

Abstract
Hepatitis C virus (HCV) is a major human pathogen that causes serious illness including acute and chronic hepatitis, cirrhosis, and hepatocellular carcinoma. Using a mass spectrometry-based proteomics approach, we have identified 175 proteins from a cell culture supernatant fraction containing HCV JFH1 virus, among which fatty acid synthase (FASN), the multifunctional enzyme catalyzing the de novo synthesis of fatty acids, was confirmed to be highly enriched. Subsequent studies showed that FASN expression increased in the human hepatoma cell line, Huh7 or its derivative, upon HCV infection. Blocking FASN activity by a pharmacological inhibitor C75 led to decreased HCV production. Reduction of FASN by RNA interference (RNAi) suppressed viral replication in both replicon and infection systems. Remarkably, FASN appeared to be selectively required for the expression of claudin-1 (CLDN1), a tight junction (TJ) protein that was recently identified as an entry co-receptor for HCV (1), but not for the expression of another HCV co-receptor, CD81. The decrease in CLDN1 expression resulting from FASN inhibition was accompanied by a decrease in transepithelial electric resistance (TER) of Huh7 cells, implying a reduction in the relative tightness of the cell monolayer. Consequently, the entry of HIV-HCV pseudotypes (HCVpp) was significantly inhibited in C75 treated Huh7 cells. Conclusion: As far as we know, this is the first line of evidence that demonstrates that HCV infection directly induces FASN expression, and thus suggests a possible mechanism by which HCV infection alters the cellular lipid profile and causes diseases such as steatosis.
Keywords: Hepatitis C virus, fatty acid synthase, viral replication, viral entry, Claudin-1
Nearly 200 million people worldwide are infected by the Hepatitis C virus (HCV), resulting in serious complications such as liver diseases and cancer. This enveloped, positive-stranded RNA virus is classified in the hepacivirus genus of the Flaviviridae family (2). The 10 kb viral genome comprises a large open reading frame encoding a precursor polyprotein of approximately 3000 amino acids. Co- and posttranslational cleavages of this polyprotein generate at least ten viral proteins, including a core protein, two glycoproteins (GP), E1 and E2, p7, and a number of nonstructural proteins (3, 4). Nearly all of these viral proteins form complexes with host factors in order to carry out the viral life cycle. It is also believed that these viral-host interactions contribute via several complex mechanisms to such maladies as inflammation, steatosis, fibrosis, altered lipid metabolism, insulin resistance, and hepatocellular carcinoma (HCC) (5).
Enveloped viruses can incorporate both viral and host proteins into their membrane(s) or inside the envelope during virus assembly and budding. Infection of host cells by viruses is also likely to alter the profile of secreted proteins (secretome). Knowledge of the protein composition of the infectious viral particle and the secretome provides important information for functional studies because subsequent characterization can be designed to focus on specific targets. In recent years, liquid chromatography (LC)-coupled online with tandem mass spectrometry (MS/MS), has been successfully applied in several cases to analyze compositions of purified virions, leading to the identification of many previously unknown components of viral particles (6). In this study we collected a supernatant fraction_containing infectious JFH1 HCV virions (HCVcc) following ultracentrifugation and profiled the protein content by LC-MS/MS. 175 proteins were identified, including FASN which was verified to be upregulated during HCV infection of Huh7.5.1 cell line. Subsequent functional analysis revealed a critical role of FASN in regulating HCV infection.
Materials and Methods
Cells and Reagents
The human kidney epithelial cell line HEK293T (CRL-11268) was purchased from the American Type Culture Collection (ATCC). The human liver cell line Huh7 was obtained from Apath Inc., with the permission of Dr. Charles Rice (Rockefeller University). The Huh7.5.1 line which was generated from a cured HCV replicon cell line was kindly provided by Dr. Francis Chisari (Scripps Research Institute). The full-length HCV Genotype 1b-replicon-containing cell line (2-3+) and the cured cell line (2-3c) were generous gifts from Dr. Stanley Lemon (University of Texas Medical Branch, Galveston, TX) (7). Genotype 2a replicon cells were established by transfecting Huh7 cells with the full-length genomic luciferase-JFH1 RNA (obtained from Dr. Ralf Bartenschlager, University of Heidelberg, Germany) and selected for clones that only supported viral replication but not the production of infectious virus. All cell lines were maintained in DMEM supplemented with 1% penicillin and streptomycin (Invitrogen, Carlsbad, CA), 1% NEAA, and 10% fetal bovine serum (Hyclone, Logan, Utah) and routinely checked to ensure they were free of mycoplasma contamination. Antibodies were obtained from Zymed/Invitrogen (anti-CLDN1, clones 2H10D10 and JAY.8, anti-ZO-1, clone ZO1-1A12, and FITC- or Rhodamine-conjugated secondary antibodies), Affinity BioReagents (HCV Core, Golden, CO), BD Biosciences (FASN, San Diego, CA) and Sigma (β-actin, Saint Louis, MO). C75 was purchased from Axxora LLC (San Diego, CA) and dissolved in DMSO. Orlistat and lentiviral-based shRNAi constructs targeting human FASN were purchased from Sigma. Draq5 was purchased from Biostatus (Shepshed, Leicestershire, United Kingdom).
Production of pseudotyped virus
In order to produce the HCVpp and VSVpp, HEK293T cells were transfected using lipofectamine 2000 (Invitrogen) with a DNA transfection mixture composed of pNL-4.3-Luc-E-R-, phCMV-HCV E1E2, or pHEF-VSV-G (expressing the VSV-G Env protein) using a procedure described elsewhere (8).
Quantification of HCV RNA by Real-time RT-PCR
RNA was isolated from the cytosol or supernatants of infected cells using Qiagen RNAeasy kit following the manufacturer’s instructions. Five microliters of RNA was analyzed using a real-time RT-PCR assay at the Division of Molecular Diagnostics of the University of Pittsburgh Medical Center using an internally developed protocol. Random primers were used for initial reverse-transcription, and pairs of gene specific primers covering a 50bp region in the 5’ IRES were used for real-time PCR. The results are interpreted as international units/mL (IU/ml) for RNA derived from supernatant or international units/million cells for cytoplasmic RNA. This number is divided by 1.6, giving the equivalent copy number of HCV genome RNA.
Transepithelial electric resistance measurement
One thousand Huh7 or Caco-2 cells were seeded on a 24-well transwell plate (Corning, 6.5-mm membrane diameter, 0.4-μm pore size). After treatments with inhibitors, resistance was measured daily following a published procedure (33).
Flow cytometric analysis
Cells stained with CD81 monoclonal antibody were analyzed on a Coulter XL flow cytometer. Acquired data were analyzed and plotted using WinMDI 2.9 software.
Results
Identification of proteins from the HCV virion-containing supernatant fraction
In order to purify HCV virions, we produced virus in large volumes and then passed the virus containing supernatant through a concentrator column to remove proteins smaller than 100 kDa. Concentrated viruses were pelleted by ultracentrifugation over a 25% sucrose cushion. For the negative control, the same amount of culture medium from uninfected cells was prepared in parallel. Virus and control pellets were separately dissolved in Laemmli sample buffer and resolved by one-dimensional SDS-PAGE. After staining, a dozen distinct bands were noticed exclusively in the HCV containing sample (Fig. 1A). These bands were excised and “in-gel” digested by trypsin. Following LC-MS/MS analysis of the extracted peptides, 175 proteins were positively identified by strict criteria, including a requirement of observing two peptides per protein matching the database (Supplementary Table 1). Analysis of the gene ontologic classifications of the identified proteins revealed that most were involved in cellular metabolisms, biogenesis, transport, etc (Fig. 1B). In order to validate the MS results, we performed western blotting and confirmed that both FASN and apolipoprotein E (ApoE) were indeed highly enriched in the sample containing HCV (Fig. 1C). ApoE is a known virion-associated protein and is required for the production of infectious virus (9). It should be pointed out that these data represent only a partial proteomics analysis. A subtractive analysis comparing the protein profiles of both samples may likely yield more protein identifications.
Figure 1. Identification of supernatant proteins that co-fractionated with HCV virions.

(A) Equal volumes of supernatant collected from HCVcc-infected and mock-infected Huh7.5.1 cells were subjected to filtration concentration and sucrose cushion ultracentrifugation. The pellets were dissolved in Laemmli buffer and resolved on a 10% one-dimensional SDS-PAGE gel. Eight representative protein bands differentially expressed between the samples are indicated with arrows and the red arrow points to the position of FASN. (B) Classification by gene ontology of the co-purified proteins with HCV virions in terms of their biological function and subcellular localization. (C) Protein pellets prepared from A were analyzed by western blotting and the presence of ApoE, FASN, and HCV Core protein was only found in the supernatant of HCVcc-infected cells.
FASN was upregulated following HCV infection
Our initial functional characterization was focused on FASN, the critical enzyme that catalyzes de novo lipogenesis. FASN is known to be upregulated in many cancers, implying an interesting role in tumorigenesis (10). The FASN gene encodes a polypeptide that comprises seven functional domains including an acyl-carrier protein (ACP) that work together to catalyze fatty acid synthesis (11). The primary products of FASN activity are the 16-carbon fatty acid palmitate, as well as the 14-carbon myristate and the 18-carbon stearate fatty acids (FAs). FAs have previously been implicated in the regulation of HCV production (12, 13). Since HCV infection is known to be associated with accumulation of fat in liver cells (i.e. steatosis), we first profiled FASN expression during HCV infection. Infection of Huh7 cells with the JFH1 virus (genotype 2a) for seven days led to considerable increase in FASN protein abundance (Fig. 2A). The level of FASN expression was also weakly increased in a replicon system that harbors a full-length genotype 1b genome (Fig. 2B). To our knowledge, this is the first evidence that HCV infection directly upregulates FASN.
Figure 2. Upregulation of FASN in HCVcc infected cells and replicon cells.
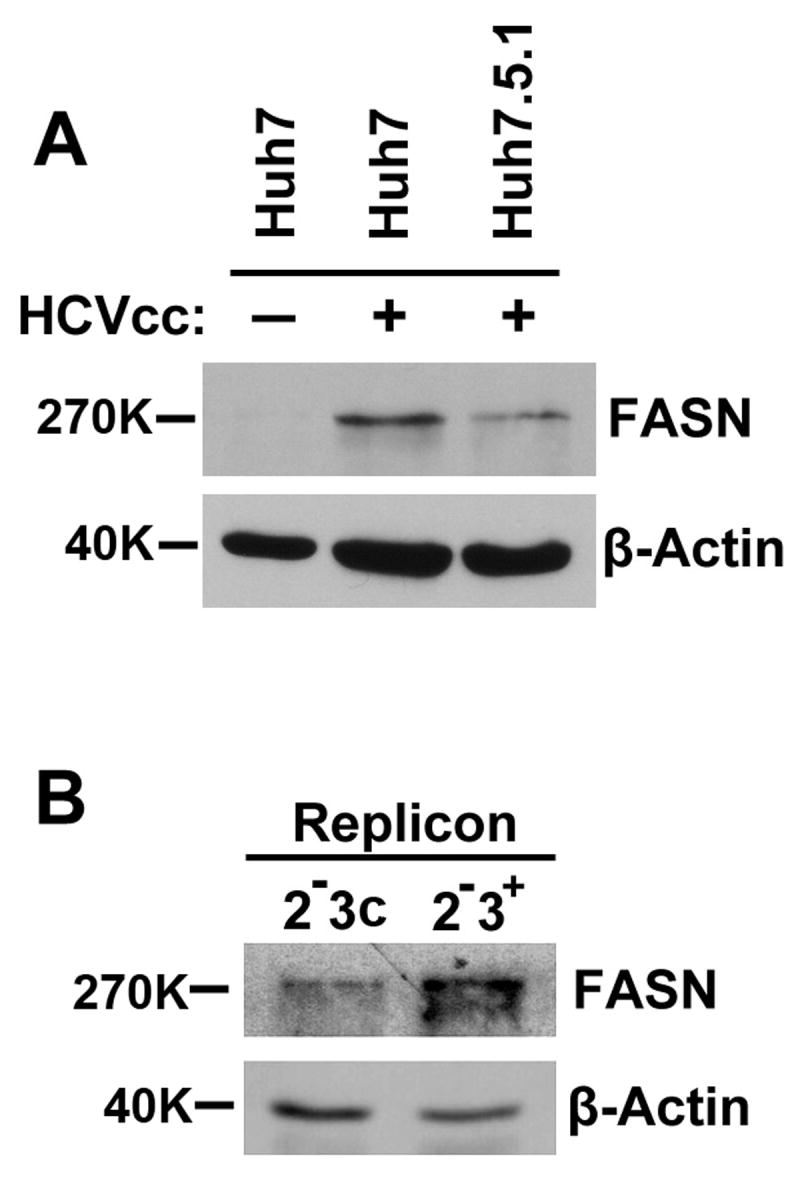
Cell lysates were prepared from Naïve Huh7 and HCVcc infected Huh7 or Huh7.5.1 cells (A) and from HCV replicon cells harboring a full-length genotype 1b genome (2-3+) or the cured cells (2-3c) (B). 20 μg cell lysates (30 μg for replicon cells) were evaluated by Western blotting using an anti-FASN monoclonal antibody. Detection of β-actin was indicative of protein loading. The basal level of FASN in naïve Huh7.5.1 was virtually undetectable (data not shown) by western blotting.
FASN is required for effective viral replication
To better understand the role of FASN in the regulation of HCV infection, we used a highly specific β-ketoacyl synthase domain FASN inhibitor C75 to block its function (14). Because C75 treatment at concentrations equal or lower than 75 μM did not cause significant cell death (Supplementary Table 2), we chose 50 μM as the working concentration throughout this study as this dose is comparable to what has been reported to specifically inhibit the FASN activity (15). Treatment of JFH1 infected cells with C75 reduced the cellular HCV core protein production by 3-4 folds (Fig. 3A). The inhibitory effect was observed also in HCV genotype 1b and 2a replicon systems at both protein and RNA levels (Fig. 3B&C), indicating that C75 treatment suppresses the viral replication (Fig. 3C). Furthermore, we observed a significant drop in the infectious titer of the released virus from C75-treated Huh7 cells that have been infected by the JFH1 virus (Fig. 3D&E). Together, these results demonstrated that C75 treatment inhibited HCV production in cell culture systems.
Figure 3. FASN is required for effective viral replication and production.
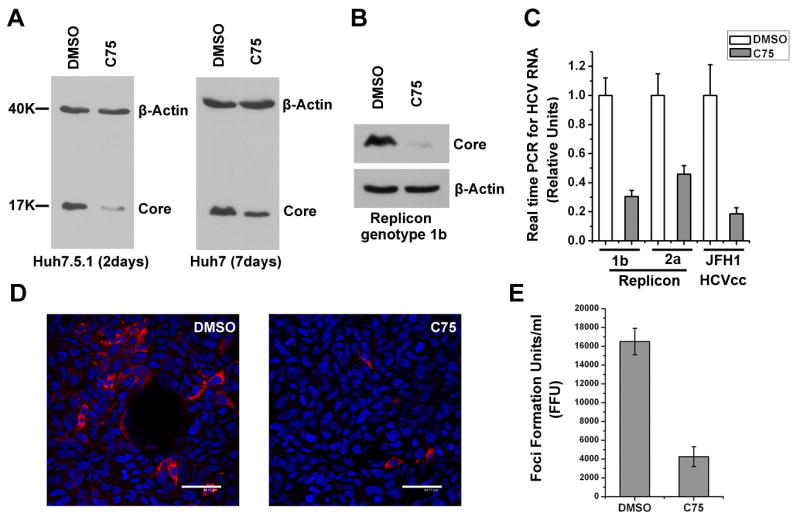
(A) Huh7 and Huh7.5.1 cells were infected with HCVcc for 7 days or 2 days respectively, and then treated with 50μM C75 or the same volume of DMSO as the negative control for indicated time periods. Cell lysates were prepared for quantification of HCV core expression by Western blotting. Detection of β-actin served as the equal loading control. (B) HCV replicon cells (2-3+) were treated with 50μM C75 or DMSO for 12 h and the Core protein abundance was quantified by Western blotting. (C) Genotype 1b and 2a replicon cells as well as HCVcc-infected Huh7 cells were treated with 50μM C75 or DMSO for 12 h. Intracellular HCV RNA was quantified by real time RT-PCR and relative international units were calculated and plotted by arbitrarily setting that of DMSO treated samples to 1. (D) Cell culture supernatants from DMSO or C75 treated HCVcc-producing Huh7 cells were collected and used to infect naïve Huh7.5.1 cells, which were then fixed and stained for viral Core protein after 48 hours. The nucleus was stained with Draq5 and shown in blue (scale bar = 44 μm). (E) Quantification of foci formation units was plotted in bar graph based on the results from D. Error bars represent standard deviations.
Next, we sought to repeat the above experiments using another FASN inhibitor, the FDA approved drug Orlistat. When tested, this drug did not lead to cell death at all at the indicated doses (data not shown). Addition of Orlistat to the media was also able to suppress the HCV core protein production in infection system (Supplementary Fig. 1). Surprisingly, Orlistat weakly enhanced the viral replication in the genotype 1b replicon system at either RNA or protein level (Supplementary Fig. 1). Because the pharmacological inhibitors used could have had side effects, we next knocked down the endogenous FASN expression using short-hairpin based interfering RNA (shRNAi) in both replicon and infection systems and found that both HCV core protein and RNA synthesis were indeed suppressed (Fig. 4).
Figure 4. Knockdown of FASN inhibits HCV replication.
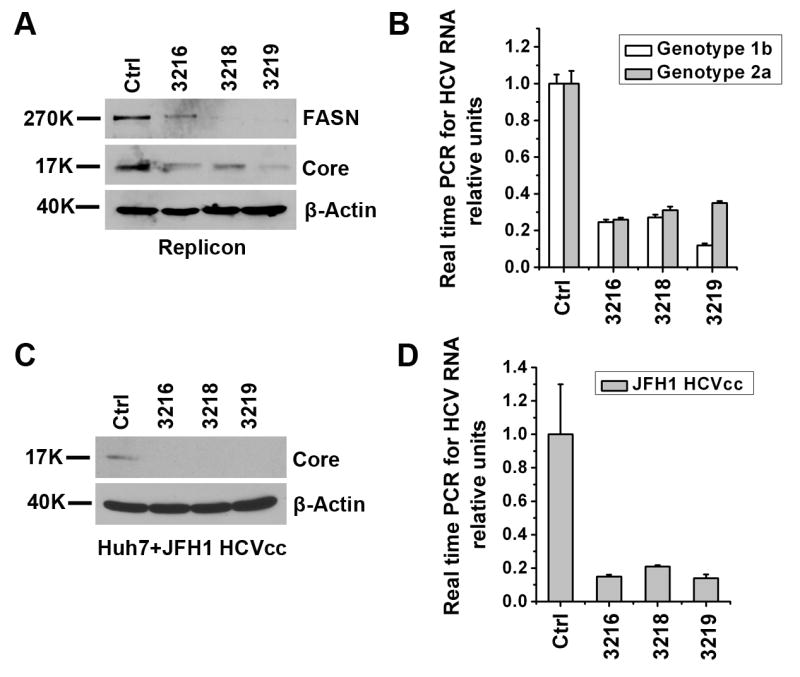
Three short hairpin RNA vectors (3216, 3218 and 3219) targeting FASN and the control vector (Ctrl) were introduced into genotype 1b replicon cells by lentiviral transduction. Forty-eight hours post transduction the cells were lysed and split into two equivalent sets, one of which was used for western analysis of HCV Core and FASN expressions (A). The other set was used for cellular HCV genomic RNA quantification using quantitative RT-PCR (B). Similar experiments were repeated in a genotype 2a replicon cell line. (C, D) Naïve Huh7 cells were transduced with above shRNAi lentiviral particles and then infected with JFH1 HCVcc (MOI 0.1) for 3 days. HCV Core protein production was determined by Western blotting (C) and viral RNA was quantified by real time RT-PCR and plotted as relative units (D). Error bars represent standard deviations.
FASN regulates CLDN1 expression and viral entry
To investigate whether FASN may affect steps of viral life cycle other than replication, we performed infection assays and found that C75 treatment had a strong inhibitory effect on the HCVpp entry but not on VSVpp infection (Fig. 5A&B). Again, the same observation could be repeated using shRNAi to knock down endogenous FASN expression (Fig. 5C&D). The inhibition on HCVpp entry is possibly due to the downregulation of the newly identified entry co-receptor CLDN1 because CLDN1 but not CD81 expression decreased significantly following C75 treatment (Fig. 6A, B, C). Same results could be said with the FASN silenced cells (data not shown). By contrast, the expression of another TJ protein ZO-1 did not appear to decrease as much (Fig. 6C). Since CLDN1 is an integral component forming the TJ strand and we have previously shown that Huh7 cell monolayers exhibited considerable transepithelial electric resistance (TER) (8), we then measured the TER in the presence or absence of inhibitors. Not surprisingly, C75 treated Huh7 and Caco-2 cells both displayed marked reductions in TER, implying the loss of the gate function of TJ (Fig. 6 D&E and Supplementary Fig. 2). On the contrary, Orlistat treatment did not exert any inhibitory effect.
Figure 5. Inhibition of FASN blocks HCVpp entry.
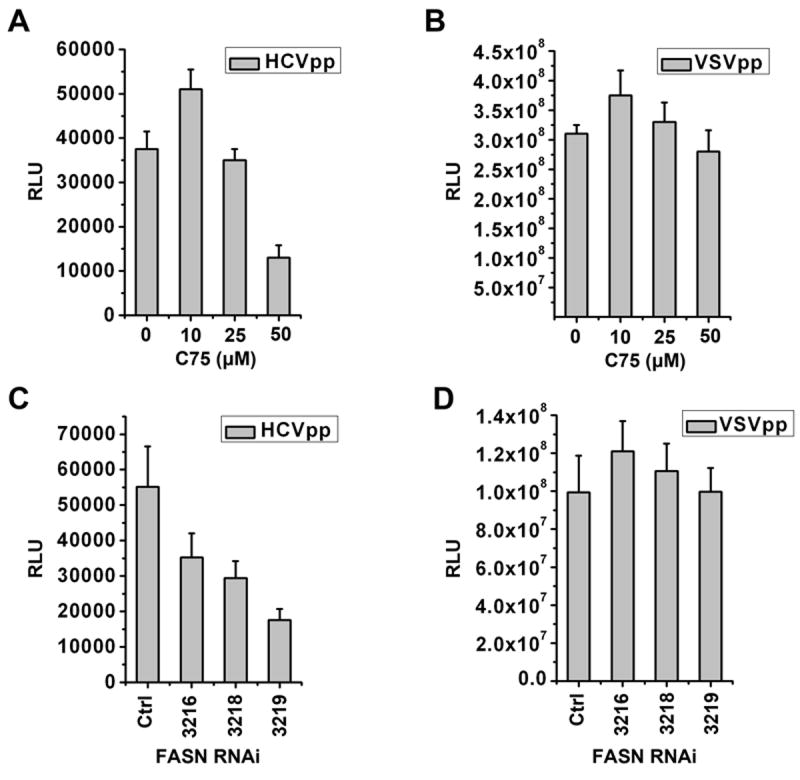
(A, B) Huh7 cells were treated with indicated concentration of C75 for 12 hours (hrs) and then spin-infected with HCVpp or VSVpp and incubated for additional 2 days followed by a luciferase assay to quantify virus entry. (C, D) Huh7 cells were transduced with 3 shRNAi lentiviruses (3216, 3218, and 3219) targeting FASN and the control shRNAi virus (Ctrl) respectively. Two days after lentiviral transduction, the cells were spin-infected with HCVpp or VSVpp and further incubated for 48 hrs prior to luciferase assay. Error bars represent standard deviations.
Figure 6. Inhibition of FASN decreases CLDN1 expression and the transepithelial electric resistance of Huh7 cells.

(A) 106 Huh7 cells were treated with DMSO or C75 (50μM) for 12 hrs and stained with either anti-CD81 (JS-81, BD Bioscience) or a control mouse IgG followed by FITC-secondary antibody staining. Positively stained cells were quantified using a Coulter XL flow cytometer. (B) Huh7 cells were treated with 50μM C75 or the same amount of DMSO for 12 hrs followed by western analysis of CLDN1 expression. The intensity of bands from Western blotting was quantified by using Scion Image version Beta 4.0.3 (scioncorp.com) and confirmed by Photoshop histogram analysis. Fold of increase was indicated by numbers below the image. The results were representative of at least three independent experiments which were normalized against β–Actin level. (C) DMSO (upper panel) or C75 (lower panel) treated Huh7 cells (12 hrs) were fixed and stained for TJ proteins CLDN1 (green) and ZO-1 (red). Nuclei were located by Draq5 staining (blue) (scale bar = 13 μm). (D) 104 Huh7 cells or Caco-2 cells (E) were seeded in a 24-well transwell plate (6.5-mm membrane diameter, 0.4-μm pore size). The cells were allowed to grow for additional 3 days after confluence to reach high TER level and then treated with DMSO, C75 (50μM) or Orlistat (10μM) for 12 hrs. TER values obtained from Day 0 post inhibitor treatment were monitored to establish a baseline resistance and were arbitrarily set to 100. Resistance data collected thereafter were normalized to the initial baseline resistance and plotted as a normalized TER.
Discussion
Determining the makeup of virions remains very challenging because the purity of the virions is always a major concern. Our proteomics profiling revealed many proteins that are worthy of further investigations regarding their functional roles in viral pathogenesis. The fact that a reported virion-associated protein, ApoE, was identified in this study validated the success of the approach. It must be noted, however, that the purification method employed in this study failed to obtain viral particles of high homogeneity. Under the current procedure, it is not possible to distinguish the virion-associated proteins from those that are merely secreted along with HCV virions. In fact, it has been reported that cellular vesicles were contained in prepared HIV virions (16, 17). Ideally, a purification scheme based on sucrose gradient ultracentrifugation and/or affinity separation would minimize contaminants. Complete proteome coverage would require identifying all the proteins contained in HCV sample, the negative sample and conducting a quantitative comparison to identify co-purified contaminants. Nevertheless, the enriched proteins in Figure 1A represent those that were co-purified in the HCV virions-containing supernatant fraction under our purification procedure and may therefore play important roles in the regulation of viral life cycle. In nature, HCV may exist in several forms: enveloped lipoprotein-free virus, enveloped lipoprotein-associated virus, non-enveloped lipoprotein-free virus, and non-enveloped lipoprotein-associated virus (18). It is therefore conceivable that compositions of these different forms of HCV virions may vary greatly.
The finding that FASN was highly enriched in the HCV-containing supernatant was quite surprising, despite the fact that we cannot confirm whether the extracellular FASN is associated within virions. Interestingly, although FASN is an intracellular protein, it was also found in breast cancer cell culture supernatants and the blood of patients with breast cancer (19, 20). It is unclear what role extracellular FASN plays or the mechanism by which FASN exits cells. It has been speculated that secreted FASN might be connected to tumor metastasis. It will be interesting to measure the extracellular FASN level in HCV patients. If it correlates with disease processes such as steatosis and carcinogenesis, the extracellular FASN level may become a potential diagnostic and prognostic marker.
The exciting discovery of FASN as a HCV-inducible gene directly links viral infection to the lipid metabolic disorder that has been widely recognized (21). Notably, during the acute infection of chimpanzees, FASN was expressed at higher levels in successfully cleared and transiently cleared animals, although the pattern was not well correlated with HCV RNA levels (22). HCV is known to cause the formation of lipid droplets with which the HCV core protein associates and recruits nonstructural proteins for viral assembly (23, 24). Our results now provide the first evidence for a causal relationship between HCV infection and FASN abundance level. It is tempting to hypothesize that up-regulation of FASN results in increased lipogenesis. If that is the case, HCV infection could directly increase lipogenesis, contributing to the formation of steatosis. Induction of FASN in HCV-infected cells could also contribute to the tumorigenesis as FASN is commonly upregulated in cancer cells. The molecular mechanism by which viral infection induces FASN expression remains unclear at this point. Two recent reports showed that overexpressions of HCV genotype 3a core protein and genotype 1a NS2 protein were able to upregulate FASN promoter activity in a luciferase-based assay (25, 26). Clinically, HCV genotype 3 is associated with more pronounced hepatic steatosis (27). It is conceivable that FASN expression may be differentially modulated by individual HCV genotypes.
Previous studies using the replicon system have demonstrated that HCV RNA replication is regulated by host geranylgeranylation and FAs (13, 22). FAs are essential constituents of all biological membrane lipids and are important substrates for energy metabolism. The endogenous biosynthesis of FAs is catalyzed by the 250-270 kd homodimeric FASN. A number of inhibitors have been reported to block one of the FASN enzymatic activities. Both C75 and Cerulenin target the KS activity of FASN and induce a rapid accumulation of malonyl-CoA. The FDA-approved drug Orlistat is an inhibitor of FASN thioesterase (TE) activity despite that its design to inhibit pancreatic lipase activity (11). Previously Cerulenin treatment was shown to block viral RNA synthesis in the replicon system (22). The disruption of membrane integrity by blocking the fatty acid synthesis pathway was thought to account for this effect because HCV replication complex is established on membranous web (28, 29). In this report, C75 was able to block viral RNA synthesis in both replicon and infection systems, as did FASN knock-down by shRNAi. Treatment of cells with Orlistat, however, had no effect in the replicon system, emphasizing the difference among inhibitors. While it is difficult to explain these differences, the reasons could potentially be attributed to efficacy and secondary targets of the inhibitors or the different cellular capabilities of compensating each individual FASN activity. In addition, varied responses to these inhibitors may also exist among different HCV genotypes.
C75 treatment also selectively inhibited CLDN1 expression. CLDN1 was recently identified by Evans et.al. as the third entry co-receptor for HCV (1). Like CD81, CLDN1 spans the plasma membrane four times and is a component of the tight junction barrier that restricts the free exchange of ions and aqueous molecules between cells (30, 31). We have demonstrated previously that CLDN1 exhibited a TJ-like distribution pattern in Huh7 cells and disruption of this pattern by TNF-α treatment inhibited HCV entry (8). In light of the finding that depletion of cholesterol decreased CD81 but increased SR-BI cell surface expression (32), it is reasonable to assume that C75 treatment does not deplete membrane cholesterol because it did not change the surface level of CD81. Interestingly, regulation of TJ permeability and occludin expression by polyunsaturated fatty acids has been reported (33). Further study is required to understand the detailed mechanism.
In conclusion, our results highlight the promise of applying functional proteomics in the study of HCV virology. FASN is not only important for productive HCV infection, but elevated FASN expression could contribute to many HCV-associated diseases processes. Our findings warrant further investigations to determine the mechanism by which HCV infection induces FASN expression and the resulted impact of that on FAs production as well as on the alteration of cell phenotypes.
Supplementary Material
Acknowledgments
The authors wish to thank Drs. C. Rice, F. Chisari, S. Lemon, F. Cosset, R. Bartenschlager, K. Li, C. Coyne, and T. Wakita for providing cell lines and constructs. We thank R. Busch, S. Dami, and M. Melan for help with RT-PCR. We also thank Dr. Leland Yee for critical reading of this manuscript.
Funded by:
NIH 1R21AI068784-01A1 grant (to T.W.)
University of Pittsburgh Central Research Development Funds (to T.W.)
Abbreviations
- FASN
fatty acid synthase
- FAs
fatty acids
- HCVpp
HIV particles pseudotyped with HCV envelope proteins
- VSVpp
HIV particles pseudotyped with vesicular stomatitis virus envelope protein G
- HCVcc
cell culture grown HCV
- CLDN1
Claudin-1
- TJ
tight junction
Footnotes
Potential conflict of interest: Nothing to report.
References
- 1.Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wolk B, Hatziioannou T, et al. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature. 2007;446:801–805. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- 2.Lindenbach BD, Rice CM. Molecular biology of flaviviruses. Adv Virus Res. 2003;59:23–61. doi: 10.1016/s0065-3527(03)59002-9. [DOI] [PubMed] [Google Scholar]
- 3.Lindenbach BD, Rice CM. Unravelling hepatitis C virus replication from genome to function. Nature. 2005;436:933–938. doi: 10.1038/nature04077. [DOI] [PubMed] [Google Scholar]
- 4.Penin F, Dubuisson J, Rey FA, Moradpour D, Pawlotsky JM. Structural biology of hepatitis C virus. Hepatology. 2004;39:5–19. doi: 10.1002/hep.20032. [DOI] [PubMed] [Google Scholar]
- 5.Bjornsson E, Angulo P. Hepatitis C and steatosis. Arch Med Res. 2007;38:621–627. doi: 10.1016/j.arcmed.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 6.Maxwell KL, Frappier L. Viral proteomics. Microbiol Mol Biol Rev. 2007;71:398–411. doi: 10.1128/MMBR.00042-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scholle F, Li K, Bodola F, Ikeda M, Luxon BA, Lemon SM. Virus-host cell interactions during hepatitis C virus RNA replication: impact of polyprotein expression on the cellular transcriptome and cell cycle association with viral RNA synthesis. J Virol. 2004;78:1513–1524. doi: 10.1128/JVI.78.3.1513-1524.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang W, Qiu C, Biswas N, Jin J, Watkins SC, Montelaro RC, Coyne CB, et al. Correlation of the tight junction-like distribution of claudin-1 to the cellular tropism of HCV. J Biol Chem. 2008;283(13):8643–8653. doi: 10.1074/jbc.M709824200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang KS, Jiang J, Cai Z, Luo G. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J Virol. 2007;81:13783–13793. doi: 10.1128/JVI.01091-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7:763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- 11.Kridel SJ, Lowther WT, Pemble CWt. Fatty acid synthase inhibitors: new directions for oncology. Expert Opin Investig Drugs. 2007;16:1817–1829. doi: 10.1517/13543784.16.11.1817. [DOI] [PubMed] [Google Scholar]
- 12.Huang H, Sun F, Owen DM, Li W, Chen Y, Gale M, Jr, Ye J. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc Natl Acad Sci U S A. 2007;104:5848–5853. doi: 10.1073/pnas.0700760104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kapadia SB, Chisari FV. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc Natl Acad Sci U S A. 2005;102:2561–2566. doi: 10.1073/pnas.0409834102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuhajda FP, Pizer ES, Li JN, Mani NS, Frehywot GL, Townsend CA. Synthesis and antitumor activity of an inhibitor of fatty acid synthase. Proc Natl Acad Sci U S A. 2000;97:3450–3454. doi: 10.1073/pnas.050582897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orita H, Coulter J, Lemmon C, Tully E, Vadlamudi A, Medghalchi SM, Kuhajda FP, et al. Selective inhibition of fatty acid synthase for lung cancer treatment. Clin Cancer Res. 2007;13:7139–7145. doi: 10.1158/1078-0432.CCR-07-1186. [DOI] [PubMed] [Google Scholar]
- 16.Bess JW, Jr, Gorelick RJ, Bosche WJ, Henderson LE, Arthur LO. Microvesicles are a source of contaminating cellular proteins found in purified HIV-1 preparations. Virology. 1997;230:134–144. doi: 10.1006/viro.1997.8499. [DOI] [PubMed] [Google Scholar]
- 17.Gluschankof P, Mondor I, Gelderblom HR, Sattentau QJ. Cell membrane vesicles are a major contaminant of gradient-enriched human immunodeficiency virus type-1 preparations. Virology. 1997;230:125–133. doi: 10.1006/viro.1997.8453. [DOI] [PubMed] [Google Scholar]
- 18.Diedrich G. How does hepatitis C virus enter cells? Febs J. 2006;273:3871–3885. doi: 10.1111/j.1742-4658.2006.05379.x. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Kuhajda FP, Li JN, Pizer ES, Han WF, Sokoll LJ, Chan DW. Fatty acid synthase (FAS) expression in human breast cancer cell culture supernatants and in breast cancer patients. Cancer Lett. 2001;167:99–104. doi: 10.1016/s0304-3835(01)00464-5. [DOI] [PubMed] [Google Scholar]
- 20.Wang YY, Kuhajda FP, Li J, Finch TT, Cheng P, Koh C, Li T, et al. Fatty acid synthase as a tumor marker: its extracellular expression in human breast cancer. J Exp Ther Oncol. 2004;4:101–110. [PubMed] [Google Scholar]
- 21.Weinman SA, Belalcazar LM. Hepatitis C: a metabolic liver disease. Gastroenterology. 2004;126:917–919. doi: 10.1053/j.gastro.2003.01.001. [DOI] [PubMed] [Google Scholar]
- 22.Su AI, Pezacki JP, Wodicka L, Brideau AD, Supekova L, Thimme R, Wieland S, et al. Genomic analysis of the host response to hepatitis C virus infection. Proc Natl Acad Sci U S A. 2002;99:15669–15674. doi: 10.1073/pnas.202608199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi ST, Polyak SJ, Tu H, Taylor DR, Gretch DR, Lai MM. Hepatitis C virus NS5A colocalizes with the core protein on lipid droplets and interacts with apolipoproteins. Virology. 2002;292:198–210. doi: 10.1006/viro.2001.1225. [DOI] [PubMed] [Google Scholar]
- 24.Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, et al. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol. 2007;9:1089–1097. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- 25.Jackel-Cram C, Babiuk LA, Liu Q. Up-regulation of fatty acid synthase promoter by hepatitis C virus core protein: genotype-3a core has a stronger effect than genotype-1b core. J Hepatol. 2007;46:999–1008. doi: 10.1016/j.jhep.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 26.Oem JK, Jackel-Cram C, Li YP, Zhou Y, Zhong J, Shimano H, Babiuk LA, et al. Activation of sterol regulatory element-binding protein 1c and fatty acid synthase transcription by hepatitis C virus non-structural protein 2. J Gen Virol. 2008;89:1225–1230. doi: 10.1099/vir.0.83491-0. [DOI] [PubMed] [Google Scholar]
- 27.Rubbia-Brandt L, Quadri R, Abid K, Giostra E, Male PJ, Mentha G, Spahr L, et al. Hepatocyte steatosis is a cytopathic effect of hepatitis C virus genotype 3. J Hepatol. 2000;33:106–115. doi: 10.1016/s0168-8278(00)80166-x. [DOI] [PubMed] [Google Scholar]
- 28.Aizaki H, Lee KJ, Sung VM, Ishiko H, Lai MM. Characterization of the hepatitis C virus RNA replication complex associated with lipid rafts. Virology. 2004;324:450–461. doi: 10.1016/j.virol.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 29.Egger D, Wolk B, Gosert R, Bianchi L, Blum HE, Moradpour D, Bienz K. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J Virol. 2002;76:5974–5984. doi: 10.1128/JVI.76.12.5974-5984.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Itallie CM, Anderson JM. The molecular physiology of tight junction pores. Physiology (Bethesda) 2004;19:331–338. doi: 10.1152/physiol.00027.2004. [DOI] [PubMed] [Google Scholar]
- 31.Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol. 2004;286:C1213–1228. doi: 10.1152/ajpcell.00558.2003. [DOI] [PubMed] [Google Scholar]
- 32.Kapadia SB, Barth H, Baumert T, McKeating JA, Chisari FV. Initiation of hepatitis C virus infection is dependent on cholesterol and cooperativity between CD81 and scavenger receptor B type I. J Virol. 2007;81:374–383. doi: 10.1128/JVI.01134-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang WG, Bryce RP, Horrobin DF, Mansel RE. Regulation of tight junction permeability and occludin expression by polyunsaturated fatty acids. Biochem Biophys Res Commun. 1998;244:414–420. doi: 10.1006/bbrc.1998.8288. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.