

Abstract
Toll-like receptor (TLR)/MyD88 activation is important in host resistance to Toxoplasma gondii during i. p. infection, but the function of this signaling pathway during oral infection, in which mucosal immunity assumes a predominant role, has not been examined. Here, we show that MyD88−/− mice fail to control the parasite and succumb within two weeks of oral infection. Early during infection, T cell IFN-γ production, recruitment of neutrophils and induction of p47 GTPase Irgm3/IGTP in the intestinal mucosa were dependent upon functional MyD88. Unexpectedly, these responses were MyD88-independent later during acute infection. In particular, CD4+ T cell IFN-γ reached normal levels independently of MyD88, despite continued absence of IL-12 in these animals. Intraperitoneal vaccination of MyD88−/− mice with an avirulent T. gondii uracil axotroph elicited robust IFN-γ responses and protective immunity to challenge with a high virulence T. gondii strain. Our results demonstrate that MyD88 is required to control Toxoplasma infection, but that the parasite can trigger adaptive immunity without the need for this TLR adaptor molecule.
Keywords: Parasitic-protozoan, signal transduction, cytokines
Introduction
Toll-like receptors (TLR)4 have emerged as a major family of pattern recognition molecules involved in sensing infectious nonself molecules and possibly endogenous molecules of the host. There are 11−13 TLR in mice and humans that recognize diverse molecules including bacterial lipopolysaccharide, lipopeptides, unmethylated CpG oligodinucleotides, as well as single and double-stranded RNA (1, 2). Signaling through TLR is complex, but all TLR (with the exception of TLR3) use the adaptor molecule MyD88 to initiate intracellular signal transduction (3-5). Most prominent among these signaling pathways are NFκB and mitogen-activated protein kinase (MAPK) cascades leading to induction of proinflammatory cytokines such as IL-12 and TNF-α. MyD88 is also used to relay signals emanating from IL-1 and IL-18 receptors. As such, MyD88 represents a bottleneck through which most TLR, as well as IL-1R/IL-18R, initiated signals must pass.
The importance of MyD88 in infectious disease models has been firmly established using genetically engineered MyD88−/− mice. Animals lacking MyD88 display decreased resistance to bacteria such as Mycobacterium tuberculosis, Staphyloccus aureus, Listeria monocytogenes, and Brucella abortus (6-9). It is also becoming clear that TLR and MyD88 are important in the innate immune response to parasitic protozoans (10). Thus, MyD88 KO mice are increased in susceptibility to infections with Toxoplasma gondii, Plasmodium berghei, Leishmania major and Trypanosoma cruzi (11-15). TLR ligands have been identified for several protozoan species, including T. gondii, Plasmodium, and T. cruzi (16-18). Nevertheless, while knock out of MyD88 may have dramatic effects on resistance to bacterial and protozoan infection, inactivation of genes encoding individual TLR often has little or no impact. This has led to the view that the response to any given pathogen is likely to involve multiple TLR that act in concert to provide optimal resistance to infection (19-21).
There is evidence that TLR-MyD88 signaling is important, not only in innate immune responses, but also in initiation of acquired immunity (22). For example, MyD88 transduced signaling leads to upregulation of costimulatory molecules on DC that is required to activate naïve T cells, and T cell antigen-specific responses are optimized when microbial peptides are presented within the context of TLR ligands (23, 24). Nevertheless, the role of TLR-MyD88 in initiation of adaptive immunity during infection with complex pathogens is less certain. In some cases MyD88 is not necessary, such as in the generation of Listeria monocytogenes-specific CD8+ T cell responses and during acquired immunity to Borrelia burgdorferi (25-27). In other cases, MyD88 seems to play a prominent role in adaptive immunity, for example in the humoral immune response to polyoma virus infection and the antiviral CD8+ response to lymphocytic choriomeningitis virus (28, 29).
In this study we focus on infection with the intracellular protozoan T. gondii, a parasite known for its ability to elicit strong protective Th1 responses, but that causes serious disease in immunocompromised individuals (30, 31). It has previously been shown that MyD88−/− animals rapidly succumb to Toxoplasma in a model involving i. p. injection of parasites (14). TLR signaling is strongly implicated in this situation because mice defective in IL-1β-converting enzyme display normal resistance to infection despite an inability to produce functional IL-1 or IL-18 (15). Tachyzoite profilin and glycosylinositolphosphatidyl lipid moieties have recently been identified as parasite ligands of TLR11 and TLR2/4, respectively (32-34). Furthermore, for the case of profilin there is evidence that signaling through TLR11 promotes an immunodominant T cell response to peptides derived from this molecule (35).
Here, we show that MyD88 is required to survive oral infection with Toxoplasma. Early during infection MyD88−/− animals displayed defects in neutrophil recruitment to mucosal tissues and induction of Irgm3/IGTP, a molecule required to survive acute infection. Surprisingly, T cell-derived IFN-γ production, a key event in adaptive immunity to the parasite, reached normal levels later during acute infection, although initial production was delayed. To test the ability of MyD88 KO mice to generate protective immunity to T. gondii we employed a genetically engineered avirulent parasite strain that invades but cannot replicate in host cells. Vaccination with this mutant elicited fully functional Th1 cell differentiation and immunity to challenge infection. Our results show that during T. gondii infection MyD88 signaling is essential for host microbicidal function, but that it is dispensable for the adaptive immune response to this intracellular pathogen.
Materials and Methods
Mice
Female mice (6−12 wks of age) were used throughout these studies. IL-12p40−/− mice and C57BL/6 controls were purchased from The Jackson Laboratory. MyD88+/+ and MyD88−/− mice (generated by S. Akira, Osaka University, and kindly provided by Dr. E. Pearlman, Case Western Reserve University) were generated by crosses of heterozygous mice. To genotype F1 litters, tail snips were lysed in the Lysis Reagent with Proteinase K according to the manufacturer's instructions (Viagen Biotech, Los Angeles, CA). DNA in digests were subjected to RT-PCR analysis to determine the mouse genotype using a protocol kindly supplied by Dr. E. Pearlman (Case Western Reserve University). Primers employed for sequence amplification were 5’-TGG-CAT-GCC-TCC-ATC-ATA-GTT-AAC-C-3’ (MyD88F), 5’-GTC-AGA-AAC-AAC-CAC-CAC-CAT-GC-3’ (MyD88R), and 5’-ATC-GCC-TTC-TAT-CGC-CTT-CTT-GAC-G-3’ (MyD88Neo). WT and KO status of the animals was confirmed after the mice were euthanized in experiments. Littermate female age-matched mice between 6−12 wk of age were used for experiments. The mice were housed under specific pathogen-free conditions at the Transgenic Mouse Facility, College of Veterinary Medicine, Cornell University overseen by the Institutional Animal Care and Use Committee.
Parasites and Infections
Cysts of the low virulence ME49 strain were obtained from brains of chronically infected Swiss-Webster mice. Oral infections were carried out by intubation with a blunt-ended needle. Except where noted, 20 cysts in a volume of 0.2 ml PBS were administered. Tachyzoites of the RH and cps1−1 strain were maintained in vitro on human foreskin fibroblasts. For the case of cps1−1, 0.3 mM uracil was added to the growth medium. Vaccination with cps1−1 was accomplished by i. p. injections of 2 × 104, 2 × 105 and 2 × 105 tachyzoites at biweekly intervals. Two weeks after the final injection, animals were challenged by subcutaneous injection of 2000 RH strain tachyzoites.
Histopathology
Tissues were collected and preserved in 10% neutral buffered formalin and submitted to the Histology Unit, College of Veterinary Medicine, Cornell University to be processed into paraffin embedded blocks for H & E staining. Lesions were scored in blind manner and graded based on severity of inflammation (minimal, mild, moderate, severe), number of inflammatory foci, distribution of inflammation, type of inflammatory reaction and severity of necrosis.
Immunohistochemistry
Sections of formalin-fixed paraffin-embedded tissues were stained with a rabbit anti-Toxoplasma antiserum by the Histology Unit, College of Veterinary Medicine, Cornell University. Other sections were stained with rabbit anti-MPO or rabbit anti-Irgm3/IGTP (36) using a standard immunoperoxidase staining protocol. Briefly, sections were de-paraffinised and rehydrated by serial immersion in xylene and graded alcohols, then finally water. Exogenous peroxidase activity was quenched by incubation in 0.5% H202 followed by microwave treatment in citrate buffer (0.1 M citric acid in PBS, pH 6.0) to unmask antigens. Sections were blocked in 10% normal serum in casein blocking reagent (Vector Labs) (20 min, 20°C). Primary antibody was diluted in PBS with casein blocking reagent, and applied to the tissue (2 hrs, 37°C). Sections were washed 4 times in PBS with 0.4% Brij® (Sigma). A biotinylated secondary antibody was diluted in PBS and incubated with the tissue (20 min, 20°C). Sections were washed 3 times (PBS with 0.4% Brij®). Biotin was detected using streptavidin-conjugated peroxidase (20 min, 20°C). Slides were subsequently washed 3 times (PBS with 0.4% Brij®) and visualized with 3-amino-9-ethylcarbazole (AEC) chromagen (Vector Labs) (10 min, 20°C). Sections were rinsed in water, counterstained in Gill's Hematoxylin (Vector Labs), mounted and examined by bright-field microscopy. The specificity of Irgm3/IGTP staining was confirmed by examination of tissues from Irgm3/Igtp−/− mice.
Flow cytometry
For phenotypic analysis of splenocyte and mesenteric lymph node cells, cell surface staining was accomplished as previously described (37) using the following fluorochrome conjugated mAb: anti-CD11c-PE, anti-Gr-1-PerCP-Cy5.5, anti-B220-PE, anti-CD8-PE-Cy7, anti-CD4-FITC, anti-CD44-APC (all from BD Pharmingen), anti-F480-APC, anti-CD3-APC (all from Invitrogen), anti-CD62L-PE, anti-CD25-PE, anti-CD69-PE (all from eBioscience). Intracellular cytokine staining was accomplished by incubating single cell suspensions in cDMEM (DMEM supplemented with 10% bovine growth serum, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, 0.05 mM β-mercaptoethanol, 100 U/ml penicillin, 100 μg/ml streptomycin, and 30 mM HEPES) with or without STAg or cps1−1. Golgiplug (BD Biosciences) was added for the last 4 hr of culture. Cells were washed and surface stained for CD4 and CD8 (20 min, 4°C). Cells were then fixed and permeabilized in cytofix/cytoperm solution (BD Biosciences) according to the manufacturer's instructions. Cells were washed in Perm Wash (BD Biosciences) and resuspended in intracellular staining buffer containing 10% normal mouse serum, 10% rat IgG and optimal concentrations of fluorochrome-tagged anti-cytokine antibodies in Perm Wash (60 min, 4°C). Cells were washed twice in Perm Wash, resuspended in FACS buffer and immediately analyzed on a FACScalibur flow cytometer (BD Biosciences) collecting at least 50,000 events per sample. Data were subsequently analyzed using FlowJo software (Tree Star).
Cell culture
Single cell suspensions were prepared and red blood cells were removed by using RBC lysis buffer according to manufacturer's protocol (Sigma-Aldrich). Cell debris was removed by filtering through a 40 μm cell strainer. Cells were washed with PBS and resuspended in cDMEM. Viable cell number was determined by trypan blue exclusion. For cytokine protein measurement, cells were plated in a 96-well plate (107/ml). Soluble tachyzoite antigen (STAg) or cps1−1 tachyzoites were added and cells were incubated at 37°C in humidified 5% CO2 for 48 and 72 hrs, then supernatants were recovered for ELISA.
In vitro infection assay
Bone marrow-derived macrophages were prepared as described (38). Cells were plated for 2 hr and incubated with or without 100 ng/ml recombinant IFN-γ (Peprotech) in cDMEM overnight. Cells were then infected with type II PTG T. gondii tachyzoites at a multiplicity of infection of 0.5 to 1. After 12 hr, cells were harvested and resuspended in 10% normal mouse serum. Surface staining was carried out using anti-F4/80-APC (Invitrogen). Cells were fixed and permeabilized with cytofix/cytoperm solution (BD Biosciences) according to the manufacturer's protocol. After washing, anti p30-FITC Ab (Argene) was used to stain T. gondii. Cells were washed twice and resuspended for flow cytometric analysis.
IL-12 in vivo depletion
Mice were administered 0.5 mg rat IgG (Jackson Immunoresearch) or anti-IL-12 C17.8 antibody by i. p. injection on day -1, day 0, day 2, day 4, and day 6 post oral infection with 20 cyst ME49 cysts. Serum and spleens were collected on day 4 and day 7 post infection. Splenocytes were cultured with 3 μg/ml soluble tachyzoite antigen for 72 h.
ELISA
Cytokine ELISA for IFN-γ and IL-12/23p40 were carried out as previously described (39).
Statistical analyses
Student's t test was employed to analyze statistical differences between groups. P values < 0.05 were considered significant. Survival curves were analyzed using a logrank test. All experiments were repeated a minimum of 2−3 times.
Results
MyD88 is required to survive oral infection with T. gondii
It was previously shown that genetic inactivation of Toll/IL-1R adaptor molecule MyD88 rendered mice highly susceptible to i. p. infection with Toxoplasma (14, 15). We sought to determine if lack of MyD88 also resulted in susceptibility during infection initiated by the natural route of infection, namely, through oral inoculation. As shown in Fig. 1A, MyD88−/− mice, but not MyD88+/+ littermates, uniformly succumbed to infection after oral administration of low virulence ME49 cysts. Interestingly, in parallel groups of mice i. p. infected with the same cyst number, MyD88−/− mice succumbed at a slightly, but significantly (p = 0.0015), accelerated rate (Fig. 1B). In H & E staining of gut and Peyer's patches we failed to detect major differences in pathology of WT and KO mice, but in livers we found MyD88-dependent inflammatory infiltrates (data not shown).
FIGURE 1.
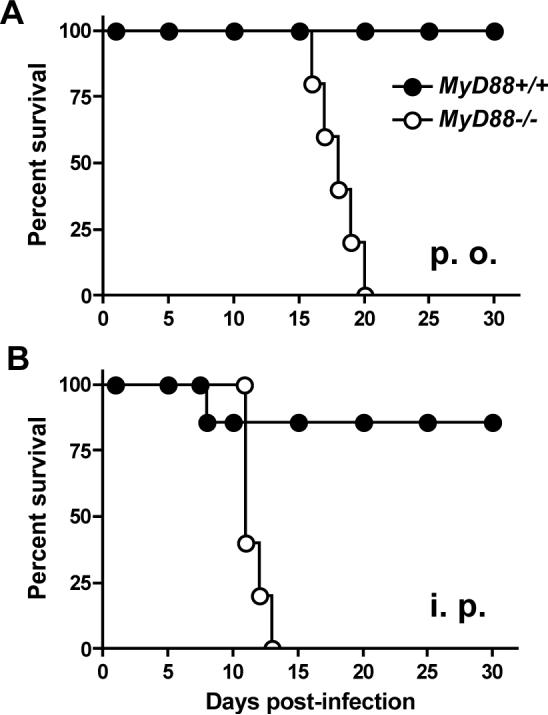
MyD88 is required to survive oral infection with Toxoplasma gondii. Groups of MyD88−/− and MyD88+/+ littermates (5 per strain) were orally and i. p. infected at the same time with 20 cysts of the ME49 strain. Panel A, oral infection; panel B, i. p. infection. Closed symbols, MyD88+/+ mice; open symbols, MyD88−/− mice.
Death during i. p. infection of MyD88−/− animals is associated with uncontrolled tachyzoite replication and dissemination (14, 15). Likewise, we found greater numbers of parasites during low dose oral infection of MyD88−/− relative to MyD88+/+ littermates. In WT mice, low numbers of tachyzoites were present in lamina propria (Fig. 2A) and Peyer's patches (Fig. 2C) of the small intestine. In contrast, high parasite numbers were present at these locations in KO animals (Fig. 2B and D, respectively). Regions of lymphocyte depletion were also apparent in Peyer's patches of KO mice (Fig. 2D) and these areas were markedly less extensive in WT animals (Fig. 2C).
FIGURE 2.
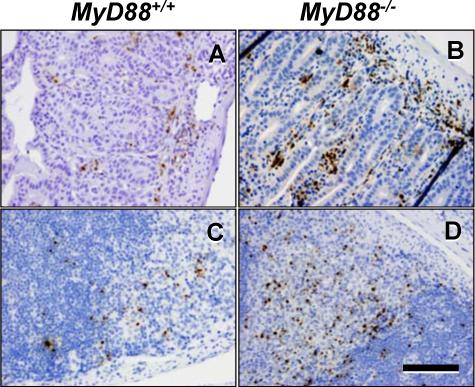
Parasite number in gut mucosal tissue during oral infection is increased in the absence of MyD88. Wild-type (A and C) and knockout (B and D) mice were infected with 20 ME49 cysts and gut tissues were collected 7 days later. Samples were subjected to immunohistochemical staining using an anti-Toxoplasma antiserum. A and B, villi and underlying lamina propria. B and D, Peyer's patches. The bar in D denotes 100 μm.
We assessed in vitro infection in the presence and absence of MyD88 using IFN-γ-activated bone marrow-derived macrophages from WT and KO strains. As shown in Fig. 3, cells from MyD88 KO mice were equally able to limit infection relative to WT macrophages. Thus, in WT cells there was a 74% reduction in infection, compared to 69% in MyD88 KO macrophages after overnight incubation in the presence of IFN-γ.
FIGURE 3.
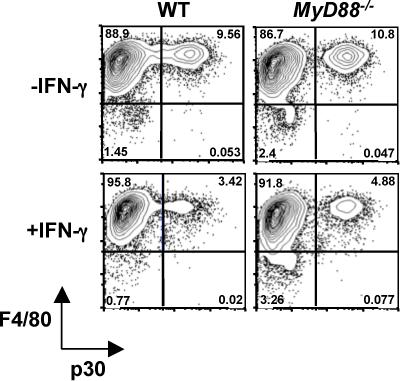
IFN-γ-activated macrophages efficiently control infection in the absence of MyD88. Bone marrow-derived macrophages were subjected to 18 hr activation with IFN-γ or incubation in medium alone. Cells were subsequently infected with PTG strain tachyzoites, and infection levels were determined 12 hr later by flow cytometry, assessing levels of the tachyzoite surface p30 antigen in F4/80-positive macrophages.
Early but not late neutrophil recruitment is dependent upon MyD88
We next examined mesenteric lymph node and spleen cell populations of WT and KO mice undergoing oral T. gondii infection. In mesenteric lymph nodes of Day 4-infected animals, there was a striking decrease in Gr-1+ neutrophil recruitment in KO relative to WT animals, but populations of macrophages (MØ), DC, as well as B and T lymphocytes were normal (Fig. 4A). Nevertheless, neutrophil numbers in mesenteric lymph nodes recovered in MyD88−/− mice by day 7 post-infection (Fig. 4B). Similarly, neutrophil numbers in spleens of Day 4-infected KO animals were less than in WT littermates (Fig. 4C), but this difference was minimal at day 7 post-infection (Fig. 4D). We next stained sections of small intestine with antibody to myeloperoxidase (MPO), an enzyme that is expressed at high level in PMN. We found an influx of MPO+ cells into the lamina propria of WT (Fig. 4E) but not MyD88 KO (Fig. 4F) animals at day 4 post-infection. In day 7-infected mice, lamina propria neutrophils were recruited in greater number in WT mice relative to day 4 (Fig. 4G), and this response did not require functional MyD88 (Fig. 4H). We, and others, previously reported rapid recruitment of Gr-1+ neutrophils into the peritoneal cavity early during i. p. infection with high virulence RH strain tachyzoites (40, 41). In Fig. 4I we show that neutrophil recruitment in this model is also highly MyD88-dependent.
FIGURE 4.
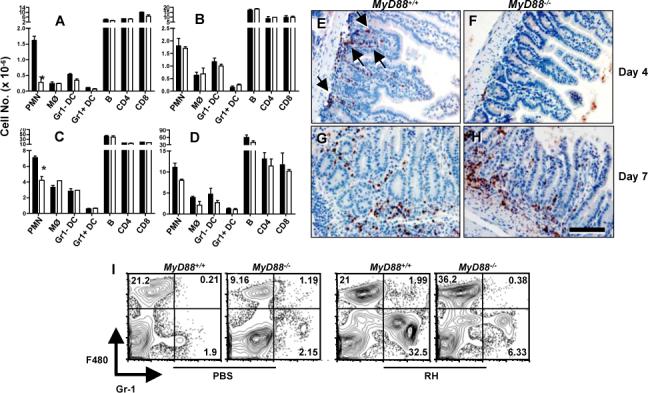
Neutrophil recruitment depends upon MyD88 during early infection. Mesenteric lymph node (A and B) and spleen (C and D) cells were prepared from MyD88−/− and MyD88+/+ littermates on Day 4 (A and C) and Day 7 (B and D) post oral infection with ME49 (20 cysts). Cells were subjected to flow cytometric analysis and total cell numbers were subsequently calculated (2 mice per group). Closed bars, MyD88+/+; open bars, MyD88−/−. PMN, Gr-1+F480− cells; MØ, Gr-1−F480+ cells; DC, CD11c+ cells; B, B220+ cells. *, p <0.05. In panels E-F, small intestines were collected from MyD88+/+ (E and G) and MyD88−/− (F and H) mice at Days 4 (E and F) and 7 (G and H) after oral ME49 infection. Sections were subsequently subjected to immunohistochemical staining with an anti-myeloperoxidase (MPO) antiserum. Arrows in panel E point to areas of infiltrating MPO+ cells. Bar denotes 100 μm. In I, wild-type and knockout mice (n = 2 per strain) were infected by i. p. injection of 106 RH strain tachyzoites. Peritoneal cells were collected 6 hr later for flow cytometric analysis.
IL-12/23p40 production is MyD88-dependent, whereas IFN-γ production is delayed but reaches normal levels in the absence of MyD88
During in vitro parasite antigen stimulation of splenocytes (Fig. 5A-C) and mesenteric lymph node cells (Fig. 5D-F), production of IL-12/23p40 was highly MyD88-dependent using as a cell source noninfected (A and D), Day 4 (B and E) and Day 7 (C and F) orally infected mice. Nevertheless, in splenocytes at all time points low but detectable levels of MyD88-independent p40 could be detected, and at day 7 post-infection residual IL-12 was apparent in mesenteric lymph node cultures from MyD88−/− animals.
FIGURE 5.

Production of IL-12/23p40 during infection is highly MyD88 dependent. Spleen (A-C) and mesenteric lymph nodes (D-F) were collected from noninfected, Day 4 and Day 7 orally infected mice (20 ME49 cysts). Single cell suspensions were prepared and cells were cultured with the indicated amounts of soluble tachyzoite lysate antigen (STAg). Closed bar, cells from wild-type mice; open bar, cells from knockout animals.
Next, we measured production of IFN-γ, a cytokine long recognized as the major mediator of resistance to T. gondii (42). As shown in Fig. 6, antigen-induced IFN-γ release by splenocytes (A) and mesenteric lymph node cells (B) was strictly dependent upon MyD88 at Day 4 post-infection. This result was confirmed and extended by intracellular cytokine staining of soluble tachyzoite lysate (STAg)-stimulated cells (Fig. 6C). Thus, in the spleen (SPL), a small proportion CD4+ T cells produced IFN-γ in dependence upon MyD88 (0.4 vs. 0.03% for WT and KO, respectively). Interestingly, there was a strong MyD88-dependent IFN-γ response by splenic CD8+ T cells under the same conditions (10.0 vs. 0.57% for WT and KO, respectively). However, in mesenteric lymph node (MLN) cultures, the STAg-induced IFN-γ response was relatively minor, and most of the MyD88-dependent IFN-γ derived from CD4+ T lymphocytes. It is of further interest to note evidence for a default to the Th2 pathway in MyD88−/− mesenteric lymph node CD4+ T cells (0.09 vs. 0.56% IL-4-positive in WT and KO, respectively) that was not apparent in splenic cultures (Fig. 6C). Thus, as previously reported in an i. p. model of Toxoplasma soluble antigen injection (43), there is an increase in the Th2 response in the absence of MyD88, but here we show this response is weak and is restricted to mesenteric lymph nodes during oral infection.
FIGURE 6.
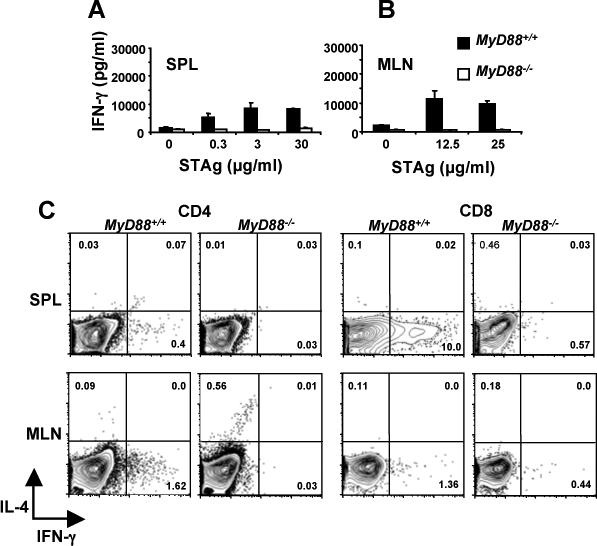
Early acute production of IFN-γ during oral Toxoplasma infection requires MyD88. Spleen cells (A) and mesenteric lymph node cells (B) from mice orally infected 4 days previously with 20 ME49 cysts were cultured with the indicated amounts of tachyzoite lysate antigen. Closed bar, MyD88+/+ cells; open bar, MyD88−/− cells. (C) Spleen and mesenteric lymph node cells from Day 4 orally infected animals were subjected to CD4 and CD8 surface staining and intracellular IFN-γ and IL-4 staining 72 hr after STAg restimulation (25 μg/ml). Cytokine expression was evaluated after gating on CD4+ and CD8+ T cells. The results show one representative mouse out of a total of three from each strain.
When we examined IFN-γ responses at day 7 post-oral infection, strikingly different results were obtained compared to the Day 4 response. Thus, at Day 7, splenocytes (Fig. 7A) and mesenteric lymph node cells (Fig. 7B) released high levels of IFN-γ independently of MyD88 and without the need for further in vitro antigen stimulation. In the presence of parasite antigen, these responses were only modestly increased. In Fig. 7C, we determined the source of IFN-γ by ex vivo staining splenocytes and mesenteric lymph node cells without further antigen stimulation. In this case, there was a vigorous MyD88-independent splenic CD4+ T cell response (Fig. 7C; 6.56 and 6.87% for WT and KO, respectively). In mesenteric lymph nodes, there was also a strong CD4+ T lymphocyte IFN-γ response, although this was partially MyD88 dependent (7.49 vs. 3.07% for WT and KO, respectively). In both spleen and mesenteric lymph nodes, the IFN-γ response by CD8+ T cells was extremely weak (data not shown).
FIGURE 7.
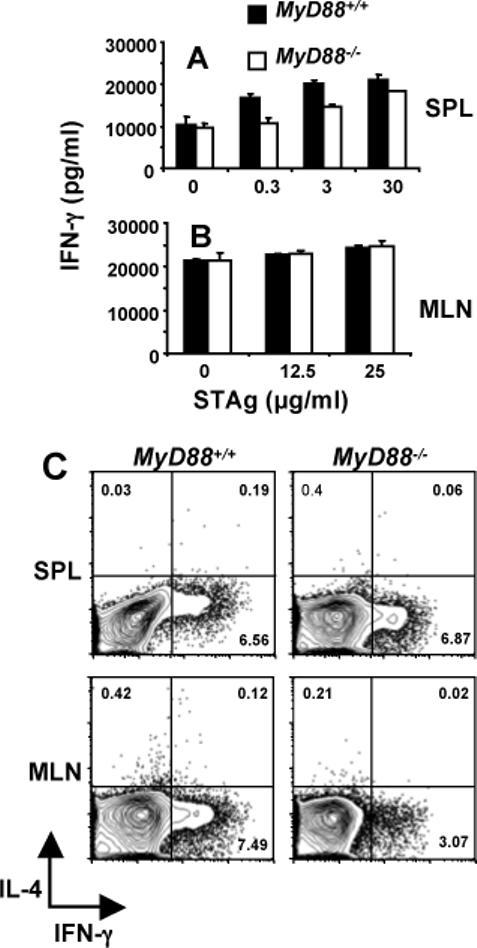
Delayed IFN-γ production and emergence of Th1 T cells does not require MyD88. Spleen (A) and mesenteric lymph node (B) cells from Day 7 orally infected wild-type (closed bars) and knockout (open bars) mice were cultured with the indicated amounts of parasite lysate antigen. (C) MyD88+/+ and MyD88−/− cells from spleen and mesenteric lymph nodes of Day 7 orally infected mice were cultured for 6 hr without further antigen stimulation in the presence of Brefeldin A. The cells were subsequently stained for CD4 and intracellular IFN-γ and IL-4. The results show one representative mouse out of a total of three from each strain.
To determine whether the IFN-γ responses we measured at Day 4 and Day 7 depended upon endogenous IL-12 production, we administered depleting IL-12p40 Ab and examined IFN-γ production over the course of infection. As shown in Fig. 8, the MyD88-dependent Day 4 response was abrogated by IL-12 depletion. In marked contrast, the MyD88-independent Day 7 response was normal in the presence of Ab. The latter result suggests that emergence of IFN-γ-producing Th1 cells at Day 7 does not require MyD88 or IL-12.
FIGURE 8.
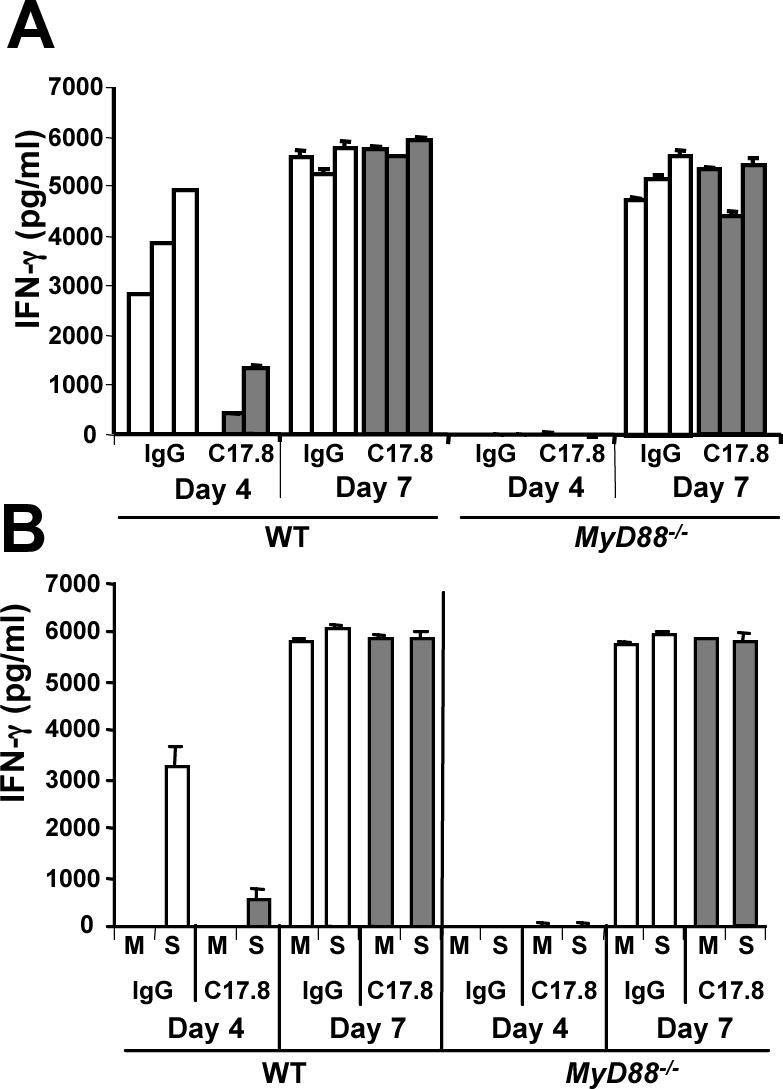
Depletion of IL-12 prevents early but not delayed IFN-γ production. Orally infected mice were subjected to IL-12p40 depletion using C17.8 Ab then serum IFN-γ production was assessed (panel A). Additionally, splenocytes were cultured in medium (M) or soluble parasite lysate (S; 3 μg/ml) then supernatants were assessed for IFN-γ production (panel B).
We also examined activation markers on CD4+ and CD8+ T cells during infection. There were no significant differences in expression of CD69 (Fig. 9) or CD25 (data not shown) over the course of infection.
FIGURE 9.
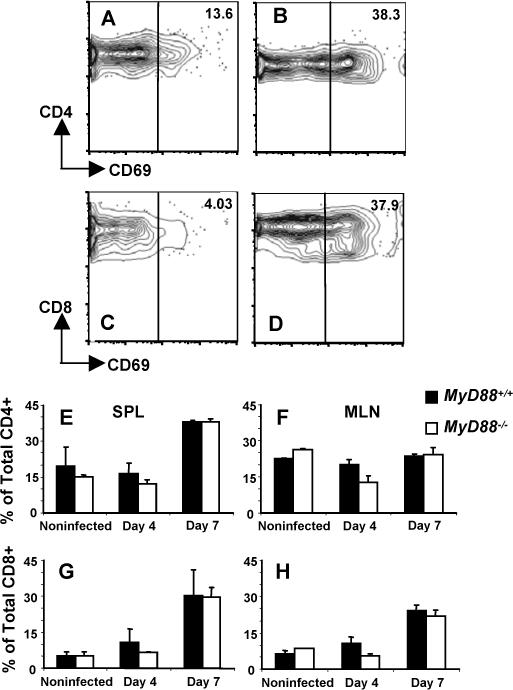
Expression of activation marker CD69 is equivalent in MyD88+/+ and MyD88−/− mice. Representative levels of CD69 are shown in noninfected CD4+ (A) and CD8+ (C) splenic WT T cells. Panels B and D show CD69 expression levels 7 days post-infection in CD4+ and CD8+ splenic T cells, respectively. Panels E-H shows mean values obtained from n=3 mice per group in WT and KO splenic (SPL) and mesenteric lymph node (MLN) T cells. E and F, splenic and mesenteric lymph node CD4+ T cells, respectively. G and H, splenic and mesenteric lymph node CD8+ T cells, respectively.
MyD88−/− mice display a defect in Irgm3 (IGTP) expression in gut mucosa at Day 4 but not Day 7 post infection
Previously it was found that IFN-γ-dependent expression of p47 GTPase family member Irgm3/IGTP is necessary to survive Toxoplasma infection (44). Here, we determined the expression pattern of this molecule in small intestines of orally inoculated MyD88−/− and MyD88+/+ mice. Interestingly, at day 4 post-infection we detected expression of Irgm3 in endothelial cells in basement membrane-submucosal regions of WT mice (Fig.10A and C) and this response was absent in mice lacking MyD88 (Fig. 10B and D). However, at day 7 after infection, small intestine epithelial cells of WT mice were strongly positive for Irgm3, and this response was identical in tissues from MyD88 KO animals (Fig. 10E and F). In sum, during early (day 4) oral infection, IL-12/23p40 and T cell IFN-γ expression was dependent upon MyD88. Similarly, neutrophil recruitment and Irgm3 expression, two responses previously implicated in resistance to Toxoplasma (44, 45), also relied on functional MyD88. Nevertheless, with the exception of IL-12/23p40 expression, each of these immune responses reached normal levels without the need for MyD88 by day 7 after infection.
FIGURE 10.
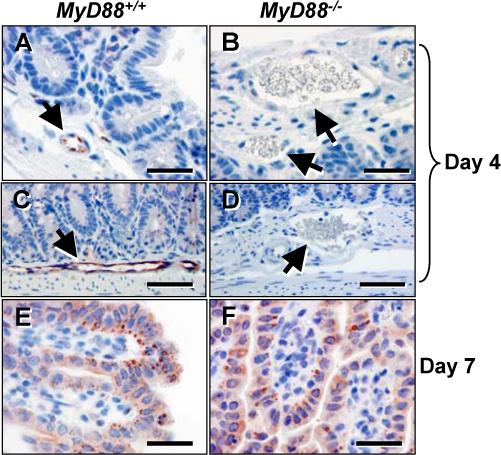
Early endothelial cell expression of Irgm3/IGTP is dependent upon MyD88. MyD88+/+ (Panels A, C and E) and MyD88−/− (panels B, D and F) were orally infected and small intestine tissue was collected 4 (panels A-D) and 7 (panels E and F) days later. Endothelial cells in WT (arrows in panels A and C) but not KO (arrows, panels B and D) stained positive for Irgm3/IGTP. At day 7 post-infection, epithelial cells of both WT (panel E) and KO (panel F) were Irgm3/IGTP positive. The results show one representative mouse out of a total of three from each strain. In panels A, B, E and F the bar denotes 20 μm. In panels C and D the bar indicates 40 μm.
MyD88 is not required for protective immunity to Toxoplasma
Although the delayed T cell IFN-γ response did not require MyD88 during oral infection (Fig. 7), and although CD4+ and CD8+ T lymphocyte expression of activation marker CD69 was identical in the presence and absence of MyD88 (Fig. 9), KO mice were unable to control infection and the animals succumbed to Toxoplasma (Fig. 1). This precluded us from rigorously testing whether MyD88−/− mice developed a protective immune response to challenge infection, which is known to depend upon T cell production of IFN-γ in normal mice (30). To circumvent this limitation, we employed Toxoplasma mutant cps1−1. This RH-derived parasite strain is a uracil auxotroph that invades but does not replicate in host cells. Infection of mice with cps1−1 results in nonpersistent infection that fails to cause pathology associated with wild type T. gondii infection (46). Importantly, cps1−1 vaccination has been shown to induce protective immunity to challenge infection (46, 47). As shown in Fig. 11A, both MyD88+/+ and MyD88−/− mice vaccinated with cps1−1 developed protective immunity to challenge with RH strain parasites. In contrast, nonvaccinated mice succumbed within two weeks of infection with this highly virulent parasite strain. We also vaccinated IL-12/23 p40−/− mice with cps1−1 (Fig. 11B). In this case, the animals failed to develop a protective immunity to RH challenge. Finally, in Fig. 11C, we orally challenged cps1−1-vaccinated mice and found that here too, MyD88 was not required for resistance to lethal ME49 infection.
FIGURE 11.
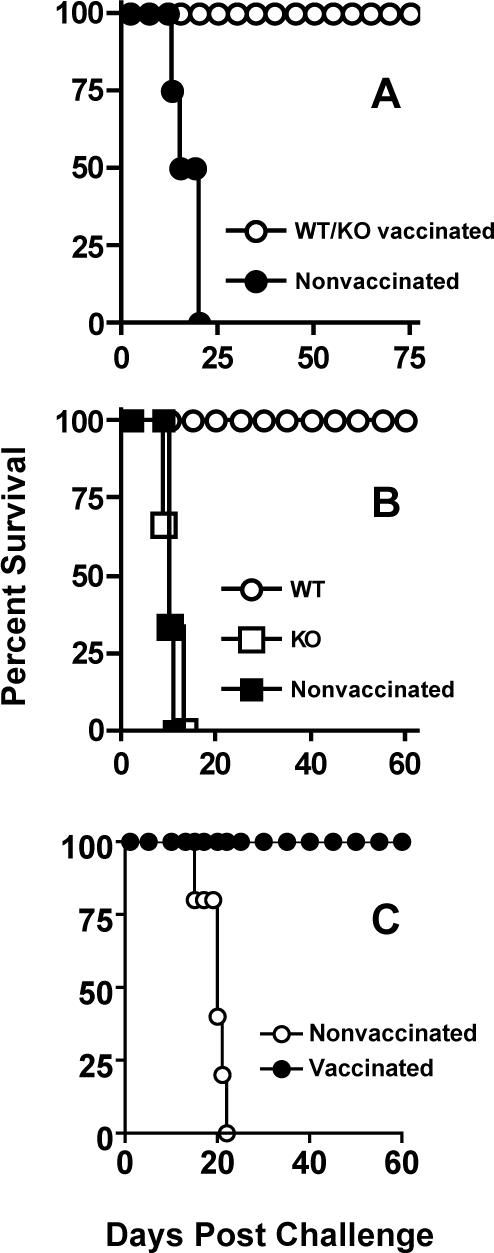
Adaptive immunity to T. gondii is functional in the absence of MyD88. Mice were vaccinated by i. p. injection of Toxoplasma uracil auxotroph cps1−1 then challenged two weeks later by subcutaneous injection of RH strain tachyzoites. In panel A, MyD88 WT and MyD88 KO animals were used. In B, IL-12p40 WT and IL-12p40 KO mice were subjected to vaccination and challenge. In graph C, cps-1−1-vaccinated MyD88 KO animals were subjected to oral infection with 100 ME49 cysts.
We examined naïve (CD62LhiCD44lo), Effector/Effector Memory (CD62LloCD44hi), and Central Memory (CD62LhiCD44hi) splenic T cell populations after cps1−1 vaccination of MyD88+/+ and MyD88−/− animals. As shown in Fig. 12A, there were no major differences between WT and KO populations amongst CD4+ (a and b) or CD8+ (c and d) T cell populations. To distinguish between Effector and Effector Memory populations that both possess a CD62LloCD44hi surface phenotype, we assessed CCR7 that is expressed at high level on Effector but not Effector Memory subpopulations. Here again, we found no difference on these populations amongst WT and KO T cells (Fig. 12B).
FIGURE 12.
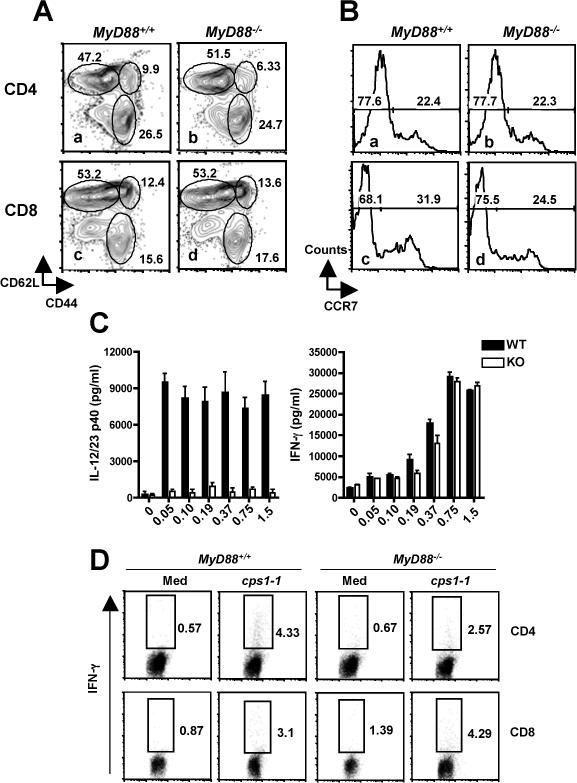
T cell but not IL-12 responses are intact in cps1−1-vaccinated MyD88−/− mice. Panel A, two weeks following cps1−1 vaccination, splenic CD4+ (a and b) and CD8+ (c and d) T lymphocytes from WT (a and c) and MyD88 KO (b and d) animals were assessed for expression of CD62L and CD44 to identify populations of naïve (CD62LhiCD44lo), Effector/Effector Memory (CD62LloCD44hi), and Central Memory (CD62LhiCD44hi) cells. In panel B, levels of CCR7 were evaluated in CD62loCD44hiCD4 and CD8 cells (a and b, and c and d, respectively) from WT (a and c) and KO (b and d) mice. In panel C, two weeks after vaccination with cps1−1, splenocytes from MyD88+/+ and MyD88−/− mice were subjected to in vitro stimulation with the indicated multiplicity of infection (MOI) of cps1−1. Supernatants were assessed for production of IL-12/23p40 and IFN-γ. In panel D, WT and KO splenocytes from vaccinated mice were cultured in medium alone or with cps1−1 tachyzoites (0.5 parasites per cell) for 48 hr. During the last 6 hr, Brefeldin A was added then cells were collected for IFN-γ and CD4/CD8 staining.
In splenocyte cultures from cps1−1 vaccinated mice, production of IL-12/23p40 was barely detectable in cells from MyD88 KO compared to the response of WT cells (Fig. 12C). In contrast, the IFN-γ recall response of MyD88−/− cells was indistinguishable from that of cells from WT littermates (Fig. 12C). We further probed the source of IFN-γ employing intracellular cytokine staining, and found that both CD4+ and CD8+ T cells from KO and WT mice contributed to IFN-γ production during in vitro recall with cps1−1 parasites (Fig. 12D). There was no significant differences between responses of WT compared to KO when responses of several mice were compared. Thus, cps1−1 vaccination of MyD88−/− mice results in protective adaptive immunity that, by the parameters measured, appears identical to that induced in MyD88+/+ animals.
Discussion
TLR-MyD88 signaling is required for resistance to several protozoan pathogens, and parasite ligands that possess TLR activating capability are increasingly being identified (10). For the case of Toxoplasma, parasite profilin and tachyzoite surface GPI anchors are recognized by TLR11 and TLR2/4 respectively (18, 32, 33). In vitro studies using models of i. p. infection or i. p. injection of soluble tachyzoite lysate have shown that MyD88 is required to control infection, and that the immune response to the parasite deviates from a Th1 to a Th2 response in the absence of this TLR adaptor (14, 43).
Here, we examined how lack of MyD88 signaling impacts host defense in animals undergoing oral infection with T. gondii. As during i. p. infection, orally infected MyD88−/− mice displayed early death associated with uncontrolled parasite replication. Like other studies employing the i. p. infection model, the IL-12 response was severely curtailed during oral infection of MyD88 KO mice. Early during oral infection of MyD88−/− animals, we found that neutrophil recruitment to mucosal tissues was defective, as was production of T cell IFN-γ and induction of Irgm3/IGTP, an IFN-γ-inducible p47 GTPase required to survive acute T. gondii infection (44). Importantly, later during acute infection MyD88-independent Th1 responses emerged. The appearence of an apparently intact, albeit delayed, Th1 response in the absence of MyD88 prompted us to examine whether we could elicit protective immunity in the absence of this signaling adaptor. Accordingly, we vaccinated mice with the avirulent cps1−1 Toxoplasma strain. This treatment stimulated protective immunity and strong IFN-γ production independently of MyD88. Taken together, our data suggest MyD88 signaling exerts anti-Toxoplasma activity at a critical early time in infection, but that it is not required to induce strong Th1-based immunity to this parasitic pathogen.
Our results were unexpected because it has been reported that MyD88 signaling plays a role in shaping adaptive immunity to T. gondii. For example, during i. p. Toxoplasma infection, defective T cell IFN-γ production was observed during acute infection (14). Following i. v. injection of soluble tachyzoite lysate antigen, the immune response in MyD88−/− animals deviates from a Th1 to Th2 cytokine response following i. p. injection of soluble tachyzoite lysate antigen (43). In addition, i. p. injection of parasite profilin has been shown to trigger a TLR11/MyD88-dependent Th1 response to the profilin molecule itself (35). Recently, it was found that MyD88 expression in T cells was required for IFN-γ production and survival during i. p. T. gondii infection (48). We do not completely understand why MyD88−/− animals undergoing oral T. gondii infection develop strong, albeit delayed, Th1 responses, when other routes of infection and antigen delivery elicit MyD88-dependent Th1 responses. It seems possible that animals succumb with insufficient time to generate a Th1 response during i. p. infection. Another possibility is that DC populations in mucosal tissues, or possibly signals elicited by gut flora in combination with Toxoplasma, play a role in triggering MyD88-independent Th1 immunity during oral infection (49, 50). For the case of protective immunity induced by cps1−1 vaccination, it is might be that multiple exposure to low doses of nonvirulent tachyzoites induces MyD88-independent adaptive immunity that is not elicited by i. p. infection with a single dose of parasites.
Our results suggest that the major role for MyD88 during oral Toxoplasma infection is in anti-microbial effector activity, either by mediating recruitment of microbicidal effector cells or by direct antimicrobial function. This is in line with several other groups. For example, T cell-dependent antibody responses induced by a panel of adjuvants proceeded normally in the absence of MyD88 and TRIF signaling (51). Similarly, MyD88-deficient mice mount normal protective antibody responses and inflammatory arthritis during Borrelia burgdorferi infection (27). During infection with the fungal pathogen Aspergillus fumigatus Th1 responses in infected airways do not require MyD88, although T-bet induction was enhanced by MyD88 signaling in the draining lymph nodes (52). It was also found that Listeria infection induces a protective CD8+ T cell response in the absence of MyD88 signaling (25, 26). During aerosol infections with Mycobacterium tuberculosis, there is evidence both for and against a role for MyD88 in adaptive immunity, depending on the study (9, 53-56). Regardless, in most of these cases, and as during oral Toxoplasma infection, while acquired immunity could proceed without MyD88, this adaptor molecule was clearly required for pathogen control during innate immunity.
Of relevance to the present work is a recent study that examined responses to oral Toxoplasma infection in the absence of TLR9, a receptor whose signaling pathway depends upon MyD88. In that study, mice were more susceptible to infection in the absence of TLR9, and this was associated with decreased T cell IFN-γ responses (57). While IL-12 was not examined, it is interesting to note that in the previous study, while IFN-γ responses were lower in the absence of TLR9, substantial levels of this cytokine were nevertheless still produced.
At present we do not know why MyD88−/− mice are unable to control T. gondii infection. Recruitment of neutrophils to mucosal tissues was defective in the knockout mice during early stages of infection, and this possibly underlies the susceptibility of these animals. Others have also found that neutrophil recruitment during infection relies upon MyD88-dependent signaling (58, 59). Previously, we reported that depletion of neutrophils with an anti-Gr-1 antibody resulted in susceptibility to oral T. gondii infection (45). Therefore, it is possible that defective neutrophil recruitment during early infection allows increased parasite survival in the intestine, ultimately leading to death of the animals. Nevertheless, we interpret results obtained using anti-Gr-1 mAb with caution, since the Gr-1 epitope is also expressed by other types of myeloid cells (60).
Our data for the first time show tissue localization of IFN-γ-inducible Irgm3/IGTP, a molecule required to survive acute Toxoplasma infection (44). Interestingly, Irgm3/IGTP was expressed in endothelial cells of the intestinal submucosa in an MyD88-dependent manner during early infection. Later, this p47 GTPase was highly expressed in epithelial cells of the small intestine in both MyD88+/+ and MyD88−/− mice. It is difficult to understand how early MyD88-dependent Irgm3/IGTP expression confined to endothelial cells could contribute to resistance to T. gondii infection, which is uniformly spread throughout mucosal tissues. Although the function of Irgm3/IGTP and other p47 GTPases is not well understood, these molecules have been implicated in autophagic destruction of the parasitophorous vacuole in tachyzoite-infected macrophages and astrocytes (61, 62). However, we have so far not detected differences in IFN-γ-mediated macrophage killing of tachyzoites in the presence and absence of MyD88.
Strong MyD88-independent T cell IFN-γ responses were elicited during both oral ME49 infection and cps1−1 vaccination. Inasmuch as production of IL-12 in both cases was minimal, this raises the question of how T cell IFN-γ responses are generated in these cases. Two related possibilities are that residual MyD88-independent IL-12 is sufficient to drive Th1 differentiation, or that more robust MyD88-independent IL-12 responses occur at sites other than the draining lymph nodes and spleen that were examined in this study. It is also possible that Th1 responses are triggered independently of IL-12 in the absence of MyD88. Indeed, when we administered depleting anti-IL-12 Ab, while the Day 4 MyD88-dependent IFN-γ response was severely curtailed, there was no effect on the Day 7 MyD88-independent response. Nevertheless, at least for the case of cps1−1-induced immunity, absence of IL-12/23p40 resulted in lack of a protective response, a result that argues for IL-12 as a mediator of protection in cps1−1 vaccinated MyD88−/− animals. In this regard, we have found that bone marrow-derived macrophages infected with the RH Toxoplasma strain produce IL-12 independently of signaling through MyD88 (63), yet the same parasite strain clearly triggers MyD88-dependent IL-12 in splenic DC (our unpublished results and refs. (14, 18). Further work will be required to determine host mediators involved in generating MyD88-independent Th1 responses during Toxoplasma infection.
In sum, our results argue that MyD88-dependent signaling is crucial in anti-microbial effector function against T. gondii. Nevertheless, the view that this adaptor molecule plays an essential role in bridging innate and adaptive immunity to Toxoplasma may require revision. Other signaling molecules and recognition systems are also likely to be important determinants of adaptive immunity to this parasite. Identifying such molecules will provide important insight into immune recognition of Toxoplasma and other microbial pathogens.
Acknowledgements
We are grateful to S. Akira for supplying us with MyD88−/− mice, and P. Fisher for assistance with immunohistochemistry.
Footnotes
Publisher's Disclaimer: This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2−3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of an author. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
This work was supported by PHS grants AI47888 (EYD), AI57831 and a VA Merit Review grant (GAT), AI14193 (DJB), and a fellowship from the King Anandamahidol Foundation (WS).
Abbreviations used in this paper: KO, Knockout; MPO, myeloperoxidase; STAg, soluble tachyzoite antigen; TLR, Toll-like receptor; WT, wild-type.
References
- 1.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 2.Uematsu S, Akira S. Toll-Like receptors (TLRs) and their ligands. Handb Exp Pharmacol. 2008:1–20. doi: 10.1007/978-3-540-72167-3_1. [DOI] [PubMed] [Google Scholar]
- 3.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- 4.Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 5.O'Neill L,A, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 6.Edelson BT, Unanue ER. MyD88-dependent but Toll-like receptor 2-independent innate immunity to Listeria: no role for either in macrophage listericidal activity. J Immunol. 2002;169:3869–3875. doi: 10.4049/jimmunol.169.7.3869. [DOI] [PubMed] [Google Scholar]
- 7.Macedo GC, Magnani DM, Carvalho NB, Bruna-Romero O, Gazzinelli RT, Oliveira SC. Central role of MyD88-dependent dendritic cell maturation and proinflammatory cytokine production to control Brucella abortus infection. J Immunol. 2008;180:1080–1087. doi: 10.4049/jimmunol.180.2.1080. [DOI] [PubMed] [Google Scholar]
- 8.Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol. 2000;165:5392–5396. doi: 10.4049/jimmunol.165.10.5392. [DOI] [PubMed] [Google Scholar]
- 9.Scanga CA, Bafica A, Feng CG, Cheever AW, Hieny S, Sher A. MyD88-deficient mice display a profound loss in resistance to Mycobacterium tuberculosis associated with partially impaired Th1 cytokine and nitric oxide synthase 2 expression. Infect Immun. 2004;72:2400–2404. doi: 10.1128/IAI.72.4.2400-2404.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gazzinelli RT, Denkers EY. Protozoan encounters with Toll-like receptor signalling pathways: implications for host parasitism. Nat Rev Immunol. 2006;6:895–906. doi: 10.1038/nri1978. [DOI] [PubMed] [Google Scholar]
- 11.Adachi K, Tsutsui H, Kashiwamura S, Seki E, Nakano H, Takeuchi O, Takeda K, Okumura K, Van Kaer L, Okamura H, Akira S, Nakanishi K. Plasmodium berghei infection in mice induces liver injury by an IL-12- and toll-like receptor/myeloid differentiation factor 88-dependent mechanism. J Immunol. 2001;167:5928–5934. doi: 10.4049/jimmunol.167.10.5928. [DOI] [PubMed] [Google Scholar]
- 12.Campos MA, Closel M, Valente EP, Cardoso JE, Akira S, Alvarez-Leite JI, Ropert C, Gazzinelli RT. Impaired production of proinflammatory cytokines and host resistance to acute infection with Trypanosoma cruzi in mice lacking functional myeloid differentiation factor 88. J. Immunol. 2004;172:1711–1718. doi: 10.4049/jimmunol.172.3.1711. [DOI] [PubMed] [Google Scholar]
- 13.Muraille E, De Trez C, Brait M, De Baetselier P, Leo O, Carlier Y. Genetically resistant mice lacking MyD88-adapter protein display a high susceptibility to Leishmania major infection associated with a polarized Th2 response. J Immunol. 2003;170:4237–4241. doi: 10.4049/jimmunol.170.8.4237. [DOI] [PubMed] [Google Scholar]
- 14.Scanga CA, Aliberti J, Jankovic D, Tilloy F, Bennouna S, Denkers EY, Medzhitov R, Sher A. Cutting edge: MyD88 is required for resistance to Toxoplasma gondii infection and regulates parasite-induced IL-12 production by dendritic cells. J. Immunol. 2002;168:5997–6001. doi: 10.4049/jimmunol.168.12.5997. [DOI] [PubMed] [Google Scholar]
- 15.Hitziger N, Dellacasa I, Albiger B, Barragan A. Dissemination of Toxoplasma gondii to immunoprivileged organs and role of Toll/interleukin-1 receptor signalling for host resistance assessed by in vivo bioluminsecence imaging. Cell. Micro. 2005;6:837–848. doi: 10.1111/j.1462-5822.2005.00517.x. [DOI] [PubMed] [Google Scholar]
- 16.Campos MAS, Almeida IC, Takeuchi O, Akira S, Valente EP, Procópio DO, Travassos LR, Smith JA, Golenbock DT, Gazzinelli RT. Activation of Toll-Like Receptor-2 by glycosylphosphatidylinositol anchors from a protozoan parasite. J. Immunol. 2001;167:416–423. doi: 10.4049/jimmunol.167.1.416. [DOI] [PubMed] [Google Scholar]
- 17.Parroche P, Lauw FN, Goutagny N, Latz E, Monks BG, Visintin A, Halmen KA, Lamphier M, Olivier M, Bartholomeu DC, Gazzinelli RT, Golenbock DT. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc Natl Acad Sci U S A. 2007;104:1919–1924. doi: 10.1073/pnas.0608745104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yarovinsky F, Zhang D, Anderson JF, Bannenberg GL, Serhan CN, Hayden MS, Hieny S, Sutterwala FS, Flavell RA, Ghosh S, Sher A. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science. 2005;308:1626–1629. doi: 10.1126/science.1109893. [DOI] [PubMed] [Google Scholar]
- 19.Bafica A, Santiago HC, Goldszmid R, Ropert C, Gazzinelli RT, Sher A. Cutting edge: TLR9 and TLR2 signaling together account for MyD88-dependent control of parasitemia in Trypanosoma cruzi infection. J Immunol. 2006;177:3515–3519. doi: 10.4049/jimmunol.177.6.3515. [DOI] [PubMed] [Google Scholar]
- 20.Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat Immunol. 2005;6:769–776. doi: 10.1038/ni1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defence. Nat Rev Immunol. 2007;7:179–190. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- 22.Kabelitz D, Medzhitov R. Innate immunity--cross-talk with adaptive immunity through pattern recognition receptors and cytokines. Curr Opin Immunol. 2007;19:1–3. doi: 10.1016/j.coi.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 23.Pasare C, Medzhitov R. Toll-dependent control mechanisms of CD4 T cell activation. Immunity. 2004;21:733–741. doi: 10.1016/j.immuni.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 24.Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R. Toll-like receptors control activation of adaptive immune responses. Nat Immunol. 2001;2:947–950. doi: 10.1038/ni712. [DOI] [PubMed] [Google Scholar]
- 25.Way SS, Kollmann TR, Hajjar AM, Wilson CB. Cutting edge: protective cell-mediated immunity to Listeria monocytogenes in the absence of myeloid differentiation factor 88. J Immunol. 2003;171:533–537. doi: 10.4049/jimmunol.171.2.533. [DOI] [PubMed] [Google Scholar]
- 26.Kursar M, Mittrucker HW, Koch M, Kohler A, Herma M, Kaufmann SH. Protective T cell response against intracellular pathogens in the absence of Toll-like receptor signaling via myeloid differentiation factor 88. Int Immunol. 2004;16:415–421. doi: 10.1093/intimm/dxh047. [DOI] [PubMed] [Google Scholar]
- 27.Bolz DD, Sundsbak RS, Ma Y, Akira S, Kirschning CJ, Zachary JF, Weis JH, Weis JJ. MyD88 plays a unique role in host defense but not arthritis development in Lyme disease. J Immunol. 2004;173:2003–2010. doi: 10.4049/jimmunol.173.3.2003. [DOI] [PubMed] [Google Scholar]
- 28.Guay HM, Andreyeva TA, Garcea RL, Welsh RM, Szomolanyi-Tsuda E. MyD88 is required for the formation of long-term humoral immunity to virus infection. J Immunol. 2007;178:5124–5131. doi: 10.4049/jimmunol.178.8.5124. [DOI] [PubMed] [Google Scholar]
- 29.Zhou S, Kurt-Jones EA, Mandell L, Cerny A, Chan M, Golenbock DT, Finberg RW. MyD88 is critical for the development of innate and adaptive immunity during acute lymphocytic choriomeningitis virus infection. Eur J Immunol. 2005;35:822–830. doi: 10.1002/eji.200425730. [DOI] [PubMed] [Google Scholar]
- 30.Denkers EY, Gazzinelli RT. Regulation and function of T cell-mediated immunity during Toxoplasma gondii infection. Clin. Microbiol. Rev. 1998;11:569–588. doi: 10.1128/cmr.11.4.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peterson E, Liesenfeld O. Clinical disease and diagnostics. In: Weiss LM, Kim K, editors. Toxoplasma gondii. The model apicomplexan: Perspectives and methods. Academic Press; Amsterdam: 2007. pp. 81–100. [Google Scholar]
- 32.Debierre-Grockiego F, Campos MA, Azzouz N, Schmidt J, Bieker U, Resende MG, Mansur DS, Weingart R, Schmidt RR, Golenbock DT, Gazzinelli RT, Schwarz RT. Activation of TLR2 and TLR4 by glycosylphosphatidylinositols derived from Toxoplasma gondii. J Immunol. 2007;179:1129–1137. doi: 10.4049/jimmunol.179.2.1129. [DOI] [PubMed] [Google Scholar]
- 33.Plattner F, Yarovinsky F, Romero S, Didry D, Carlier MF, Sher A, Soldati-Favre D. Toxoplasma profilin is essential for host cell invasion and TLR dependent induction of interleukin-12. Cell Host and Microbe. 2008;14:77–87. doi: 10.1016/j.chom.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 34.Yarovinsky F, Sher A. Toll-like receptor recognition of Toxoplasma gondii. Int J Parasitol. 2006;36:255–259. doi: 10.1016/j.ijpara.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 35.Yarovinsky F, Kanzler H, Hieny S, Coffman RL, Sher A. Toll-like receptor recognition regulates immunodominance in an antimicrobial CD4+ T cell response. Immunity. 2006;25:655–664. doi: 10.1016/j.immuni.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 36.Taylor GA, Jeffers M, Largaespada DA, Jenkins NA, Copeland NG, Woude GF. Identification of a novel GTPase, the inducibly expressed GTPase, that accumulates in response to interferon gamma. J Biol Chem. 1996;271:20399–20405. doi: 10.1074/jbc.271.34.20399. [DOI] [PubMed] [Google Scholar]
- 37.Sukhumavasi W, Egan CE, Denkers EY. Mouse neutrophils require JNK2 MAPK for Toxoplasma gondii-induced IL-12p40 and CCL2/MCP-1 release. J Immunol. 2007;179:3570–3577. doi: 10.4049/jimmunol.179.6.3570. [DOI] [PubMed] [Google Scholar]
- 38.Kim L, Butcher BA, Denkers EY. Toxoplasma gondii interferes with lipopolysaccharide-induced mitogen-activated protein kinase activation by mechanisms distinct from endotoxin tolerance. J. Immunol. 2004;172:3003–3010. doi: 10.4049/jimmunol.172.5.3003. [DOI] [PubMed] [Google Scholar]
- 39.Gavrilescu LC, Butcher BA, Del Rio L, Taylor GA, Denkers EY. STAT1 is essential for antimicrobial function but dispensable for gamma interferon production during Toxoplasma gondii infection. Infect. Immun. 2004;72:1257–1264. doi: 10.1128/IAI.72.3.1257-1264.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bliss SK, Butcher BA, Denkers EY. Rapid recruitment of neutrophils with prestored IL-12 during microbial infection. J. Immunol. 2000;165:4515–4521. doi: 10.4049/jimmunol.165.8.4515. [DOI] [PubMed] [Google Scholar]
- 41.Mordue DG, Sibley LD. A novel population of Gr-1+-activated macrophages induced during acute toxoplasmosis. J. Leuk. Biol. 2003;74:1–11. doi: 10.1189/jlb.0403164. [DOI] [PubMed] [Google Scholar]
- 42.Suzuki Y, Orellana MA, Schreiber RD, Remington JS. Interferon-γ: The major mediator of resistance against Toxoplasma gondii. Science. 1988;240:516–518. doi: 10.1126/science.3128869. [DOI] [PubMed] [Google Scholar]
- 43.Jankovic D, Kullberg MC, Hieny S, Caspar P, Collazo CM, Sher A. In the absence of IL-12, CD4+ T cell responses to intracellular pathogens fail to default to a Th2 pattern and are host protective in an IL-10−/− setting. Immunity. 2002;16:429–439. doi: 10.1016/s1074-7613(02)00278-9. [DOI] [PubMed] [Google Scholar]
- 44.Taylor GA, Collazo CM, Yap GS, Nguyen K, Gregorio TA, Taylor LS, Eagleson B, Secret L, Southon EA, Reid SW, Tessarollo L, Bray M, McVicar DW, Komschlies KL, Young HA, Biron CA, Sher A, Vande Woude GF. Pathogen-specific loss of host resistance in mice lacking the IFN-γ-inducible gene IGTP. Proc. Natl. Acad. Sci. USA. 2000;97:751–755. doi: 10.1073/pnas.97.2.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bliss SK, Zhang Y, Denkers EY. Murine neutrophil stimulation by Toxoplasma gondii antigen drives high level production of IFN-γ-independent IL-12. J. Immunol. 1999;163:2081–2088. [PubMed] [Google Scholar]
- 46.Fox BA, Bzik DJ. De novo pyrimidine biosynthesis is required for virulence of Toxoplasma gondii. Nature. 2002;415:926–929. doi: 10.1038/415926a. [DOI] [PubMed] [Google Scholar]
- 47.Shaw MH, Freeman GJ, Scott MF, Fox BA, Bzik DJ, Belkaid Y, Yap GS. Tyk2 negatively regulates adaptive Th1 immunity by mediating IL-10 signaling and promoting IFN-gamma-dependent IL-10 reactivation. J Immunol. 2006;176:7263–7271. doi: 10.4049/jimmunol.176.12.7263. [DOI] [PubMed] [Google Scholar]
- 48.Larosa DF, Stumhofer JS, Gelman AE, Rahman AH, Taylor DK, Hunter CA, Turka LA. T cell expression of MyD88 is required for resistance to Toxoplasma gondii. Proc Natl Acad Sci U S A. 2008;105:3855–3860. doi: 10.1073/pnas.0706663105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heimesaat MM, Bereswill S, Fischer A, Fuchs D, Struck D, Niebergall J, Jahn HK, Dunay IR, Moter A, Gescher DM, Schumann RR, Gobel UB, Liesenfeld O. Gram-Negative Bacteria Aggravate Murine Small Intestinal Th1-Type Immunopathology following Oral Infection with Toxoplasma gondii. J Immunol. 2006;177:8785–8795. doi: 10.4049/jimmunol.177.12.8785. [DOI] [PubMed] [Google Scholar]
- 50.Kelsall BL, Leon F. Involvement of intestinal dendritic cells in oral tolerance, immunity to pathogens, and inflammatory bowel disease. Immunol Rev. 2005;206:132–148. doi: 10.1111/j.0105-2896.2005.00292.x. [DOI] [PubMed] [Google Scholar]
- 51.Gavin AL, Hoebe K, Duong B, Ota T, Martin C, Beutler B, Nemazee D. Adjuvant-enhanced antibody responses in the absence of toll-like receptor signaling. Science. 2006;314:1936–1938. doi: 10.1126/science.1135299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rivera A, Ro G, Van Epps HL, Simpson T, Leiner I, Sant'Angelo DB, Pamer EG. Innate immune activation and CD4+ T cell priming during respiratory fungal infection. Immunity. 2006;25:665–675. doi: 10.1016/j.immuni.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 53.Bafica A, Scanga CA, Feng CG, Leifer C, Cheever A, Sher A. TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J Exp Med. 2005;202:1715–1724. doi: 10.1084/jem.20051782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fremond CM, Yeremeev V, Nicolle DM, Jacobs M, Quesniaux VF, Ryffel B. Fatal Mycobacterium tuberculosis infection despite adaptive immune response in the absence of MyD88. J Clin Invest. 2004;114:1790–1799. doi: 10.1172/JCI21027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Holscher C, Reiling N, Schaible UE, Holscher A, Bathmann C, Korbel D, Lenz I, Sonntag T, Kroger S, Akira S, Mossmann H, Kirschning CJ, Wagner H, Freudenberg M, Ehlers S. Containment of aerogenic Mycobacterium tuberculosis infection in mice does not require MyD88 adaptor function for TLR2, -4 and -9. Eur J Immunol. 2008;38:680–694. doi: 10.1002/eji.200736458. [DOI] [PubMed] [Google Scholar]
- 56.Reiling N, Ehlers S, Holscher C. MyDths and un-TOLLed truths: Sensor, instructive and effector immunity to tuberculosis. Immunol Lett. 2008;116:15–23. doi: 10.1016/j.imlet.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 57.Minns LA, Menard LC, Foureau DM, Darche S, Ronet C, Mielcarz DW, Buzoni-Gatel D, Kasper LHM. TLR9 Is Required for the Gut-Associated Lymphoid Tissue Response following Oral Infection of Toxoplasma gondii. J Immunol. 2006;176:7589–7597. doi: 10.4049/jimmunol.176.12.7589. [DOI] [PubMed] [Google Scholar]
- 58.Lebeis SL, Bommarius B, Parkos CA, Sherman MA, Kalman D. TLR signaling mediated by MyD88 is required for a protective innate immune response by neutrophils to Citrobacter rodentium. J Immunol. 2007;179:566–577. doi: 10.4049/jimmunol.179.1.566. [DOI] [PubMed] [Google Scholar]
- 59.Rodriguez N, Fend F, Jennen L, Schiemann M, Wantia N, Prazeres da Costa CU, Durr S, Heinzmann U, Wagner H, Miethke T. Polymorphonuclear neutrophils improve replication of Chlamydia pneumoniae in vivo upon MyD88-dependent attraction. J Immunol. 2005;174:4836–4844. doi: 10.4049/jimmunol.174.8.4836. [DOI] [PubMed] [Google Scholar]
- 60.Egan CE, Sukhumavasi W, Bierly AL, Denkers EY. Understanding the multiple functions of Gr-1+ cell subpopulations during microbial infection. Immunologic Res. 2008;40:35–48. doi: 10.1007/s12026-007-0061-8. [DOI] [PubMed] [Google Scholar]
- 61.Halonen SK, Taylor GA, Weiss LM. Gamma interferon-induced inhibition of Toxoplasma gondii in astrocytes is mediated by IGTP. Infect Immun. 2001;69:5573–5576. doi: 10.1128/IAI.69.9.5573-5576.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ling YM, Shaw MH, Ayala C, Coppens I, Taylor GA, Ferguson DJ, Yap GS. Vacuolar and plasma membrane stripping and autophagic elimination of Toxoplasma gondii in primed effector macrophages. J Exp Med. 2006;203:2063–2071. doi: 10.1084/jem.20061318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim L, Butcher BA, Lee CW, Uematsu S, Akira S, Denkers EY. Toxoplasma gondii genotype determines MyD88-dependent signaling in infected macrophages. J. Immunol. 2006;177:2584–2591. doi: 10.4049/jimmunol.177.4.2584. [DOI] [PubMed] [Google Scholar]