

Abstract
Histone modifications modulate chromatin structure and function. A posttranslational modification-randomized, combinatorial library based on the first twenty-one residues of histone H4 was designed for systematic examination of proteins that interpret a histone code. The 800-member library represented all permutations of most known modifications within the N-terminal tail of histone H4. To determine its utility in a protein-binding assay, the on-bead library was screened with an antibody directed against phosphoserine 1 of H4. Among the hits, 59/60 sequences were phosphorylated at S1, while 30/30 of those selected from the non-hits were unphosphorylated. A 512-member version of the library was then used to determine the binding specificity of the double tudor domain of hJMJD2A, a histone demethylase involved in transcriptional repression. Global linear least squares fitting of modifications from the identified peptides (40 hits and 34 non-hits) indicated that methylation of K20 was the primary determinant for binding, but that phosphorylation/acetylation on neighboring sites attenuated the interaction. To validate the on-bead screen, isothermal titration calorimetry was performed with thirteen H4 peptides. Dissociation constants ranged from 1 mM - 1μM and corroborated the screening results. The general approach should be useful for probing the specificity of any histone-binding protein.
Keywords: histone code, chromatin, JMJD2A, tudor domain, posttranslational modifications, one-bead, one-compound, combinatorial peptide library
Histone proteins package DNA into chromatin and regulate the accessibility of DNA in processes such as transcription, repair and replication (1). Control of chromatin structure and function is mediated by reversible posttranslational modifications (PTMs) of histones. The most prevalent histone modifications occur on the unstructured N-terminal “tails” and include acetylation, methylation and phosphorylation, but others such as citrullination, ubiquitylation, sumoylation, ADP-ribosylation and biotinylation have been described (1). Mounting evidence suggests that particular modification states modulate histone-histone, histone-DNA and histone-non-histone protein interactions.
Recently, many specialized protein domains (histone-binding modules) have been identified and shown to display binding preferences for a particular modified amino acid side-chain. Examples of such modules include bromodomains, members of the Royal superfamily (e.g. chromodomains, tudor domains, MBT domains), PHD fingers and 14-3-3 proteins, which recognize acetylated lysine, methylated lysine and/or arginine, methylated lysine and phosphorylated serine and/or threonine, respectively (2). Given the complexity of possible modifications and the existence of modular protein domains that recognize modified forms of amino acid side-chains, a “histone code” has been postulated. One histone code hypothesis states that the combinatorial modification pattern on a histone can result in a particular biological response (3). These biological outcomes are achieved by the recruitment of protein complexes, which initiate specific downstream events. An alternative view is a signaling network model of chromatin, which asserts that multiple modifications combine to confer switch-like behavior characterized by bistability, robustness and adaptability (4).
Regardless of model, the problem reduces to a molecular understanding of how these protein modules bind and recognize modified histones with the appropriate binding affinity to initiate a response. Here, we will refer to the histone code as the histone modification state that is read and interpreted, in terms of binding selectivity and affinity, by histone binding proteins/modifying enzymes. In a recent example of a protein reading the histone code, the double tudor domain (DTD) of the histone demethylase transcriptional repressor, jumonji domain containing 2A (JMJD2A), was shown to preferentially bind to di- and trimethylated versions of H4K20 or H3K4 through a cation-π interaction (5-7). Targets identified for JMJD2A demethylase activity include trimethylated H3-K9/K36 (8, 9). Various lines of evidence suggest that JMJD2A functions as a transcriptional repressor (10, 11). In one study, JMJD2A was found to utilize the nuclear receptor corepressor (N-CoR) complex to trigger gene-specific repression, and importantly, this activity was dependent on the double tudor domain of JMJD2A (10). In another study, JMJD2A mediated repression of E2F-regulated promoters by recruitment of histone deacetylases (HDACs) and the retinoblastoma gene (Rb), implicating a role in cell proliferation and oncogenesis (11). It should be noted that the function of JMJD2A may be context-specific as it has also been implicated as a transcriptional co-activation of androgen receptor genes (12). Given the modification-specific manner in which JMJD2A operates, the double tudor domain is believed to play an important role in targeting. However, how combinatorial histone modification patterns affect the binding preferences of the DTD remains unexplored.
Although much work has been devoted to antibody (13, 14) - and mass spectrometry (15, 16) - based indexing of histone modifications, relatively few efforts have sought to understand the consequences of the enormous combinatorial complexity achieved through multiple histone modifications (5, 17). Several isolated cases have illustrated how the interplay of histone modifications can regulate biological outcomes (18). However, no study has addressed the problem of systematically surveying how histone modification patterns influence proteins that recognize covalent modifications on histones. This unmet need is underscored by the presence of potentially hundreds of histone-binding modules (2) and an ever-increasing number of documented histone modifications (1). A recent study identifying 74 unique histone H4 modification patterns in differentiating embryonic stems cells highlights the diversity of modifications that occur in vivo in a single cell type (19). Dissecting the interactions between histone-binding modules and modified histones is critical toward understanding the lexicon of the histone code. Unbiased exploration of the histone modification patterns read by histone-binding modules will require tools capable of representing the combinatorial complexity of modified histones.
In this study, we have developed a method for addressing the histone code using a one-bead, one-compound (OBOC) (20) combinatorial library based on an N-terminal histone sequence. The design, synthesis and utilization of a library possessing all possible permutations of most modifications at known PTM sites within the 21 N-terminal amino acids of histone H4 (Fig. 1) is described. PTMs included were phosphorylation, acetylation, citrullination, and all possible methylation states at lysines (mono-, di-, tri-) and arginines (mono-, symmetric di-, asymmetric di-) known to be methylated (1). Citrulline (U), a product of arginine deiminase, was included at position 3 due to recent findings linking this modification to gene regulation (21). The construction and characterization of an 800-member library was followed by an initial validation screen of a modification-specific antibody against histone H4. A 512-member version of the library was used to elucidate the binding preferences of the double tudor domain of the human demethylase JMJD2A (hJMJD2A). The resulting data indicated that the hJMJD2A DTD “reads” the methylation status of K20 and that modifications at neighboring sites influence overall binding affinity. The resulting combinatorial modification patterns suggest a rheostat-like mechanism of binding, where binding affinity can exist anywhere along a continuum between 1 μM and 1 mM and is defined by the coexistence of multiple PTMs. The general approach should be applicable to interrogating the specificity of histone-binding proteins/enzymes.
Figure 1.

The H4 histone tail library corresponds to the first 21 amino acids of human histone H4 and is attached to a linker composed of two β-alanines (B) and a methionine. Citrulline is demarcated by U and N-termini are acetylated (Ac). Sites of variation are above/below X’s.
Experimental Procedures
General Methods
All chemical and biochemical reagent were purchased from commercial suppliers. The α-phos (S1) H4 antibody serum was a gift from the laboratory of Prof. David Allis. The antibody was raised against the H4 sequence: (Sph)GRGKGGKG(14). A plasmid for the double tudor domain of JMJD2A was obtained from the laboratory of Prof. Rui-Ming Xu (purification protocol furnished by Dr. Ying Huang). Peptides were synthesized on a Symphony synthesizer from Protein Technologies (Tucson, AZ). Analytical gradient HPLC was performed on a Shimadzu series 2010C HPLC with a Vydac C18 column (10 μm, 4.6 × 250 mm). Mass spectrometry was executed on an Applied Biosystems 4800 instrument. Statistical analysis was performed with MathWorks software (Natick, MA) and R (22).
Library Construction
The combinatorial histone H4 peptide library was constructed on TentaGel Macrobead NH2 resin (280- 320 μm, 0.21 mmol/g loading, 65,550 beads/g) using the split-pool approach (20) for sites of variability. Sites of variability include positions 20 (K, Kac, Kme, Kme2, Kme3), 16 (K, Kac), 12 (K, Kac), 8 (K, Kac), 5 (K, Kac), 3 (R, Rme1, Rme2s, Rme2a, citrulline) and 1 (S, Sph). All amino acids (at least 5 equivalents/ coupling) were double coupled for two hours with standard Fmoc/tBu chemistry (23). Prior to the partially randomized histone H4 sequence, a three amino acid linker, BBM (B = β-alanine) was synthesized. N-termini of all peptides were acetylated with acetic anhdyride. A 50 mg (13.5 μmol) portion of the library was deprotected for five hours with Reagent K (TFA/EDT/thioanisole/water/phenol: 82.5%, 2.5%, 5%, 5%, 5%) (24) prior to use. Synthesis of the reduced loading capacity library was performed as above except that it was on resin that had been reacted with 0.9 equivalents of N-acetylimidzole (final loading capacity = 0.02 mmol/g). In addition, asymmetric dimethyl arginine at position 3 and acetyl lysine at position 20 were not included.
On-bead Library Screen and of α-phos (S1) H4 Screen
Prescreen
Fifty milligrams (13.5 μmol) of the peptide library was added to a 4 mL filter column and washed thoroughly with DCM, MeOH, ddH2O and PBST buffer (25 mM NaPi pH 7.2, 150 mM NaCl, 0.1 % Tween 20). The resin was swelled for one hour with gentle rocking prior to drainage and one hour of blocking with 3 % BSA in PBST. After draining the blocking solution to the resin bed, 1 mL of 50 nM biotinylated goat-anti-rabbit antibody in PBST containing 3 % BSA was added. Following one hour of rocking, the solution was drained to the resin bed and washed 3 × 1 mL PBST. The resin was then rocked with 1 mL of a 25 nM solution of Q-dot 605 streptavidin conjugate in PBST for two hours. Following drainage to the resin bed, the resin was washed 10 × 2 mL PBST. At this point, the resin was resuspended in PBST and viewed under a fluorescent microscope and any fluorescent beads could be removed from the library (none were observed).
Screen
After prescreening the library for nonspecific interactions with the secondary antibody or the quantum dots, a screen was performed. The only difference from the prescreen was a one hour incubation with 1 mL of a 100:1 dilution of α-phos (S1) H4 in PBST with 3 % BSA after the swell step and washing 3 × 1 mL PBST prior to addition of the secondary antibody. When viewed under the microscope, a number of fluorescent and non-fluorescent beads were manually selected.
hJMJD2A DTD Purification
(Protocol and plasmid courtesy of the laboratory of Dr. Rui-Ming Xu). Glutathine-S-Transferase (GST) tagged hJMJD2A (895-1011) was provided on an AMP resistant, pGEXKG construct with a PreScission Protease (GE Healthcare, U.K.) cut site (LFQ/GP). Transformed BL21(DE3) cells were grown to an OD of 0.6 at 37° C prior to addition of IPTG a final concentration of 0.4 mM. The temperature was lowered to 18° C and the cells were induced overnight. The cell pellet was thawed on ice in buffer S (20 mM Tris, pH 8.0, 500 mM NaCl, 0.1% βME) and sonicated. The cell lysate was centrifuged, the supernatant was collected and run on a glutathione sepharose column (GE Healthcare, U.K.). After eluting with 10 mM reduced glutathione, the GST tag was removed with PreScission protease during dialysis into Buffer B (20 mM Tris, pH 8.0, 100 mM NaCl, 0.1% βME). Finally, the protein was passed through another glutathione sepharose column followed by an anion exchange (Q) column (GE Healthcare, U.K.). Protein concentration was determined by absorbance at 280 nm (ε = 13.610 mM-1cm-1) and purity was assessed by SDS-PAGE.
Biotinylation of hJMJD2A DTD
hJMJD2A DTD was chemically biotinylated with the EZ-Link NHS-Chromogenic Biotin Reagent (Pierce, Rockford, Il). Biotinylation reactions were optimized to yield ∼1 biotin/ molecule of hJMJD2A. Biotin incorporation was determined by measurement at 354 nm. To ensure that hJMJD2A was functional after biotinylation, isothermal titration calorimetry (ITC) was performed with the biotinylated version and an unbiotinylated version of hJMJD2A using a test peptide trimethylated at K20. Typical ITC experiments are described below.
On-bead Library hJMJD2A Double Tudor Domain (DTD) BCIP Screen
Thirty-eight milligrams (10.3 μmol) of the peptide library was added to a 1.5 mL filter column and washed thoroughly with DCM, MeOH, ddH2O and HBST buffer (30 mM Hepes pH 7.5, 150 mM NaCl, 0.1 % Tween 20). The resin was swelled for one hour with HBST and incubated for an additional hour with 0.1 % BSA in HBST. After draining the blocking solution to the resin bed, 800 μL of 50 nM biotinylated hJMJD2A (or in the case of the increased hJMJD2A screen, 2 μM) in HBST containing 0.1 % BSA was added and the mixture was allowed to rock gently for two hours. A procedure for bead color development was adapted from a recent study by Sweeney et al. (25). Following 20 minutes of incubation with BCIP (bromo-4-chloro-indolyl phosphate) and streptavidin-conjugated alkaline phosphatase, beads were thoroughly washed with ddH2O and sorted by color intensity into dark blue or colorless categories as judged by eye.
Peptide Sequencing with MALDI-TOF/TOF MS
Beads that were selected under the microscope were incubated with 200 μL of 8 M guanidinium hydrochloride prior to washing 3 × 500 μL ddH2O and drying. Peptides were cyanogen bromide cleaved from each bead and desalted before sequencing with MALDI-TOF/TOF MS.
Statistical Analysis of Data
Global linear least squares fitting was performed by assigning a numerical value to each type of modification. All unmodified/methylated lysines, unmodified/methylated arginines and serines were given a value of 0, while acetylated lysines, citrullines and phosphorylated serines were given a value of 1 for the α-phos (S1) H4 antibody fit. The numerical assignments of the modifications were chosen to differentiate amino acids on the basis of charge (positive vs. neutral/negative). Identical assignments were used in the hJMJD2A DTD fit except at K20, unmodified lysine and monomethylated lysine were given values of 0, while di- and trimethylated lysine were given values of 1. Because all of the modification states at K20 in the hJMJD2A DTD screen were positively charged, we chose the aforementioned designations to reflect the distinct grouping of tri- and dimethyllysine in the hit pool and mono- and unmethylated lysine in the non-hit pool. These designations simply served to account for empirical observations in our fitting protocol. In the linear fit, the values were fitted to the equation Yi = α1Xi1 + α2Xi2 + ... + αkXik. Here Yi =1 indicates that the i-th sample belongs to the hit pool, Yi =0 indicates that the i-th sample belongs to the non-hit pool, and Xik has the value 0 or 1 given by the k-th modification of the i-th sample, as described above.
Standard multiple linear regression was performed to estimate the values of the parameters α1, α2, ..., αk, as well as the statistical significance of these values, based on computing their t-statistics and their corresponding p-values. Correlation matrices for the hJMJD2A DTD were constructed by first calculating the correlation coefficients for each pool of peptides (using the same numbering scheme as for the linear least squares fit). Colors were applied to the correlation coefficients based on their sign and magnitude, using the MATLAB default colormap jet.
Synthesis of Peptides for ITC Studies
All peptides used in the ITC studies were based on the 21 N-terminal amino acids of H4 (no linker). Each peptide was synthesized on a 25 μmol scale on amide resin using standard Fmoc/tBu chemistry. N-termini were acetylated with acetic anhydride and peptides were deprotected with cocktail B (92.5 % TFA; 5 % thioanisole; 2.5 % ethanedithiol) and triturated in diethyl ether. All peptides were HPLC purified (average purity of 91 %) over a C18 column and were characterized by MALDI-TOF MS prior to ITC experiments.
Isothermal Titration Calorimetry
ITC experiments were recorded at 25° C with a VP-ITC titration calorimeter (MicroCal, Northhampton, MA). Protein concentrations ranged from 18-40 μM and peptide concentrations ranged from 250-500 μM. Peptide concentrations were determined by the masses of their trifluoroacetic acid salts and were normalized to each other by absorbance at 214 nm with RP-HPLC. All experiments were performed in HBS (30 mM sodium phosphate pH 7.5, 150 mM NaCl). In a typical experiment, 40 injections of peptide (1 × 1 μL, 6 × 4 μL and 33 × 8 μL) were delivered at 120-second intervals to a 1.4 mL solution of protein. The initial data point was routinely discarded. Data was fitted by Lavenberg-Marquardt nonlinear regression with Origin 7.0 software using the one-site model. On average, n values were 0.91 ± 0.25.
Results
Library design and validation
A combinatorial library based on PTMs of the first twenty-one amino acids of histone H4 was constructed with a split-pool synthetic strategy (20). In this library, posttranslational randomization occurred at positions 1 (S, Sph), 3 (R, Rme, Rme2s, Rme2a, citrulline), 5 (K, Kac), 8 (K, Kac), 12 (K, Kac), 16 (K, Kac) and 20 (K, Kac, Kme1, Kme2, Kme3), while all other amino acids were held constant (Fig. 1). Therefore, the library was composed of 800 distinct species. Sites chosen for PTM-randomization were based on previous observations of positions that are known to be modified in vivo (1). Furthermore, all possible methylation states were included at sites known to be methylated (positions 3 and 20). While it is not clear whether both symmetric and asymmetric dimethylation of R3 occur in vivo, both modifications have been shown to occur in vitro upon action of PRMT5 and PRMT1, respectively (26). Therefore we chose to include both types of modification in the library. In rat liver, phosphorylation of H4R18 has been observed (27), but was not included in the library because this acid labile-modification was not expected to survive the acidic deprotection conditions during the construction of the library. In addition we chose not to include H4 sumoylation or biotinylation to simplify peptide synthesis and to avoid interference with the on-bead assay (which requires a biotin-steptavidin interaction), respectively. A three amino acid linker of two β-alanines and methionine was included C-terminal of the H4 sequence to afford flexibility and a peptide cleavage site, respectively. To our knowledge, the resulting library of twenty-four total amino acids is the longest OBOC peptide library reported and is the first to be based exclusively on randomization of diverse posttranslational modifications.
Evaluation of the integrity of the library was performed on randomly selected beads using mass spectrometry and RP-HPLC analysis. A tandem mass spectral (MS/MS) strategy was adopted for peptide sequencing. Isomeric dimethyl arginines were differentiated based on neutral losses of 31 and 70 for the symmetric and 45 for the asymmetric versions, respectively (28). Acetylation and trimethylation at position 20 were not distinguished. To ensure the presence of expected library members, cyanogen bromide-cleaved peptides from ten randomly selected beads were sequenced by MALDI-TOF/TOF MS (Supplementary Table 1, Supplementary Fig. 1). RP-HPLC of the cleavage products from ten additional randomly selected beads revealed peptides of high purity (typically 80-90%, Supplementary Fig. 2). Thus, analysis of the cleaved peptides indicated that the library was composed of the expected products, in high purity and amenable to MS/MS sequencing.
To examine the utility of the on-bead peptide library for protein binding assays, the library was screened with a modification-specific antibody, α-phos (S1) H4, which recognizes the phosphorylated form of S1 of histone H4 (14). In the screening experiment, detection of bound antibody was achieved with a biotinylated goat-anti-rabbit secondary antibody and streptavidin-coated quantum dots. To account for non-specific binding of the secondary antibody or quantum dots, the library was prescreened using these reagents but without addition of the primary antibody. No detectable fluorescence was observed in this control experiment. Following the α-phos (S1) H4 antibody screen, beads from the library were examined under a fluorescent microscope (Supplementary Fig. 3). Approximately half the beads exhibited variable but detectable levels of quantum dot-associated fluorescence. Ninety individual beads were manually selected and classified as either fluorescent or non-fluorescent at 605 nm. Peptides from individual beads were cleaved with cyanogen bromide and sequenced by tandem mass spectrometry. A summary of individual sequences can be found in Supplementary Table 2. Importantly, 98 % (59/60) of the sequences from the fluorescent pool were phosphorylated at S1, while none of peptides from the non-fluorescent pool (30/30) bore this modification. Consequently, using our screening method, we observed no “false negatives” and were able to unequivocally identify the site of interaction between H4 and the antibody. These results suggested that the H4 library was well suited for an on-bead protein binding assay and that the conditions might be adapted to facilitate screening experiments with physiological histone binding proteins.
Library screen with hJMJD2A double tudor domain
Using the H4 tail library, we next sought to characterize the binding specificity of a physiological histone-binding protein in the context of an ensemble of histone modifications. The double tudor domain (DTD) of the histone demethylase, hJMJD2A, was selected as a target due to recent findings that it binds methylated versions of K4 of histone H3, and K20 of histone H4 (5-7) and is involved in JMJD2A-mediated transcriptional repression (10, 11). We also reasoned that the hJMJD2A DTD would provide a stringent test of the utility of the H4 library, because unlike the α-phos (S1) H4 antibody, the hJMJD2A DTD was expected to bind near the C-terminus (i.e., in close proximity to the bead) and with ∼10,000 fold-lower affinity. After an extensive survey of screening conditions to minimize false positives, to increase sensitivity, to optimize selection and to reduce cost, a number of alterations were made to the library and the screening methods. In the revised assay format, we used a version of the library at 10 % of the original loading capacity (final loading capacity = 0.02 mmol/g; see Supplementary Note 1). In this second-generation library, asymmetric dimethyl arginine at position 3 and acetylated lysine at position 20 were not included in order to expedite MS-based peptide sequencing. Sequencing was simplified due to the fact that it was no longer necessary to differentiate asymmetric dimethyl arginine from symmetrically dimethylated arginine and acetylated lysine from trimethylated lysine. It should be noted that acetylation at K20 has not been observed in humans in vivo (29). With the reduced loading capacity, an enzyme-linked colorimetric assay (25) was adopted to improve sensitivity and minimize cost. This assay relies on the turnover of bromo-4-chloro-indolyl phosphate (BCIP) by streptavidin-linked alkaline phosphatase, which deposits a blue precipitate onto beads bearing peptides that bind to the target protein (Fig. 2). In this case, the protein was directly biotinylated to render the assay readily adaptable to any histone-binding protein without the need for a protein-specific antibody. Protein biotinylation conditions were optimized to afford a 1:1 stoichiometry of hJMJD2A DTD to biotin. Binding of hJMJD2A DTD to trimethylated K20 was not compromised by biotinylation (data not shown).
Figure 2.
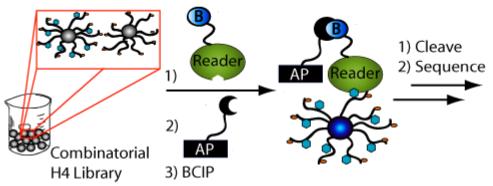
Generalized on-bead assay scheme. To determine histone code reader (i.e. histone binding protein) specificity, the biotinylated protein is incubated with the library prior to addition of streptavidin-conjugated alkaline phosphatase, which recruits and turns over bromo-4-chloro-indolyl phosphate (BCIP) in the vicinity of beads bearing interacting peptide sequences. Turnover of BCIP leaves a blue precipitate on these beads.
To establish the binding preferences of the hJMJD2A DTD in the presence of hundreds of possible histone H4 modification patterns, we performed H4 library screening experiments at 50 nM and 2 μM hJMJD2A. At the more stringent concentrations (50 nM), the screen would facilitate selection of the highest affinity peptides (intensely blue beads), whereas screening for the colorless beads at 2 μM screen should yield peptides that exhibit the weakest binding. In this way, we were able to interrogate the entire binding continuum of the hJMJD2A DTD. Varying shades of blue were observed on beads in the 50 nM biotinylated hJMJD2A DTD screening experiment (Supplementary Fig. 4). Sequencing of the cleavage products from thirty intensely blue beads revealed, as expected, a distinct preference for multiple methylation at K20 (Fig. 3a and Supplementary Table 3). Eighty-three percent (25/30) of the peptides identified in this screen were di- or trimethylated at K20. Of the thirty sequences, we noted an average of 1.7 acetyl groups per peptide, with half of the peptides being acetylated at K5. Approximately half of the sequences were phosphorylated at S1 and 73 % (22/30) were citrullinated or monomethylated at R3. In stark contrast, the thirty-four colorless beads selected in the 2 μM screen bore peptides that were never trimethylated and were un- or monomethylated at K20 with an 85 % (29/34) frequency (Fig. 3b and Supplementary Table 3). Of these sequences, we observed an average of 2.9 acetyl groups per peptide and in this case, K8 and K12 were each acetylated 76 % (26/34) of the time. Besides the increase in acetylation, 85 % (29/34) of the peptides were phosphorylated at S1. At R3, 76 % (26/34) of these sequences were citrullinated or symmetrically dimethylated. Furthermore, all dimethylated peptides from the colorless pool (5/34) were phosphorylated and at least triacetylated. Consistent with these observations, ten additional blue beads from the 2 μM screen were analyzed, and importantly, all ten peptides were trimethylated at K20 (Supplementary Table 3).
Figure 3.
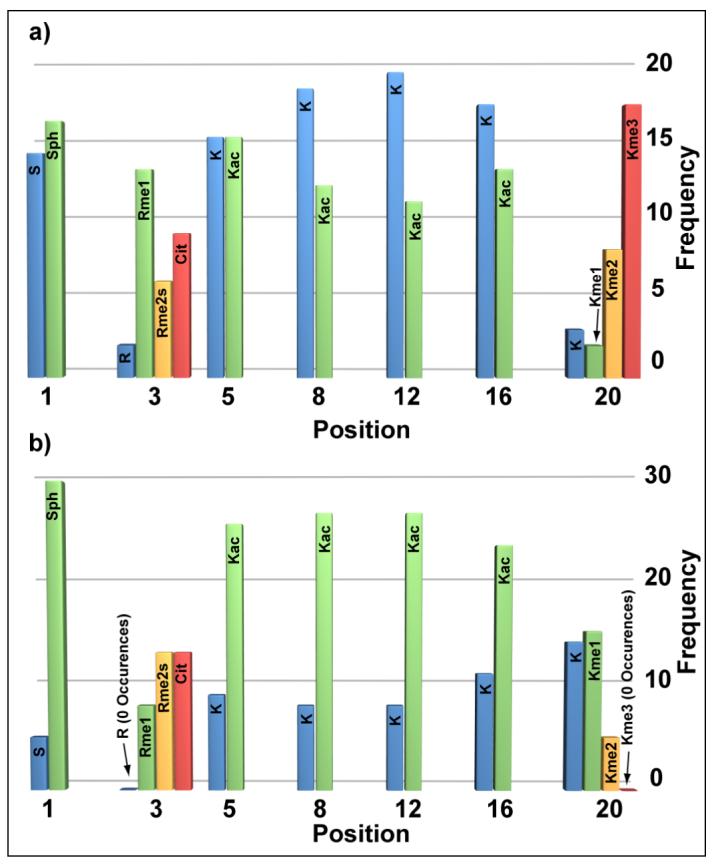
Frequency of PTMs observed from the (a) intensely blue and (b) colorless populations of beads at amino acid positions 1, 3, 5, 8, 12, 16 and 20 when the combinatorial H4 tail library was screened with hJMJD2A DTD at 50 nM and 2 μM concentrations, respectively.
Statistical analysis of hJMJD2A screening results
To address the statistical significance of the observed binding trends for hJMJD2A DTD, global linear least squares fitting was performed (Supplementary Table 4) (30). In the global fit (n = 64), we compared sequences from the intensely blue beads in the 50 nM screen to those from the colorless beads in the 2 μM screen. A value of 0 was assigned to all unmodified lysines, unmodified/methylated arginines and serines and a value of 1 was assigned to acetylated lysines, citrullines and phosphorylated serines. These assignments were meant to differentiate the amino acids based on charge. A value of 0 was assigned to un- and monomethylated lysine at position 20, while a value of 1 was assigned to di- and trimethylated lysine at this position. These designations were chosen to reflect the observed grouping of di- and trimethylation at K20 among the sequences from the blue beads and un- and monomethylation at K20 among the colorless beads. The global linear least squares fitting results indicated that the modification state at K20 (p-value = 1.2×10-7) had an enormous influence on binding to the hJMJD2A DTD, while modification at K12 (p-value = 0.0078), K16 (p-value = 0.047) and S1 (p-value = 0.0058) had more subtle effects. Modification at position 3 did not have an appreciable effect on binding (p-value = 0.41; Supplementary Note 2). Importantly, the coefficients obtained from the fit were negative in all cases except for that corresponding to the K20 term. This result suggested that modification of K20 (i.e.-methylation) had a positive influence on binding to the hJMJD2A DTD while modification at all other positions (i.e.- phosphorylation, citrullination, acetylation), had a negative influence. The observation that modification at sites other than K20 influence binding is supported by the fact that five K20 dimethylated peptides identified in the colorless pool (Supplementary Table 3) were phosphorylated and tri-or tetraacetylated acetylated.
Next, correlation matrices were used to examine the combinatorial interplay between modifications at multiple sites. We will refer to correlation matrices showing the cross-correlation between modifications as determined by a histone-binding protein as “histone code fingerprints” (HCFs). In this format, the color in each square of the matrix signifies the strength and direction of the relationship between modifications at any two positions, as determined by the correlation coefficients (30). In other words, the shade of the color represents the degree to which modifications at the intersecting positions correlate (occur together) or anti-correlate (do not occur together). These matrices are especially useful for understanding the contributions to binding afforded by modifications at positions other than the primary binding site. In order to visualize the relationships among modifications from the hJMJD2A DTD screening experiments, correlation coefficients for positions 1, 3, 5, 8, 12 and 16 were calculated using the same number assignment scheme as in the global linear least squares fit and displayed as correlation matrices (Fig. 4). Correlations occur in cases where modifications occur together (e.g.-K, K or Kac, Kac) while anti-correlations occur in instances of inverse modification (e.g.-K, Kac or Kac, K). In the HCF for sequences from intensely blue beads in the 50 nM screen, the degree of correlation or anti-correlation is rather weak (Fig. 5a). Because S1 and S1ph occur at approximately equal frequencies, the negative correlations observed at row or column 1 can be attributed to inverse relationships to either of these modifications. These inverse relationships occur when serine is found with citrulline/acetylated lysine or when phosphoserine is found with lysine/(methylated)arginine. The positive correlation between positions 12 and 16 is due to the fact that K, K (12/30) and Kac, Kac (6/30) combine to occur at a 60 % frequency. The HCF resulting from sequences from colorless beads in the 2 μM hJMJD2A DTD screening experiment shows significantly stronger relationships among modifications. In this case, since phosphorylation at S1 occurs at an 85 % (29/34) frequency, the positive correlation observed along row 1 and column 1 is most reflective of co-occurrence of citrullination at R3 and acetylation at K5, K8, K12 and K16. Indeed, when S1 is phosphorylated, K5, K8, K12 and K16 are acetylated at 79 %, (23/29) 79 % (23/29), 76 % (22/29) and 72 % (21/29) levels, respectively. The positive correlation between R3 and K16 is primarily attributed to the fact that 85 % (11/13) of the time when R3 is citrullinated, acetylation occurs at K16. Finally, positive correlation between positions 8 and 12 can be rationalized by the fact that when K8 is acetylated, K12 is acetylated at an 85 % (22/26) frequency. Taken together, these observations suggest that “modification crosstalk” at positions other than the primary site of binding can contribute to the interaction of the histone H4 tail with the hJMJD2A DTD. More specifically the results suggest that the net phosphorylation/acetylation/citrullination state of an H4 tail has a negative effect on binding to the hJMJD2A DTD.
Figure 4.
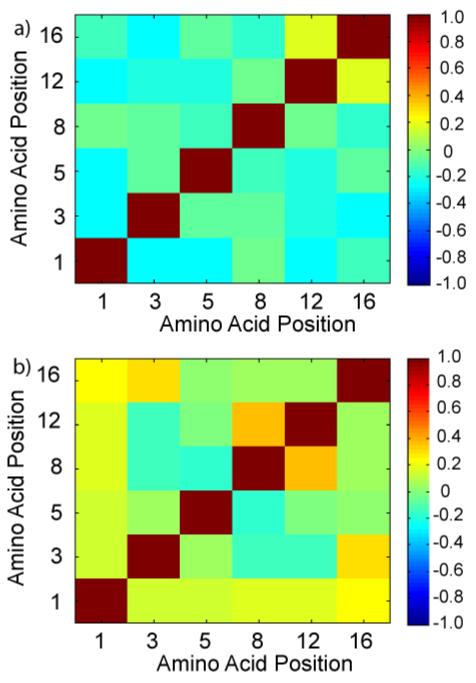
Histone code fingerprints (HCFs) depicting the combinatorial interplay of PTMs (at sites other than K20) observed for binding of the hJMJD2A DTD to histone H4 tails. The HCF’s are correlation matrices of the PTMs observed from a) sequences from intensely blue beads in the 50 nM screen b) sequences from colorless beads in the 2 μM screen. Numerical values for each square were determined by calculating the correlation coefficients for matrices consisting of each grouping of peptides (numerical assignments for each modification can be found in the main text). The color of each square signifies the strength and direction of the relationship between any two amino acid positions. Positive values on the colorbar represent correlations while negative values represent anti-correlations.
Figure 5.

Sample plot of ITC of JMJD2A double tudor domain with 8. Raw titration data and integrated heats are shown in the top and bottom panels respectively. Different volumes were injected during the course of the experiment (1 × 1 μL, 6 × 4 μL and 33 × 8 μL injection). Dissociation constant (Kd = 3.5 ± 0.1 μM) and stoichiometry of binding (n = 0.9) were determined by Lavenberg-Marquardt nonlinear regression.
Validation of hJMJD2A DTD screening results with isothermal titration calorimetry
To validate the binding trends obtained from the histone H4 library screens and statistical analysis, individual binding constants were determined for several N-terminal histone H4 peptides with the hJMJD2A DTD. Specifically, five peptides corresponding to hit sequences (from intensely blue beads), four peptides corresponding to non-hits (from colorless beads) and four additional peptides to examine secondary-site trends were synthesized and binding constants for the hJMJD2A DTD were obtained with ITC (Table 1). Hit peptides bound to the hJMJD2A DTD with dissociation constant (Kd) values ranging from ∼1-8 μM, while non-hit sequences bound with Kd values spanning from 25->500 μM. Hit sequence 4, unmethylated at K20, was selected for off-bead validation because we suspected this was a “false hit”. Indeed, ITC yielded a Kd value (∼190 μM) that was well in the affinity range of non-hits. The occasional incidence of false positive hits likely arises from the presence of damaged beads, which often stain more darkly than undamaged beads, and from a small fraction of unfolded protein that can interact non-specifically with the beads.
Table 1.
Dissociation constants with standard deviations for selected peptides (residues 1-21) from the intensely blue pool (50 nM screen)a, the colorless pool (2 μM screen)b and for comparisonc as determined by ITC. Values are the averages of two or more separate assays, except for those annotated with ‘*’, in which case the error associated with the fit is reported. Dissociation constants for peptides marked with ‘x’ are approximations with relatively large errors associated with these low-affinity interactions
| Peptide | PTM Position | Kd (μM) | ||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 3 | 5 | 8 | 12 | 16 | 20 | ||
| 1a | S | U | Kac | K | K | K | Kme3 | 2.1 ± 0.2 |
| 2a* | Sph | R | Kac | K | K | K | Kme3 | 1.4 ± 0.1 |
| 3a | S | MeR | Kac | K | Kac | Kac | Kme2 | 7.5 ± 1.1 |
| 4a*x | S | MeR | K | Kac | Kac | K | K | 190 ± 32 |
| 5b* | Sph | Rme2s | Kac | Kac | Kac | Kac | Kme1 | 74.4 ± 31.5 |
| 6bx | Sph | Rme2s | Kac | Kac | Kac | K | K | >500 |
| 7b | Sph | U | K | Kac | Kac | Kac | K | N.D. |
| 8c | S | Rme2s | K | K | K | K | Kme2 | 3.8 ± 0.4 |
| 9a | Sph | Rme2s | K | K | K | K | Kme2 | 5.8 ± 1.5 |
| 10c | S | Rme2s | K | Kac | Kac | K | Kme2 | 6.7 ± 0.1 |
| 11c | S | Rme2s | Kac | K | K | Kac | Kme2 | 8.8 ± 0.8 |
| 12c | S | Rme2s | Kac | Kac | Kac | Kac | Kme2 | 11.6 ± 1.4 |
| 13b | Sph | Rme2s | Kac | Kac | Kac | Kac | Kme2 | 25.2 ± 3.1 |
The hJMJD2A DTD screening results and statistical analysis suggested that modifications at sites other than K20 could influence binding. In particular, the net acetylation level and phosphorylation status of the H4 peptides had the most dramatic effect on binding (Fig. 4). With the series of peptides 8-13, we were able to rigorously examine how these “secondary-site” modifications influence hJMJD2A DTD binding to a K20 dimethylated histone peptide (Table 1, Fig. 5). As predicted from the screening analysis, ITC revealed that the net reduction of positive charge (i.e. phosphorylation, acetylation) attenuated binding of H4 peptides to the DTD of hJMJD2A. While hit sequence 9 bound with a low micromolar Kd value, tetraacetylated peptide 13 (appeared twice in the non-hit pool) exhibited a four-fold decrease in affinity relative to 9. Diacetylation (10,11) resulted in a ∼2 fold decrease in binding affinity compared to the unacetylated counterpart (8). Interestingly phosphorylation of S1 did not significantly alter the binding properties of peptide 8 relative to peptide 9, but did result in a ∼2-fold decrease in binding from 12 to 13. As a whole, these results suggest that the location of secondary-site modifications plays less of a role in altering binding properties than the net level of modification, which corresponds to an overall reduction of positive charge. These observations are in agreement with the global linear least squares analysis and HCFs as well as the fact that all dimethylated peptides in the colorless pool were phosphoryated and tri- or tetraacetylated.
Discussion
Combinatorial histone tail libraries offer a unique strategy for characterizing the interactions between histone code readers and an ensemble of histone modification states. We have demonstrated the feasibility of producing high quality, combinatorial histone H4 tail libraries and developed a robust screening platform. The system performed well in control studies with a modification-specific antibody and in characterizing the binding preferences of a physiological histone-binding domain capable of discriminating among methylation states at lysine. Furthermore, we found that the hJMJD2A DTD “reads” the modification state not only at K20 but that PTMs at other positions influence affinity and contribute to a rheostat-like mechanism of binding in a charge-dependent manner. These results are supported by a recent crystal structure of an H4K20me3 peptide in complex with the double tudor domain, which suggests that the peptide binds in an extended conformation along an extremely acidic patch of the hybrid tudor 2 portion of the protein (7). Although the recognition surface for the hJMJD2A DTD is small (1039 Å2 buried surface area in the case of an H4K4me3 cocrystal (1)), we propose that remote H4 modifications have a significant influence on the overall interaction. ITC results suggest that depending on the modification state of the histone H4 tail, the hJMJD2A DTD binding affinity can fluctuate from 1 μM to 1 mM. The reported library and screening methodology should be suitable for determining the modification preferences of any N-terminal histone H4-binding protein.
From the standpoint of library design, several points merit consideration. To our knowledge, the combinatorial histone H4 tail 24-mer library is both the longest OBOC-library and the first to be comprised of diverse randomized PTMs (nine distinct species). Accordingly, the library was designed to resolve the nuanced interplay among PTMs in the context of binding any N-terminal histone H4-binding protein. This is in stark contrast to most OBOC studies, which typically seek to identify high-affinity ligands among highly diverse peptides (31) or small molecules (32). Importantly, unlike oriented peptide libraries (33), the OBOC format permits the isolation of individual peptide sequences (as opposed to “consensus sequences”) and therefore provides crucial information with regard to the context of each PTM—a critical consideration when examining histone modification patterns. A unique feature of the on-bead assay, relative to in-solution assays, is the multivalent nature of peptides displayed on the bead. Interestingly, internucleosomal tail-tail interactions are thought to make a significant contribution to chromatin fiber dynamics (34) and histone H4 in condensed chromatin exists at a concentration of ∼4 mM (35, 36). On the reduced loading capacity beads used for the hJMJD2A screens, histone H4 peptides were displayed at a concentration of ∼2 mM. Thus, to a first approximation, the on-bead format mimics condensed chromatin with respect to histone H4 tail density, a potential advantage of the assay.
As mentioned previously, both fluorescent and colorimetric screening approaches were employed in this study. The fluorescent method is attractive because it lends itself to automated bead sorting and quantitative analysis (37). However, due to the superior sensitivity of enzymatic signal amplification, we found the colorimetric version to be more useful when utilizing a reduced loading capacity library. In addition, the enzyme-linked version is tunable in that it allows control of the amount of color development on individual beads. The colorimetric screen is also less expensive and accessible to laboratories that lack a fluorescent microscope or bead sorter.
The screening results suggest a potential epigenetic mechanism for regulation of JMJD2A-mediated repression. Previous reports have suggested that monomethylation of K20 is associated with hyperacetylation of histone H4 (38) and transcriptional activation, while trimethylation of K20 is associated with H4 hypoacetylation and transcriptional repression (39, 40). Dimethylation of H4K20, on the other hand, has been observed on un- to tetraacetylated H4 in HeLa cells (41). While the methylation state of H4K20 is clearly the primary factor in the interaction with the hJMJD2A DTD, our results suggest that the H4 acetylation status appears to fine-tune the affinity (Fig. 6a). For example, in the absence of other modifications, < 1.5 increase in affinity has been noted for binding of the hJMJD2A DTD to a H4K20 trimethylated peptide relative to a dimethylated version (5). However, for a peptide (13) that is dimethylated at K20 and tetraacetylated we observe an 18-fold loss in affinity for the hJMJD2A DTD relative to a peptide (2) that is trimethylated at K20 and monoacetylated. We propose a tentative model in which hJMJD2A preferentially binds to trimethylated but unacetylated H4 via the DTD and recruits HDACs for intra- or internucleosomal deacetylation and subsequent transcriptional repression. Because K20 monomethylation is associated with hyperacetylation, only weak binding of the hJMJD2A DTD is expected (see Table 1, Peptide 5), which supports the observation that this pattern is observed with transcriptionally active chromatin (38) (Fig. 6b). This hypothesis is consistent with recent evidence that demonstrates a critical role for the DTD and HDAC recruitment in JMJD2A-mediated transcriptional repression of the ASCL2 gene (10) and the E2F transcription factor-regulated promoters (11). Interestingly, besides recruitment of HDACs, JMJD2A-mediated transcriptional repression has also been linked to demethylation of H3K9/H3K36 (8, 9). While the DTD is not necessary for enzymatic activity (8), it may serve to interpret a preexisting histone code (e.g.- hypoacetylated and multimethylated K20 on H4) and in doing so, exert spatiotemporal control over JMJD2A demethylase activity. As was noted earlier, JMJD2A has also been proposed to co-activate androgen receptor regulated genes (12). It is possible that the DTD facilitates this outcome by binding to trimethylated H3K4, which is enriched at the promoters of actively expressed genes (1). However, this remains to be confirmed experimentally.
Figure 6.
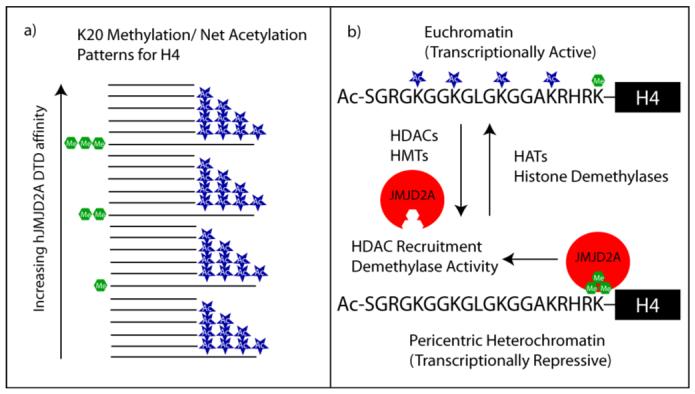
a) Representation of hJMJD2A DTD binding affinity with horizontal lines demarcating various modification states (binding differences not shown to scale). The hJMJD2A DTD: H4 interaction is primarily controlled by the methylation state of K20, but alterations in the net acetylation state result in incremental changes in affinity. b) Working model for JMJD2A-mediated transcriptional repression. Histone H4 monomethylation at K20 and hyperacetylation correlates with transcriptionally-poised chromatin while trimethylation of K20 and hypoacetylation tracks with transcriptional repression. The histone modification state is controlled by the dynamic action of HATs, HMTs (histone methyltransferases), HDACs and histone demethylases. The hJMJD2A DTD preferentially binds to transcriptionally-repressive H4 where it can recruit HDACs and potentially localize demethylase activity.
As demonstrated in the studies presented, OBOC histone tail libraries have great potential for exploring the combinatorial crosstalk involved in the histone code. By coupling screening results to rigorous statistical analysis and ITC validation, we have provided a useful framework for dissecting the interactions that contribute to a protein’s histone-code reading capacity. With this general strategy, future efforts will involve the generation of PTM libraries based on other histone sequences and the determination of binding specificity for potentially hundreds of modification-specific binding modules. Histone-binding proteins that interact with sites (e.g. -H3K4, H3K9) in close proximity to other modifiable amino acids and protein complexes containing multiple histone-binding modules will be especially interesting targets. We anticipate that each histone code reader will have unique histone code fingerprints (HCFs), which will serve to elucidate its code reading capacity and binding preferences. Moreover, these OBOC histone tail libraries should be useful in probing the specificity of histone-modifying enzymes.
Supplementary Material
Acknowledgement
We thank Dr. Greg Barrett-Wilt for assistance with MS/MS, Dr. Rui-Ming Xu and Dr. Ying Huang for the hJMJD2A plasmid and purification protocol, Dr. C. David Allis for α-phos (S1) H4, and Dr. Joshua Coon for review of the manuscript.
This work was supported by NIH Grant GM059785
The abbreviations used are
- PTM
posttranslational modification
- hJMJD2A DTD
human jumonji domain containing 2A
- PHD
plant homeodomain
- OBOC
one-bead, one-compound
- HDAC
histone deacetylase
- HAT
histone acetyltransferase
- N-CoR
nuclear receptor corepressor
- pRb
product of retinoblastoma gene
- MALDI-TOF MS
matrix-assisted laser desorption/ionization time of flight mass spectrometry
- HPLC
high performance liquid chromatography
- ITC
isothermal titration calorimetry
- Q-dot
quantum dot
- BCIP
bromo-4-chloro-indolyl phosphate
- BSA
bovine serum albumin
- PBST
phosphate buffered saline with tween
- HBST
hepes buffered saline with tween
- HCF
histone code fingerprint.
References
- 1.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 2.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 4.Schreiber SL, Bernstein BE. Signaling network model of chromatin. Cell. 2002;111:771–778. doi: 10.1016/s0092-8674(02)01196-0. [DOI] [PubMed] [Google Scholar]
- 5.Kim J, Daniel J, Espejo A, Lake A, Krishna M, Xia L, Zhang Y, Bedford MT. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep. 2006;7:397–403. doi: 10.1038/sj.embor.7400625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang Y, Fang J, Bedford MT, Zhang Y, Xu RM. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science. 2006;312:748–751. doi: 10.1126/science.1125162. [DOI] [PubMed] [Google Scholar]
- 7.Lee J, Thompson JR, Botuyan MV, Mer G. Distinct binding modes specify the recognition of methylated histones H3K4 and H4K20 by JMJD2A-tudor. Nat Struct Mol Biol. 2007 doi: 10.1038/nsmb1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klose RJ, Yamane K, Bae Y, Zhang D, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature. 2006;442:312–316. doi: 10.1038/nature04853. [DOI] [PubMed] [Google Scholar]
- 9.Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, Shi Y. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006;125:467–481. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 10.Zhang D, Yoon HG, Wong J. JMJD2A is a novel N-CoR-interacting protein and is involved in repression of the human transcription factor achaete scute-like homologue 2 (ASCL2/Hash2) Mol Cell Biol. 2005;25:6404–6414. doi: 10.1128/MCB.25.15.6404-6414.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gray SG, Iglesias AH, Lizcano F, Villanueva R, Camelo S, Jingu H, Teh BT, Koibuchi N, Chin WW, Kokkotou E, Dangond F. Functional characterization of JMJD2A, a histone deacetylase-and retinoblastoma-binding protein. J Biol Chem. 2005;280:28507–28518. doi: 10.1074/jbc.M413687200. [DOI] [PubMed] [Google Scholar]
- 12.Shin S, Janknecht R. Activation of androgen receptor by histone demethylases JMJD2A and JMJD2D. Biochem Biophys Res Commun. 2007;359:742–746. doi: 10.1016/j.bbrc.2007.05.179. [DOI] [PubMed] [Google Scholar]
- 13.Perez-Burgos L, Peters AH, Opravil S, Kauer M, Mechtler K, Jenuwein T. Generation and characterization of methyl-lysine histone antibodies. Methods Enzymol. 2004;376:234–254. doi: 10.1016/S0076-6879(03)76016-9. [DOI] [PubMed] [Google Scholar]
- 14.Barber CM, Turner FB, Wang Y, Hagstrom K, Taverna SD, Mollah S, Ueberheide B, Meyer BJ, Hunt DF, Cheung P, Allis CD. The enhancement of histone H4 and H2A serine 1 phosphorylation during mitosis and S-phase is evolutionarily conserved. Chromosoma. 2004;112:360–371. doi: 10.1007/s00412-004-0281-9. [DOI] [PubMed] [Google Scholar]
- 15.Coon JJ, Ueberheide B, Syka JE, Dryhurst DD, Ausio J, Shabanowitz J, Hunt DF. Protein identification using sequential ion/ion reactions and tandem mass spectrometry. Proc Natl Acad Sci U S A. 2005;102:9463–9468. doi: 10.1073/pnas.0503189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pesavento JJ, Mizzen CA, Kelleher NL. Quantitative analysis of modified proteins and their positional isomers by tandem mass spectrometry: human histone H4. Anal Chem. 2006;78:4271–4280. doi: 10.1021/ac0600050. [DOI] [PubMed] [Google Scholar]
- 17.Vermeulen M, Stunnenberg HG. An in vitro assay to study the recruitment and substrate specificity of chromatin modifying enzymes. Biol Proced Online. 2004;6:157–162. doi: 10.1251/bpo85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Latham JA, Dent SY. Cross-regulation of histone modifications. Nat Struct Mol Biol. 2007;14:1017–1024. doi: 10.1038/nsmb1307. [DOI] [PubMed] [Google Scholar]
- 19.Phanstiel D, Brumbaugh J, Berggren WT, Conard K, Feng X, Levenstein ME, McAlister GC, Thomson JA, Coon JJ. Mass spectrometry identifies and quantifies 74 unique histone H4 isoforms in differentiating human embryonic stem cells. Proc Natl Acad Sci U S A. 2008;105:4093–4098. doi: 10.1073/pnas.0710515105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lam KS, Salmon SE, Hersh EM, Hruby VJ, Kazmierski WM, Knapp RJ. A new type of synthetic peptide library for identifying ligand-binding activity. Nature. 1991;354:82–84. doi: 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]
- 21.Thompson PR, Fast W. Histone citrullination by protein arginine deiminase: is arginine methylation a green light or a roadblock? ACS Chem Biol. 2006;1:433–441. doi: 10.1021/cb6002306. [DOI] [PubMed] [Google Scholar]
- 22.Ihaka R, Gentleman R. R: A Language for Data Analysis and Graphics. Journal of Computational and Graphical Statistics. 1996;5:299–314. [Google Scholar]
- 23.Bodanszky M. Principles of Peptide Synthesis. 2nd ed. Springer-Verlag; Germany: 1993. [Google Scholar]
- 24.King DS, Fields CG, Fields GB. A cleavage method which minimizes side reactions following Fmoc solid phase peptide synthesis. Int J Pept Protein Res. 1990;36:255–266. doi: 10.1111/j.1399-3011.1990.tb00976.x. [DOI] [PubMed] [Google Scholar]
- 25.Sweeney MC, Wavreille AS, Park J, Butchar JP, Tridandapani S, Pei D. Decoding protein-protein interactions through combinatorial chemistry: sequence specificity of SHP-1, SHP-2, and SHIP SH2 domains. Biochemistry. 2005;44:14932–14947. doi: 10.1021/bi051408h. [DOI] [PubMed] [Google Scholar]
- 26.Wysocka J, Allis CD, Coonrod S. Histone arginine methylation and its dynamic regulation. Front Biosci. 2006;11:344–355. doi: 10.2741/1802. [DOI] [PubMed] [Google Scholar]
- 27.Chen CC, Smith DL, Bruegger BB, Halpern RM, Smith RA. Occurrence and distribution of acid-labile histone phosphates in regenerating rat liver. Biochemistry. 1974;13:3785–3789. doi: 10.1021/bi00715a026. [DOI] [PubMed] [Google Scholar]
- 28.Brame CJ, Moran MF, McBroom-Cerajewski LD. A mass spectrometry based method for distinguishing between symmetrically and asymmetrically dimethylated arginine residues. Rapid Commun Mass Spectrom. 2004;18:877–881. doi: 10.1002/rcm.1421. [DOI] [PubMed] [Google Scholar]
- 29.Garcia BA, Hake SB, Diaz RL, Kauer M, Morris SA, Recht J, Shabanowitz J, Mishra N, Strahl BD, Allis CD, Hunt DF. Organismal differences in post-translational modifications in histones H3 and H4. J Biol Chem. 2007;282:7641–7655. doi: 10.1074/jbc.M607900200. [DOI] [PubMed] [Google Scholar]
- 30.Casella G, Berger RL. Statistical Inference. Duxbury Press; 2001. [Google Scholar]
- 31.Lam KS, Lehman AL, Song A, Doan N, Enstrom AM, Maxwell J, Liu R. Synthesis and screening of “one-bead one-compound” combinatorial peptide libraries. Methods Enzymol. 2003;369:298–322. doi: 10.1016/S0076-6879(03)69017-8. [DOI] [PubMed] [Google Scholar]
- 32.Dixon S, Ziebart KT, He Z, Jeddeloh M, Yoo CL, Wang X, Lehman A, Lam KS, Toney MD, Kurth MJ. Aminodeoxychorismate synthase inhibitors from one-bead one-compound combinatorial libraries: “staged” inhibitor design. J Med Chem. 2006;49:7413–7426. doi: 10.1021/jm0609869. [DOI] [PubMed] [Google Scholar]
- 33.Songyang Z, Blechner S, Hoagland N, Hoekstra MF, Piwnica-Worms H, Cantley LC. Use of an oriented peptide library to determine the optimal substrates of protein kinases. Curr Biol. 1994;4:973–982. doi: 10.1016/s0960-9822(00)00221-9. [DOI] [PubMed] [Google Scholar]
- 34.Hansen JC. Conformational dynamics of the chromatin fiber in solution: determinants, mechanisms, and functions. Annu Rev Biophys Biomol Struct. 2002;31:361–392. doi: 10.1146/annurev.biophys.31.101101.140858. [DOI] [PubMed] [Google Scholar]
- 35.Bohrmann B, Haider M, Kellenberger E. Concentration evaluation of chromatin in unstained resin-embedded sections by means of low-dose ratio-contrast imaging in STEM. Ultramicroscopy. 1993;49:235–251. doi: 10.1016/0304-3991(93)90230-u. [DOI] [PubMed] [Google Scholar]
- 36.Leforestier A, Livolant F. Liquid crystalline ordering of nucleosome core particles under macromolecular crowding conditions: evidence for a discotic columnar hexagonal phase. Biophys J. 1997;73:1771–1776. doi: 10.1016/S0006-3495(97)78207-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garske AL, Denu JM. SIRT1 top 40 hits: use of one-bead, one-compound acetyl-peptide libraries and quantum dots to probe deacetylase specificity. Biochemistry. 2006;45:94–101. doi: 10.1021/bi052015l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Talasz H, Lindner HH, Sarg B, Helliger W. Histone H4-lysine 20 monomethylation is increased in promoter and coding regions of active genes and correlates with hyperacetylation. J Biol Chem. 2005;280:38814–38822. doi: 10.1074/jbc.M505563200. [DOI] [PubMed] [Google Scholar]
- 39.Sarg B, Helliger W, Talasz H, Koutzamani E, Lindner HH. Histone H4 hyperacetylation precludes histone H4 lysine 20 trimethylation. J Biol Chem. 2004;279:53458–53464. doi: 10.1074/jbc.M409099200. [DOI] [PubMed] [Google Scholar]
- 40.Nishioka K, Rice JC, Sarma K, Erdjument-Bromage H, Werner J, Wang Y, Chuikov S, Valenzuela P, Tempst P, Steward R, Lis JT, Allis CD, Reinberg D. PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol Cell. 2002;9:1201–1213. doi: 10.1016/s1097-2765(02)00548-8. [DOI] [PubMed] [Google Scholar]
- 41.Zhang K, Williams KE, Huang L, Yau P, Siino JS, Bradbury EM, Jones PR, Minch MJ, Burlingame AL. Histone acetylation and deacetylation: identification of acetylation and methylation sites of HeLa histone H4 by mass spectrometry. Mol Cell Proteomics. 2002;1:500–508. doi: 10.1074/mcp.m200031-mcp200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.