

Abstract
Experimental autoimmune encephalomyelitis (EAE) is a T cell-mediated, autoimmune disease of the central nervous system (CNS) that serves as a model for various cellular and molecular aspects of the human disease, multiple sclerosis (MS). Although EAE has long been considered a T cell-mediated disease, macrophages play a role in disease pathogenesis and are known to accumulate in the CNS under the control of chemokines. In the present report we demonstrate that mice induced to develop EAE were treated with a small molecular weight molecule that suppresses proinflammatory cytokine production which resulted in significantly decreased clinical EAE, CNS CCL2 expression and CNS macrophage accumulation. These results demonstrate the efficacy of a novel class of therapeutic molecules for CNS demyelinating disease.
Keywords: multiple sclerosis, proinflammatory cytokines, experimental autoimmune encephalomyelitis, neuroinflammation, pyridazine, chemokines, minozac
INTRODUCTION
Experimental autoimmune encephalomyelitis (EAE) is a T cell-mediated demyelinating disease of the central nervous system (CNS) that serves as a model for multiple sclerosis (MS) (Steinman and Zamvil, 2006). In addition to the requirement for autoreactive T cells, macrophages have been shown to be necessary for EAE development (Brosnan et al., 1981; Cua et al., 1995).
Many chemokines are expressed in the CNS of mice that develop EAE (Godiska et al., 1995). Deletion or neutralization of CCL2 (Huang et al., 2001; Kennedy et al., 1998) as well as deletion of its receptor, CCR2 (Fife et al., 2000) decreased EAE severity. CCL2 (McManus et al., 1998) has been suggested to be involved in MS. Chemokines have been postulated to regulate accumulation of inflammatory cells in the CNS during EAE/MS pathogenesis (Karpus and Ransohoff, 1998).
The development of therapeutics for MS has recently focused on preventing or reducing CNS accumulation of inflammatory cells. Anti-CD49d antibody treatment of mice had a dramatic effect of preventing accumulation of inflammatory cells in the CNS (Yednock et al., 1992). In fact, the ability of Tysabri® to dramatically improve disability in MS patients (Rice et al., 2005) underscores the desirability of this approach. To that end we investigated a novel compound, Minozac (Mzc) (Hu et al., 2007), for its ability to prevent accumulation of inflammatory cells in the CNS and inhibit EAE.
MATERIALS AND METHODS
Mice
Female SJL (H2s) mice were purchased form Harlan Sprague Dawley (Indianapolis,IN) and used as previously described (Karpus et al., 1995). Animal care and use was in accordance with the Northwestern University Animal Care and Use Committee and United States Public Health Service policies.
Antigen and antibodies
PLP139–151 (HSLGKWLGHPDKF) was purchased from Peptides International (Louisville, KY) (Karpus et al., 1995). Fluorochrome-conjugated monoclonal antibodies to murine CD4 (RM4–5), CD8a (Ly-2), CD45 (Ly-5), CD11b (Mac-1) and purified CD16/32 (Fc block, clone 2.4G2) were purchased from BD Pharmingen (San Diego, CA). Fluorochrome-conjugated monoclonal antibodies to murine-Foxp3 were purchased from eBioscience (San Diego,CA). For immunohistochemistry, anti-mouse CD11b was purchased from R&D System (Minneapolis, MN).
Induction of active EAE
For the induction of EAE, female SJL/J mice were injected subcutaneously as previously described (Karpus et al., 1995). Animals were graded according to their clinical severity using the following scale: grade 0, no abnormality; grade 1, limp tail; grade 2, limp tail and hind limb weakness; grade 3, partial hind limb paralysis; grade 4, complete hind paralysis; grade 5, death.
Flow Cytometry
Mononuclear cells were isolated from the CNS of mice perfused intracardially with 0.15 M saline solution. Spinal cords were dissected from the vertebral canal. Mononuclear cells were isolated and prepared as previously described (Hendrzak et al., 1994; Pope et al., 1996). Data collection was performed on a LSR II (Becton Dickinson, San Jose, CA) flow cytometer using FACS Diva software (Becton Dickinson) and analysis was performed offline using FCS Express software.
Histology and immunohistochemistry
Mice were anesthetized with sodium pentobarbital (Abbott Laboratories, IL) and perfused intracardially through the left ventricle with ice cold PBS. Tissues were embedded in OCT prior to cryostat sectioning. Frozen sections (8–10 µm) were blocked with 5% normal goat serum in PBS for 30 min at room temperature and incubated with anti-CD11b for 2 hrs at room temperature. Sections were treated 3% H2O2 to quench endogenous peroxidase activity and then incubated with goat secondary antibodies directly conjugated to HRP (Vectastatin ABC kit, Vector). Biotin-avidin binding was detected by diaminobenzidine (DAB) substrate (Sigma).
Statistical Analysis
Sample median, mean, standard deviation, and statistical significance were calculated using SPSS 13.0 software. Clinical disease comparisons of median disease severity over the course of time were made and statistical significance was calculated using the Mann-Whitney test for ordinal data. Single comparisons of two means were analyzed by Student’s t test. Comparisons of percent affected mice were performed using a χ2 test. P values < 0.05 were considered significant.
RESULTS AND DISCUSSION
Mzc treatment decreased incidence and severity of EAE
Mzc is a recently developed novel suppressor of proinflammatory cytokine production that is water soluble and stable, has a brain:blood peak concentration ratio of 1.5, and is efficacious in an Alzheimer’s disease model (Hu et al., 2007). We reasoned that there was significant glia-derived cytokine expression in EAE and asked whether Mzc treatment would have an effect on disease development and progression. Two groups of 12 mice were immunized with PLP139–151 in CFA and monitored for the development of EAE. The control group was given 0.2 ml PBS i.g. while the treatment group was given 15 mg/kg Mzc in PBS i.g. daily starting at the time of immunization and continuing for 21 consecutive days. The results indicate that Mzc treatment significantly reduced the median EAE severity (Figure 1A). The data in Figure 1B also demonstrate that Mzc treatment significantly reduced the disease incidence. However, treatment with Mzc did not affect the mean day of disease onset (Figure 1C) in those mice that developed disease.
Figure 1.

Minozac (Mzc) treatment attenuates EAE. Two groups of 12 SJL mice were immunized with PLP139–151 in CFA. One group was treated with a 15 mg/kg/day i.g. dose of Mzc for 21 days while the control group received a daily vehicle (PBS) administration. At each time point (x axis) the 12 mice in each group were assessed for clinical disease score (y axis). The clinical disease score data does not represent the mean of the group. The boxes (white for PBS and gray for Mzc treatment) indicate the 25th to 75th percentile of the data in each group at each time point. The horizontal lines in each box indicate the median of the data set. The error bars indicate the data set variability (range of scores throughout the groups). The Mzc group showed statistically significantly reduced disease (*, p<0.05, Mann-Whitney test for ordinal data) at days 9, 11, 15, 17 and 19 post immunization. The data are representative of three similarly performed replicates (A). Mzc significantly (p<0.05, χ2 test) inhibited the number of mice that developed clinical signs of EAE (B). Mzc treatment did not have an effect of the mean day of onset (N.S.=not significant) (C). Mzc treatment reduced CNS expression of CCL2 (D) and TNF (E). Two groups of 10 SJL mice were immunized with PLP139–151 in CFA. One group was treated with a 15 mg/kg/day i.g. dose of Mzc for 19 days while the control group received a daily vehicle (PBS) administration. The mice were monitored for the development and progression of clinical EAE. Three mice were killed and their CNS was examined individually for the presence of CCL2 and TNF by ELISA at disease onset and the peak of clinical disease (both were determined relative to disease in the control group). The data indicate the concentration of cytokine in the CNS homogenate. There was a significant decrease (p<0.05) in CCL2 concentration in the Mzc-treated CNS at both the onset and peak stages of EAE. There was a significant decrease (p<0.05) in the TNF concentration in the Mzc-treated group at the peak stage of clinical disease. The experiment is representative of three identical replicates.
Mzc treatment decreased CNS CCL2 expression
We asked whether Mzc treatment reduced expression of CCL2, postulated to regulate CNS macrophage migration (Huang et al., 2001). Spinal cords from mice treated as in Figure 1 were homogenized and assessed for CCL2 as previously described (Kennedy et al., 1998). The results shown in Figure 1D demonstrate that Mzc treatment significantly decreased expression of CCL2 in the CNS at both time points measured. The results shown in Figure 1E indicate that Mzc treatment significantly inhibited TNF production in the CNS at the peak, but not onset, of clinical disease. It is unlikely that Mzc down regulated EAE in these experiments by directly affecting T cell activation as we did not observe any differences in antigen-specific T cell proliferation or IFN-γ or IL-17 production (data not shown). Likewise, Mzc treatment did not induce an increase in Foxp3+ T regulatory cells (data not shown). Finally, there was no evidence that CNS TGF-β or IL-10 expression was increased in the Mzc-treated mice (data not shown).
Mzc treatment decreased macrophage accumulation in the CNS during EAE
To determine the mechanism of Mzc inhibition of EAE we first examined the CNS of representative mice from a similar experiment described in Figure 1. We found that there were decreased leukocytes and perivascular lesions in the spinal cords stained with H&E of Mzc-treated compared to control mice (Figure 2A and B). Furthermore, spinal cord sections stained with anti-CD11b to identify macrophages/microglia/dendritic showed numerous positive staining cells in the control-treated groups (Figure 2C) and this was decreased in Mzc-treated spinal cords (Figure 1D). In order to quantify whether Mzc treatment resulted in a decrease in CNS accumulation of CD11b+ cells, we performed a flow cytometric analysis at disease onset and peak clinical disease. The data shown in Figure 2E indicated there was relatively similar lymphocyte (CD45hiCD11b−), macrophage (CD45hiCD11b+) and microglia (CD45loCD11b+) populations between the control and Mzc-treated groups at disease onset. However, at the peak of clinical disease time point, we found a dramatic and significant decrease in the macrophage population (CD45hiCD11b+) in the Mzc-treated group compared to controls (Figure 2B, 3.5% vs. 19.3%). Quantification of 5 individual mice (Figure 2F) indicated there was a significant reduction in the number of CD45+CD11b+ macrophages in the CNS of Mzc-treated mice.
Figure 2.
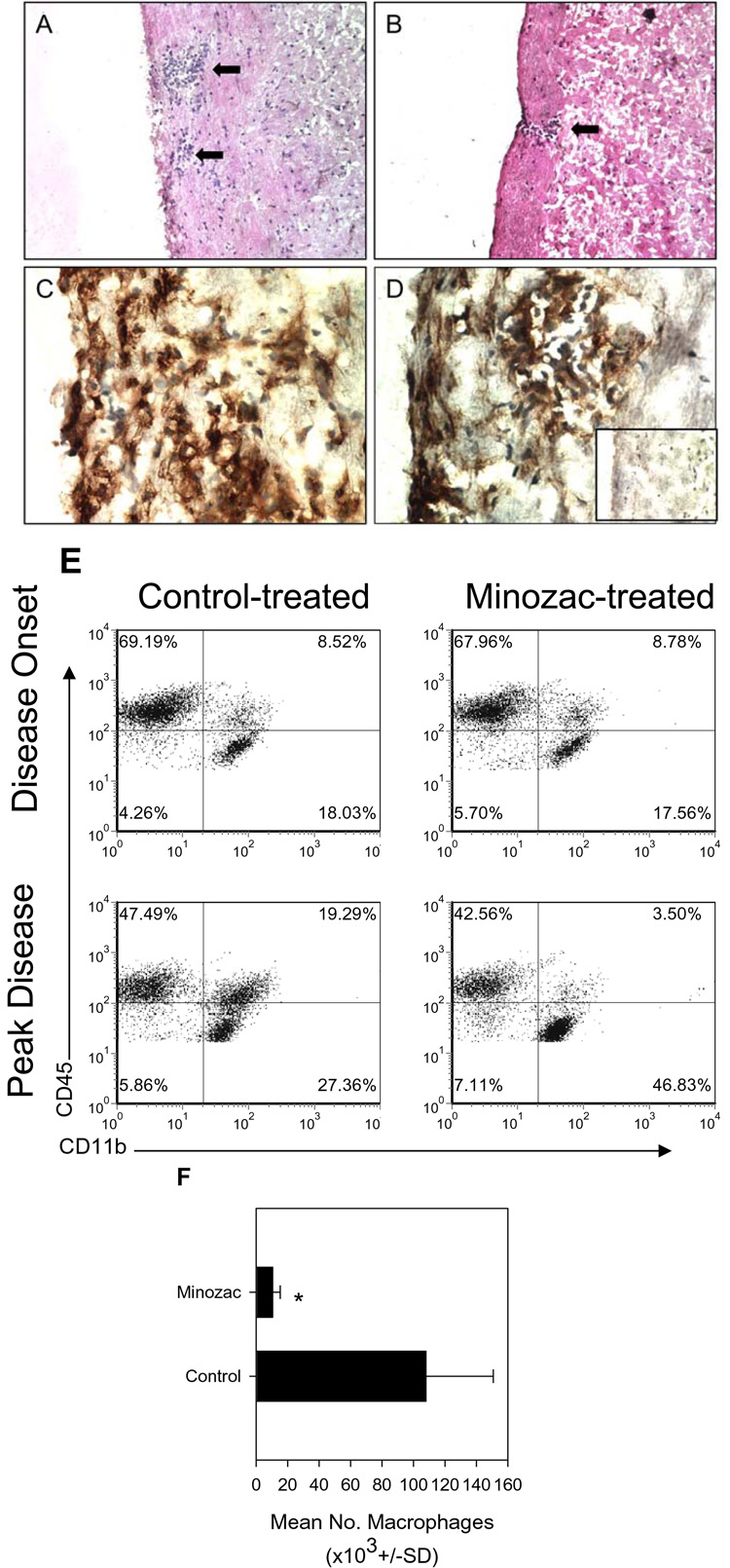
Mzc treatment reduces CNS inflammatory infiltrates. The spinal cords of PBS-treated (panel A) and Mzc-treated (panel B) mice were examined by H&E staining of frozen sections for the presence of leukocyte infiltrates. The data in Panel A indicate the control spinal cord contained prominent perivascular inflammatory infiltrates (two arrows; magnification=200X) while in Panel B a small lesion in the Mzc-treated spinal cord (single arrow; magnification=200X) was noted. Frozen spinal cord sections from PBS- (Panel C) or Mzc-treated (Panel D) mice were immunostained for the presence of CD11b+ cells. The data indicate the presence of CD11b+ cells in the spinal cords of both treatment groups (magnification=600X). Spinal cords from un-immunized, normal mice were also stained for the presence of CD11b+ cells (Panel D, inset). The results indicate few positively staining cells (brown) relative to immunized PBS- or Mzc-treated mice. The results are representative of three mice in each group and similar to two independent experimental replicates. Mzc treatment reduces accumulation of macrophages in the CNS (E). Two groups of 10 SJL mice were immunized with PLP139–151 in CFA. One group was treated with a 15 mg/kg/day i.g. dose of Mzc for 21 days while the control group received a daily vehicle (PBS) administration. The mice were monitored for the development and progression of clinical EAE. When the control group began to develop EAE (disease onset) 5 mice were killed and their CNS was examined individually for the relative proportions of lymphocytes (CD45hiCD11b−), macrophages (CD45hiCD11b+) and microglia (CD45loCD11b+). The data are derived from representative mice in each group and were identical in all mice from each group. The results indicate a difference in the proportion of CNS-infiltrating macrophages between the Mzc-treated (3.5%) and the control group (19.3%) at the peak of clinical disease. The data are representative of two identical replicate experiments. Quantitative analysis of 5 mice in each group indicated a significant (p<0.05) decrease in CD45+CD11b+ macrophage numbers in the CNS of Mzc-compared to control-treated mice (F).
Mechanism of Mzc Inhibition of EAE
EAE is often used as a model to study mechanisms of MS and to gain insights into development of new treatments for CNS autoimmune demyelinating disease (Steinman and Zamvil, 2006). At least two current MS treatments, glatiramer acetate and natalizumab (Johnson et al., 1995; Miller et al., 2003) have resulted from original observations using the EAE model (Teitelbaum et al., 1999; Yednock et al., 1992). In the present report we assessed the efficacy of a novel, brain-penetrant, small molecule compound for the ability to inhibit induction and progression of EAE. Our results suggested that Mzc treatment affected the accumulation of macrophages in the CNS, which is known to be involved in disease pathogenesis (Brosnan et al., 1981) by decreased CNS expression of CCL2 (Figure 1D). These data strongly support the conclusion that Mzc is a novel approach to the treatment of CNS inflammatory disease.
Mzc is a refined therapeutic (Hu et.al., 2007) that is an analog of the original lead compound MW01-5-188WH (188WH) that also inhibited glial production of IL-1β and TNF, was brain penetrant, and attenuated defects in a model of Alzheimer’s Disease (Ralay Ranaivo et al., 2006). The importance of glia in EAE has been well documented. Specifically, astrocytes are a major CNS source of chemokines such as CCL2 (Hayashi et al., 1995), a significant producer of TNF (Chung et al., 1991), as well as IL-12 and IL-23 (Constantinescu et al., 2005; Xu and Drew, 2007). Microglia have been shown to express a similar complement of cytokines (Becher et al., 2003; Benveniste, 1997; Gehrmann et al., 1995). Therefore, from a drug development perspective it remains an attractive approach to develop inhibitor molecules that have a high degree of brain penetrance and inhibit glia-produced inflammatory cytokines. It is possible that Mzc treatment accelerated microglia proliferation (Figure 2E) as these cells normally proliferated under CNS inflammatory circumstances (Gehrmann et al., 1995). This is an area that is currently under investigation. Given its efficacy, oral bioavailability, aqueous solubility, blood-brain barrier penetrance, and stability, Mzc and its analogs remain strong candidates for further investigation as a potential therapeutic modality in human CNS autoimmune diseases, including MS.
ACKNOWLEDGEMENTS
This work was supported by NS34510, AG028561, NS047586, and AG000260.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Becher B, Durell BG, Noelle RJ. IL-23 produced by CNS-resident cells controls T cell encephalitogenicity during the effector phase of experimental autoimmune encephalomyelitis. Journal of Clinical Investigation. 2003;112:1186–1191. doi: 10.1172/JCI19079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benveniste EN. Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. Journal of Molecular Medicine. 1997;75:165–173. doi: 10.1007/s001090050101. [DOI] [PubMed] [Google Scholar]
- Brosnan CF, Bornstein MB, Bloom BR. The effects of macrophage depletion on the clinical and pathologic expression of experimental allergic encephalomyelitis. Journal of Immunology. 1981;126:614–620. [PubMed] [Google Scholar]
- Chung IY, Norris JG, Benveniste EN. Differnetial tumor necrosis factor á expression by astrocytes from experimental allergic encephalomyelitis-susceptible and -resistant rat strains. Journal of Experimental Medicine. 1991;173:801–811. doi: 10.1084/jem.173.4.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinescu CS, Tani M, Ransohoff RM, Wysocka M, Hilliard B, Fujioka T, Murphy S, Tighe PJ, Sarma JD, Trinchieri G, Rostami A. Astrocytes as antigen-presenting cells: expression of IL-12/IL-23. J Neurochem. 2005;95:331–340. doi: 10.1111/j.1471-4159.2005.03368.x. [DOI] [PubMed] [Google Scholar]
- Cua DJ, Hinton DR, Kirkman L, Stohlman SA. Macrophages regulate induction of delayed-type hypersensitivity and experimental allergic encephalomyelitis in SJL mice. European Journal of Immunology. 1995;25:2318–2324. doi: 10.1002/eji.1830250830. [DOI] [PubMed] [Google Scholar]
- Fife BT, Huffnagle GB, Kuziel WA, Karpus WJ. CC Chemokine Receptor 2 Is Critical for Induction of Experimental Autoimmune Encephalomyelitis. Journal of Experimental Medicine. 2000;192:899–906. doi: 10.1084/jem.192.6.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrmann J, Matsumoto Y, Kreutzberg GW. Microglia: Intrinsic immuneffector cell of the brain. Brain Research Reviews. 1995;20:269–287. doi: 10.1016/0165-0173(94)00015-h. [DOI] [PubMed] [Google Scholar]
- Godiska R, Chantry D, Dietsch GN, Gray PW. Chemokine expression in murine experimental allergic encephalomyelitis. Journal of Neuroimmunology. 1995;58:167–176. doi: 10.1016/0165-5728(95)00008-p. [DOI] [PubMed] [Google Scholar]
- Hayashi M, Luo Y, Laning J, Strieter RM, Dorf ME. Production and function of monocyte chemoattractant protein- 1 and other á-chemokines in murine glial cells. Journal of Neuroimmunology. 1995;60:143–150. doi: 10.1016/0165-5728(95)00064-9. [DOI] [PubMed] [Google Scholar]
- Hendrzak JA, Wallace PD, Morahan PS. Optimizing the detection of cell surface antigens on elicited or activated mouse peritoneal macrophages. Cytometry. 1994;17:349–356. doi: 10.1002/cyto.990170412. [DOI] [PubMed] [Google Scholar]
- Hu W, Ralay Ranaivo H, Roy SM, Behanna HA, Wing LK, Munoz L, Guo L, Van Eldik LJ, Watterson DM. Development of a novel therapeutic suppressor of brain proinflammatory cytokine up-regulation that attenuates synaptic dysfunction and behavioral deficits. Bioorganic & Medicinal Chemistry Letters. 2007;17:414–418. doi: 10.1016/j.bmcl.2006.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DR, Wang J, Kivisakk P, Rollins BJ, Ransohoff RM. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. Journal of Experimental Medicine. 2001;193:713–726. doi: 10.1084/jem.193.6.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KP, Brooks BR, Cohen JA, Ford CC, Goldstein J, Lisak RP, Myers LW, Panitch HS, Rose JW, Schiffer RB. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology. 1995;45:1268–1276. doi: 10.1212/wnl.45.7.1268. [DOI] [PubMed] [Google Scholar]
- Karpus WJ, Lukacs NW, McRae BL, Strieter RM, Kunkel SL, Miller SD. An important role for the chemokine macrophage inflammatory protein-1à in the pathogenesis of the T cell-mediated autoimmune disease, experimental autoimmune encephalomyelitis. Journal of Immunology. 1995;155:5003–5010. [PubMed] [Google Scholar]
- Karpus WJ, Ransohoff RM. Chemokine regulation of experimental autoimmune encephalomyelitis: temporal and spatial expression patterns govern disease pathogenesis. Journal of Immunology. 1998;161:2667–2671. [PubMed] [Google Scholar]
- Kennedy KJ, Strieter RM, Kunkel SL, Lukacs NW, Karpus WJ. Acute and relapsing experimental autoimmune encephalomyelitis are regulated by differential expression of the CC chemokines macrophage inflammatory protein-1à and monocyte chemotactic protein-1. J Neuroimmunol. 1998;92:98–108. doi: 10.1016/s0165-5728(98)00187-8. [DOI] [PubMed] [Google Scholar]
- McManus C, Berman JW, Brett FM, Staunton H, Farrell M, Brosnan CF. MCP-1, MCP-2 and MCP-3 expression in multiple sclerosis lesions: an immunohistochemical and in situ hybridization study. Journal of Neuroimmunology. 1998;86:20–29. doi: 10.1016/s0165-5728(98)00002-2. [DOI] [PubMed] [Google Scholar]
- Miller DH, Khan OA, Sheremata WA, Blumhardt LD, Rice GP, Libonati MA, Willmer-Hulme AJ, Dalton CM, Miszkiel KA, O'Connor PW. A controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2003;348:15–23. doi: 10.1056/NEJMoa020696. [DOI] [PubMed] [Google Scholar]
- Pope JG, Karpus WJ, VanderLugt C, Miller SD. Flow cytometric and functional analyses of central nervous system-infiltrating cells in SJL/J mice with Theiler's virus-induced demyelinating disease - Evidence for a CD4 + T cell-mediated pathology. Journal of Immunology. 1996;156:4050–4058. [PubMed] [Google Scholar]
- Ralay Ranaivo H, Craft JM, Hu W, Guo L, Wing LK, Van Eldik LJ, Watterson DM. Glia as a therapeutic target: selective suppression of human amyloid-beta-induced upregulation of brain proinflammatory cytokine production attenuates neurodegeneration. J Neurosci. 2006;26:662–670. doi: 10.1523/JNEUROSCI.4652-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GPA, Hartung H-P, Calabresi PA. Anti-{alpha}4 integrin therapy for multiple sclerosis: Mechanisms and rationale. Neurology. 2005;64:1336–1342. doi: 10.1212/01.WNL.0000158329.30470.D0. [DOI] [PubMed] [Google Scholar]
- Steinman L, Zamvil SS. How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Annals of Neurology. 2006;60:12–21. doi: 10.1002/ana.20913. [DOI] [PubMed] [Google Scholar]
- Teitelbaum D, Arnon R, Sela M. Immunomodulation of experimental autoimmune encephalomyelitis by oral administration of copolymer 1. Proc.Natl.Acad.Sci.U.S.A. 1999;96:3842–3847. doi: 10.1073/pnas.96.7.3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Drew PD. Peroxisome Proliferator-Activated Receptor-{gamma} Agonists Suppress the Production of IL-12 Family Cytokines by Activated Glia. J Immunol. 2007;178:1904–1913. doi: 10.4049/jimmunol.178.3.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yednock TA, Cannon C, Fritz LC, Sanchez-Madrid F, Steinman L, Karin N. Prevention of experimental autoimmune encephalomyelitis by antibodies against à4á1 integrin. Nature. 1992;356:63–66. doi: 10.1038/356063a0. [DOI] [PubMed] [Google Scholar]