDRs, and if both effects occur simultaneously, with the expected mutual reinforcement, then even greater increases are expected. In addition, further increases are predicted if receptors cluster into higher-order oligomers.
DISCUSSION
We analyzed a model for signal transduction by SFK-dependent receptors (Fig 1c). We found that dimerization significantly stimulates receptor and SFK phosphorylation without requiring any other regulatory events. Importantly, the model does not require regulation of SFK activity either by C-terminal phosphorylation/dephosphorylation or by stabilization of the active conformation of the SFK when bound to phosphorylated receptor. The results are surprising because there are many more steps than for the RTK model (Fig. 1b), and many of the steps are inefficient. For example, only a subset of SFK molecules are in a conformation that can bind receptors; phosphorylation of SFKs in the activation loop causes only a modest activation; the scaffolding effects require ternary and quaternary protein complexes; and the cellular concentrations of phosphorylated receptors and SFKs are low relative to their mutual binding affinity. Nevertheless, receptor dimerization will strongly increase SDR phosphorylation and SFK activation, provided that the values for receptor and SFK concentrations from cells and kinetic constants from in vitro assays and certain other assumptions (see below) are accurate. The ability of the model to predict effects that are consistent with biological measurements, without recourse to detailed computational modeling, suggests that the minimal system may underlie biological reality.
We had initially expected that the greatest extent of dimerization-induced stimulation would be two-fold, with one bound SFK phosphorylating two receptor molecules instead of one. However, much greater than two-fold increases may result, depending on cellular conditions (Fig. 3c, 4b, 4d). The "something for nothing" effect does not come at no expense, but is driven by "futile" cycles of phosphorylation-dephosphorylation. Another result of the analysis is that the signaling is stimulated in direct proportion to the fraction of receptors that are dimerized. There is no element of a "switch like" (cooperative or ultra-sensitive) response. However, the same applies to RTKs, where binding of a ligand to induce dimerization of a pair of RTKs activates only that one pair. Despite this initially linear response, positive and negative feedback events downstream of the initial receptor activation can allow a switch-like cellular outcome. In the case of multichain immune recognition receptors (MIRRs), this is called kinetic proofreading or serial engagement (41).
The validity of the results relies on the validity of the underlying assumptions. To our knowledge, the assumptions are reasonable and supported by the literature. The first is that molecular flexibility allows an SFK bound to one receptor in a dimer to phosphorylate its partner. It is known that an SFK bound to one phosphotyrosine can phosphorylate other tyrosine residues in the same substrate (42, 43), and others have proposed trans-phosphorylation of clustered SDRs by associated SFKs (32, 33). Activated SFKs have few inter-domain contacts (12, 44), and molecular dynamics simulations predict flexibility (45, 46). We also suspect that SDRs are flexible, based on their lack of predicted secondary structure elements and the experimental finding that TCR proximity but not orientation is critical for activation (47). Thus it seems likely that receptor trans-phosphorylation can occur.
We also assume that SFK phosphorylation in the activation loop is intermolecular and can occur on or off membranes. This is supported by experiment (14, 34–36). Phosphorylation in solution is inefficient because the SFK concentration is too low (35, 36). However, SFKs are more likely to trans-phosphorylate when bound to dimeric receptors. Indeed, SFKs that are associated with active SDRs have more phosphotyrosine than unbound SFKs (48).
The model does not require regulation of PTPs. They are passive players that keep the phosphorylation of monomeric receptors and SFKs low and reset the system when ligand is removed. They are also important because they cannot dephosphorylate sites to which other proteins are bound (30), allowing a positive feedback effect on phosphorylated receptors. There is abundant evidence that the balance of PTP to SFK is critical for MIRR signaling (49). Although we do not require the PTP to be regulated, if it is, e.g. by displacement from dimerized SDRs, by reactive oxygen produced by active SFKs, or by active recruitment to phosphorylated SDRs (24, 26, 50), then receptor activation may be further increased.
SUMMARY AND CONCLUSIONS
We have found that dimerization can induce increasing receptor phosphorylation by SFK, and SFK activation, by mass action kinetics alone. The increasing phosphorylation upon dimerization is due to a positive feedback between receptor phosphorylation, SFK binding, and SFK activation by phosphorylation in the kinase domain. The same principles can also explain cooperative phosphorylation of proteins with multiple phosphorylation sites. They also suggest that other SH2 domain-containing tyrosine kinases may be similarly regulated, since the SFK SH3 domain and C-terminal phosphate are not involved. However, we wish to emphasize that we do not exclude roles for many other mechanisms in the system, such as changes in PTP activity, the lipid environment or kinases or PTPs that act on SFKs, but the results suggest that such changes are not necessary and may be evolutionary fine-tuning of a basically simple system.
Supplementary Material
SUPPORTING INFORMATION AVAILABLE Four files of supporting information are available free of charge via the Internet at http://pubs.acs.org. One pdf file contains the following information: Table 2 (Cellular concentrations and reaction rates for SFKs and SDRs, with literature citations), Appendix 1 (Effect of positive and negative feedback on phosphorylation of monomeric receptor), Appendix 2 (Trans-phosphorylation of receptor dimers, allowing for feedback), Appendix 3 (The effect of SFK trans-phosphorylation, independent of receptor trans-phosphorylation) and Figures S1 and S2. In addition, three Excel spreadsheets contain formulae for plotting Figure 2–Figure 4 with different input values.
ACKNOWLEDGEMENTS
We thank J. Brugge, T. Hunter, T. Miller, A. Shaw, A. Veillette, E. Warren and A. Weiss for pointing us towards relevant literature. We are grateful to T. Hunter, A. Shaw, J. Schlessinger, W.y. Shou and A. Weiss for thoughtful suggestions on various drafts of the manuscript.
This work was supported in part by Public Health Service grants CA41072 and GM068610.
Abbreviations
- FcεRI
immunoglobulin Fc receptor type εRI
- PTP
protein tyrosine phosphatase
- SDR
SFK-dependent receptor
- SFK
Src family kinase
- SH2
Src homology 2
- SH3
Src homology 3
- RTK
receptor tyrosine kinase
- TCR
T cell receptor
REFERENCES
- 1.Tamir I, Cambier JC. Antigen receptor signaling: integration of protein tyrosine kinase functions. Oncogene. 1998;17:1353–1364. doi: 10.1038/sj.onc.1202187. [DOI] [PubMed] [Google Scholar]
- 2.Sigalov A. Multi-chain immune recognition receptors: spatial organization and signal transduction. Semin. Immunol. 2005;17:51–64. doi: 10.1016/j.smim.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 3.Germain RN. T-cell signaling: the importance of receptor clustering. Curr. Biol. 1997;7:R640–R644. doi: 10.1016/s0960-9822(06)00323-x. [DOI] [PubMed] [Google Scholar]
- 4.Veillette A, Latour S, Davidson D. Negative regulation of immunoreceptor signaling. Annu. Rev. Immuno. 2002;20:669–707. doi: 10.1146/annurev.immunol.20.081501.130710. [DOI] [PubMed] [Google Scholar]
- 5.Fuller GL, Williams JA, Tomlinson MG, Eble JA, Hanna SL, Pohlmann S, Suzuki-Inoue K, Ozaki Y, Watson SP, Pearce AC. The C-type lectin receptors CLEC-2 and Dectin-1, but not DC-SIGN, signal via a novel YXXL-dependent signaling cascade. J. Biol. Chem. 2007;282:12397–12409. doi: 10.1074/jbc.M609558200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palmer A, Zimmer M, Erdmann KS, Eulenburg V, Porthin A, Heumann R, Deutsch U, Klein R. EphrinB phosphorylation and reverse signaling: regulation by Src kinases and PTP-BL phosphatase. Mol Cell. 2002;9:725–737. doi: 10.1016/s1097-2765(02)00488-4. [DOI] [PubMed] [Google Scholar]
- 7.Lahdenpera J, Kilpelainen P, Liu XL, Pikkarainen T, Reponen P, Ruotsalainen V, Tryggvason K. Clustering-induced tyrosine phosphorylation of nephrin by Src family kinases. Kidney Int. 2003;64:404–413. doi: 10.1046/j.1523-1755.2003.00097.x. [DOI] [PubMed] [Google Scholar]
- 8.Benes CH, Wu N, Elia AE, Dharia T, Cantley LC, Soltoff SP. The C2 domain of PKCdelta is a phosphotyrosine binding domain. Cell. 2005;121:271–280. doi: 10.1016/j.cell.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 9.Jaramillo ML, Afar DE, Almazan G, Bell JC. Identification of tyrosine 620 as the major phosphorylation site of myelin-associated glycoprotein and its implication in interacting with signaling molecules. J. Biol. Chem. 1994;269:27240–27245. [PubMed] [Google Scholar]
- 10.Tissir F, Goffinet AM. Reelin and brain development. Nat Rev Neurosci. 2003;4:496–505. doi: 10.1038/nrn1113. [DOI] [PubMed] [Google Scholar]
- 11.Herz J, Chen Y. Reelin, lipoprotein receptors and synaptic plasticity. Nat Rev Neurosci. 2006;7:850–859. doi: 10.1038/nrn2009. [DOI] [PubMed] [Google Scholar]
- 12.Cowan-Jacob SW, Fendrich G, Manley PW, Jahnke W, Fabbro D, Liebetanz J, Meyer T. The crystal structure of a c-Src complex in an active conformation suggests possible steps in c-Src activation. Structure. 2005;13:861–871. doi: 10.1016/j.str.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 13.Martin GS. The hunting of the Src. Nat Rev Mol Cell Biol. 2001;2:467–475. doi: 10.1038/35073094. [DOI] [PubMed] [Google Scholar]
- 14.Boggon TJ, Eck MJ. Structure and regulation of Src family kinases. Oncogene. 2004;23:7918–7927. doi: 10.1038/sj.onc.1208081. [DOI] [PubMed] [Google Scholar]
- 15.Veillette A, Bookman MA, Horak EM, Bolen JB. The CD4 and CD8 T cell surface antigens are associated with the internal membrane protein-tyrosine kinase p56lck. Cell. 1988;55:301–308. doi: 10.1016/0092-8674(88)90053-0. [DOI] [PubMed] [Google Scholar]
- 16.Shaw AS, Amrein KE, Hammond C, Stern DF, Sefton BM, Rose JK. The Lck tyrosine protein kinase interacts with the cytoplasmic tail of the CD4 glycoprotein through its unique amino-terminal domain. Cell. 1989;59:627–636. doi: 10.1016/0092-8674(89)90008-1. [DOI] [PubMed] [Google Scholar]
- 17.Veillette A, Bookman MA, Horak EM, Samelson LE, Bolen JB. Signal transduction through the CD4 receptor involves the activation of the internal membrane tyrosine-protein kinase p56lck. Nature. 1989;338:257–259. doi: 10.1038/338257a0. [DOI] [PubMed] [Google Scholar]
- 18.Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61:203–212. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]
- 19.Schlessinger J. Signal transduction. Autoinhibition control. Science. 2003;300:750–752. doi: 10.1126/science.1082024. [DOI] [PubMed] [Google Scholar]
- 20.Cochran JR, Aivazian D, Cameron TO, Stern LJ. Receptor clustering and transmembrane signaling in T cells. Trends Biochem. Sci. 2001;26:304–310. doi: 10.1016/s0968-0004(01)01815-1. [DOI] [PubMed] [Google Scholar]
- 21.Razzaq TM, Ozegbe P, Jury EC, Sembi P, Blackwell NM, Kabouridis PS. Regulation of T-cell receptor signalling by membrane microdomains. Immunology. 2004;113:413–426. doi: 10.1111/j.1365-2567.2004.01998.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montixi C, Langlet C, Bernard AM, Thimonier J, Dubois C, Wurbel MA, Chauvin JP, Pierres M, He HT. Engagement of T cell receptor triggers its recruitment to low-density detergent-insoluble membrane domains. EMBO J. 1998;17:5334–5348. doi: 10.1093/emboj/17.18.5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xavier R, Brennan T, Li Q, McCormack C, Seed B. Membrane compartmentation is required for efficient T cell activation. Immunity. 1998;8:723–732. doi: 10.1016/s1074-7613(00)80577-4. [DOI] [PubMed] [Google Scholar]
- 24.Shaw AS, Dustin ML. Making the T cell receptor go the distance: a topological view of T cell activation. Immunity. 1997;6:361–369. doi: 10.1016/s1074-7613(00)80279-4. [DOI] [PubMed] [Google Scholar]
- 25.Horejsi V. Transmembrane adaptor proteins in membrane microdomains: important regulators of immunoreceptor signaling. Immunol. Lett. 2004;92:43–49. doi: 10.1016/j.imlet.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 26.Rodgers W, Rose JK. Exclusion of CD45 inhibits activity of p56lck associated with lycolipid-enriched membrane domains. J. Cell Biol. 1996;135:1515–1523. doi: 10.1083/jcb.135.6.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bunnell SC, Singer AL, Hong DI, Jacque BH, Jordan MS, Seminario MC, Barr VA, Koretzky GA, Samelson LE. Persistence of cooperatively stabilized signaling clusters drives T-cell activation. Mol. Cell. Biol. 2006;26:7155–7166. doi: 10.1128/MCB.00507-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bunnell SC, Hong DI, Kardon JR, Yamazaki T, McGlade CJ, Barr VA, Samelson LE. T cell receptor ligation induces the formation of dynamically regulated signaling assemblies. J. Cell Biol. 2002;158:1263–1275. doi: 10.1083/jcb.200203043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strasser V, Fasching D, Hauser C, Mayer H, Bock HH, Hiesberger T, Herz J, Weeber EJ, Sweatt JD, Pramatarova A, Howell B, Schneider WJ, Nimpf J. Receptor clustering is involved in Reelin signaling. Mol. Cell. Biol. 2004;24:1378–1386. doi: 10.1128/MCB.24.3.1378-1386.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rotin D, Margolis B, Mohammadi M, Daly RJ, Daum G, Li N, Fischer EH, Burgess WH, Ullrich A, Schlessinger J. SH2 domains prevent tyrosine dephosphorylation of the EGF receptor: identification of Tyr992 as the high-affinity binding site for SH2 domains of phospholipase C gamma. EMBO J. 1992;11:559–567. doi: 10.1002/j.1460-2075.1992.tb05087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Felder S, Zhou M, Hu P, Urena J, Ullrich A, Chaudhuri M, White M, Shoelson SE, Schlessinger J. SH2 domains exhibit high-affinity binding to tyrosine-phosphorylated peptides yet also exhibit rapid dissociation and exchange. Mol. Cell. Biol. 1993;13:1449–1455. doi: 10.1128/mcb.13.3.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wofsy C, Vonakis BM, Metzger H, Goldstein B. One lyn molecule is sufficient to initiate phosphorylation of aggregated high-affinity IgE receptors. Proc. Natl. Acad. Sci. USA. 1999;96:8615–8620. doi: 10.1073/pnas.96.15.8615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pribluda VS, Pribluda C, Metzger H. Transphosphorylation as the mechanism by which the high-affinity receptor for IgE is phosphorylated upon aggregation. Proc. Natl. Acad. Sci. USA. 1994;91:11246–11250. doi: 10.1073/pnas.91.23.11246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cooper JA, MacAuley A. Potential positive and negative autoregulation of p60c-src by intermolecular autophosphorylation. Proc. Natl. Acad. Sci. USA. 1988;85:4232–4236. doi: 10.1073/pnas.85.12.4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barker SC, Kassel DB, Weigl D, Huang X, Luther MA, Knight WB. Characterization of pp60c-src tyrosine kinase activities using a continuous assay: autoactivation of the enzyme is an intermolecular autophosphorylation process. Biochemistry. 1995;34:14843–14851. doi: 10.1021/bi00045a027. [DOI] [PubMed] [Google Scholar]
- 36.Moarefi I, LaFevre-Bernt M, Sicheri F, Huse M, Lee CH, Kuriyan J, Miller WT. Activation of the Src-family tyrosine kinase Hck by SH3 domain displacement. Nature. 1997;385:650–653. doi: 10.1038/385650a0. [DOI] [PubMed] [Google Scholar]
- 37.Wofsy C, Torigoe C, Kent UM, Metzger H, Goldstein B. Exploiting the difference between intrinsic and extrinsic kinases: implications for regulation of signaling by immunoreceptors. J. Immunol. 1997;159:5984–5992. [PubMed] [Google Scholar]
- 38.Iba H, Cross FR, Garber EA, Hanafusa H. Low level of cellular protein phosphorylation by nontransforming overproduced pp60c-src. Mol. Cell. Biol. 1985;5:1058–1066. doi: 10.1128/mcb.5.5.1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burkhardt AL, Stealey B, Rowley RB, Mahajan S, Prendergast M, Fargnoli J, Bolen JB. Temporal regulation of non-transmembrane protein tyrosine kinase enzyme activity following T cell antigen receptor engagement. J. Biol. Chem. 1994;269:23642–23647. [PubMed] [Google Scholar]
- 40.Saouaf SJ, Mahajan S, Rowley RB, Kut SA, Fargnoli J, Burkhardt AL, Tsukada S, Witte ON, Bolen JB. Temporal differences in the activation of three classes of non-transmembrane protein tyrosine kinases following B-cell antigen receptor surface engagement. Proc. Natl. Acad. Sci. USA. 1994;91:9524–9528. doi: 10.1073/pnas.91.20.9524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goldstein B, Faeder JR, Hlavacek WS. Mathematical and computational models of immune-receptor signalling. Nat Rev Immunol. 2004;4:445–456. doi: 10.1038/nri1374. [DOI] [PubMed] [Google Scholar]
- 42.Mayer BJ, Hirai H, Sakai R. Evidence that SH2 domains promote processive phosphorylation by protein-tyrosine kinases. Curr. Biol. 1995;5:296–305. doi: 10.1016/s0960-9822(95)00060-1. [DOI] [PubMed] [Google Scholar]
- 43.Pellicena P, Miller WT. Processive phosphorylation of p130Cas by Src depends on SH3-polyproline interactions. J. Biol. Chem. 2001;276:28190–28196. doi: 10.1074/jbc.M100055200. [DOI] [PubMed] [Google Scholar]
- 44.Hochrein JM, Lerner EC, Schiavone AP, Smithgall TE, Engen JR. An examination of dynamics crosstalk between SH2 and SH3 domains by hydrogen/deuterium exchange and mass spectrometry. Protein Sci. 2006;15:65–73. doi: 10.1110/ps.051782206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karplus M, Kuriyan J. Molecular dynamics and protein function. Proc. Natl. Acad. Sci. USA. 2005;102:6679–6685. doi: 10.1073/pnas.0408930102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Young MA, Gonfloni S, Superti-Furga G, Roux B, Kuriyan J. Dynamic coupling between the SH2 and SH3 domains of c-Src and Hck underlies their inactivation by C-terminal tyrosine phosphorylation. Cell. 2001;105:115–126. doi: 10.1016/s0092-8674(01)00301-4. [DOI] [PubMed] [Google Scholar]
- 47.Cochran JR, Cameron TO, Stone JD, Lubetsky JB, Stern LJ. Receptor proximity, not intermolecular orientation, is critical for triggering T-cell activation. J. Biol. Chem. 2001;276:28068–28074. doi: 10.1074/jbc.M103280200. [DOI] [PubMed] [Google Scholar]
- 48.Yamashita T, Mao SY, Metzger H. Aggregation of the high-affinity IgE receptor and enhanced activity of p53/56lyn protein-tyrosine kinase. Proc. Natl. Acad. Sci. USA. 1994;91:11251–11255. doi: 10.1073/pnas.91.23.11251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hermiston ML, Xu Z, Majeti R, Weiss A. Reciprocal regulation of lymphocyte activation by tyrosine kinases and phosphatases. J. Clin. Invest. 2002;109:9–14. doi: 10.1172/JCI14794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tonks NK. Redox redux: revisiting PTPs and the control of cell signaling. Cell. 2005;121:667–670. doi: 10.1016/j.cell.2005.05.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPORTING INFORMATION AVAILABLE Four files of supporting information are available free of charge via the Internet at http://pubs.acs.org. One pdf file contains the following information: Table 2 (Cellular concentrations and reaction rates for SFKs and SDRs, with literature citations), Appendix 1 (Effect of positive and negative feedback on phosphorylation of monomeric receptor), Appendix 2 (Trans-phosphorylation of receptor dimers, allowing for feedback), Appendix 3 (The effect of SFK trans-phosphorylation, independent of receptor trans-phosphorylation) and Figures S1 and S2. In addition, three Excel spreadsheets contain formulae for plotting Figure 2–Figure 4 with different input values.