

Abstract
Herkinorin is the first μ-opioid receptor (MOP-r) selective ligand from the salvinorin A diterpenoid scaffold. Herkinorin has relative μ>κ>δ binding selectivity, and can act as an agonist at both μ- and κ-receptors, in vitro. These studies were the first in vivo evaluation of herkinorin's effects in non-human primates, using prolactin release, a neuroendocrine biomarker assay that is responsive to both μ- and κ- agonists, as well as to compounds with limited ability to cross the blood-brain barrier. In cumulative dosing studies (0.01-0.32 mg/kg, i.v.), herkinorin produced only small effects in gonadally intact males (n=4), but a more robust effect in females (n=4). Timecourse studies with herkinorin (0.32 mg/kg) confirmed this greater effectiveness in females, and revealed a fast onset after i.v., administration (e.g., by 5-15 min). Antagonism experiments with different doses of nalmefene (0.01 and 0.1 mg/kg) caused dose-dependent and complete prevention of herkinorin's effect in females. This is consistent with a principal μ-agonist effect of herkinorin, with likely partial contribution by κ-agonist effects. The peripherally selective antagonist quaternary naltrexone (1 mg/kg, s.c.) caused approximately 70% reduction in the peak effect of herkinorin (0.32 mg/kg) in females, indicating that this effect of herkinorin is prominently mediated outside the blood-brain barrier.
Introduction
Salvinorin A, a plant-derived hallucinogenic diterpene, is a highly selective κ-opioid agonist, and a novel template for semi-synthetic opioid analogs (Roth et al., 2002; Prisinzano and Rothman, 2008). One of these novel analogs is herkinorin, the first salvinorin-derived compound with μ- over κ- selectivity reported in the literature (Harding et al., 2005). Herkinorin has approximately 8-fold selectivity for μ- over κ- receptors, and approximately 98-fold selectivity for μ- over δ- receptors, in competition binding assays (Harding et al., 2005). Herkinorin acts as a high efficacy agonist at both μ- and κ- receptors, in the GTPγS assay (Harding et al., 2005), with greater relative potency at μ-receptors. Herkinorin also displays some unique features in its interactions at the μ-receptor, such as decreased agonist-induced internalization (Groer et al., 2007; Xu et al., 2007).
To date, there is no information on the in vivo effects of herkinorin in primates. The present study focuses on an initial evaluation of the in vivo opioid agonist effects of herkinorin in rhesus monkeys, using a translationally viable neuroendocrine biomarker assay, release of the anterior pituitary hormone prolactin. Different factors render this neuroendocrine biomarker a practical approach for initial evaluation of herkinorin: a) Prolactin levels are increased by both μ- and κ- agonists in mammals including humans (δ-agonists appear not be to be active) (Hoehe et al., 1988; Ur et al., 1997; Kreek et al., 1999; Bowen et al., 2002; Butelman et al., 2007), and b) Effects of these opioids are at least partially mediated by opioid receptors outside the blood-brain barrier (e.g., in particular hypothalamic nuclei) (Merchenthaler, 1991; Butelman et al., 2004; Zheng et al., 2005; Butelman et al., 2008), and may therefore be used to determine the pharmacodynamic effects of compounds with limited ability to cross the blood-brain barrier.
This study presents the first direct evaluation of the pharmacodynamic effects of herkinorin in male and female non-human primates, and also investigate potential μ- vs. κ- receptor effects of this novel compound, as well as activity outside the blood-brain barrier.
Methods
Subjects
Four captive-bred male and four female rhesus monkeys (Macaca mulatta; age range: 8-12 years old approximately; weight range: 7.0-12.5 kg), were used. All subjects were gonadally intact. Unless otherwise stated, each study was carried out with an n=4 of either males or females. Females were studied in the follicular phase, estimated as days 1-12 from the onset of visible menses. Monkeys were singly housed in colony rooms maintained at 20-22°C with controlled humidity, and a 12:12 hour light: dark cycle (lights on at 0700). Monkeys were fed approximately 12 jumbo primate chow biscuits, adjusted individually (PMI Feeds, Richmond, VA). Subjects also received appetitive treats, and multivitamins plus iron. An environmental enrichment plan was in place (e.g., music and videos). Water was freely available in home cages, via a waterspout.
Subjects had complex history of pharmacological exposure (primarily opioid), but had no history of any chronic or high frequency exposure to any agent. Consecutive experiments in the same subject were separated by at least 72 h; the order of experiments was unsystematic among subjects. All studies were carried out over the course of several months, while all subjects were in stable colony rooms.
Procedure for neuroendocrine experiments
Chair-trained monkeys were tested after repeated exposure to the experimental situation. Monkeys were chaired and transferred to the experimental room between 0945h and 1030h on each test day, approximately. An indwelling catheter (24 gauge; Angiocath, Becton Dickinson, Sandy, UT) was placed in a superficial leg vein, and secured with elastic tape. An injection port (Terumo, Elkton, MD) was attached to the catheter; port and catheter were flushed (0.3 ml of 50 U/ml heparinized saline) before use, and after blood sampling or injection. Approximately 20 min following catheter placement, two baseline blood samples of approximately 1.5 ml were collected, 5 min apart from each other (defined as -10 and -5 min, relative to the onset of drug injection). Samples were kept at room temperature until the time of spinning (3,000 rpm at 4°C) and serum separation. Serum samples were kept at -40°C until time of analysis (typically within 2 weeks of collection). Samples were analyzed in duplicate with a standard human prolactin immunoradiometric kit (DPC-Siemens Medical Solutions Diagnostics, Los Angeles CA), following manufacturer's instructions. Studies report high protein homology in human vs. rhesus monkey prolactin, as well as antibody cross-reactivity (Brown and Bethea, 1994; Pecins-Thompson et al., 1996). The reported sensitivity limit of this assay was 0.1 ng/ml; each individual kit was calibrated with known standards, in the range 2-200 ng/ml. The intra- and inter-assay coefficients of variation with this kit in the laboratory were approximately 3% and 11%, respectively.
Monkeys were tested in a cumulative dosing or timecourse procedure. Cumulative dosing procedure: A cumulative dosing procedure was employed to produce a rapid initial estimate of herkinorin's potency and effectiveness within a single session. Cumulative dosing procedures have been previously used to efficiently measure potency and effectiveness of μ- and κ- agonists in this biomarker assay (Bowen et al., 2002; Butelman et al., 2002). Cumulative dosing studies commenced with baseline pre-injection sample collection (two separate samples taken approximately 10 and 5 min before the onset of dosing), followed by four consecutive 30-min inter-injection cycles, with agonist doses increasing in 0.5 log unit steps. Blood sampling occurred 15 min after each injection. Herkinorin was studied herein up to the largest dose that could be administered due to solubility limitations (0.32 mg/kg). Herkinorin was then compared to salvinorin A (its structurally related parent compound, a selective κ-agonist) and loperamide (a peripherally selective μ-agonist), under similar conditions. A vehicle control study was also carried out, with 4 consecutive vehicle injections under identical timing conditions.
Timecourse procedure
Following baseline sample collection, a single agonist dose (e.g., of herkinorin) was administered i.v., followed by sampling at 5, 15, 30, 60, 90 and 120 min after administration. In antagonism experiments, a single dose of antagonist (s.c. nalmefene or quaternary naltrexone) was administered 30 min before an agonist, followed by testing as above. In these antagonism experiments, a single sample was also taken 20 min after administration of the antagonist alone (i.e., during the pretreatment period).
Design
Cumulative dose-effect curve studies
Cumulative dose-effect curve studies were carried out in males and females for herkinorin (0.01-0.32 mg/kg, i.v.) compared to repeated vehicle injection, for control purposes. The effects of loperamide (0.01-0.32 mg/kg males and females) and salvinorin A (0.001-0.032 mg/kg [males] and 0.00032-0.01 mg/kg [females], based on prior studies; Butelman et al., 2007; Butelman et al., 2008) were also studied under identical cumulative dosing procedures.
Timecourse studies
The timecourse effects of herkinorin were studied in male subjects, at the largest dose that could be administered, as limited by solubility (0.32 mg/kg, i.v., compared to vehicle, i.v.). This was followed by a similar determination of the effects of this dose in females.
Since males demonstrated only a small effect of herkinorin up to the largest dose that could be studied, antagonism experiments described below were only carried out in females. Nalmefene (0.01 or 0.1 mg/kg, s.c.) was thus administered as a pretreatment to herkinorin (0.32 mg/kg, i.v.). Nalmefene has relative μ- over κ– selectivity as an antagonist in primates (France and Gerak, 1994). In prior studies, the lower nalmefene dose (0.01 mg/kg) was sufficient to block the effects of selective μ-agonists (France and Gerak, 1994), whereas the larger dose (0.1 mg/kg) was necessary to block the effects of κ-agonists (Butelman et al., 2007). The effects of herkinorin (0.32 mg/kg) were also studied after pretreatment with the peripherally selective antagonist, quaternary naltrexone (1 mg/kg s.c.) (Valentino et al., 1983). In a comparison study, the effects of fentanyl (0.01 mg/kg, i.v.) were studied after analogous pretreatment with a μ-selective dose of nalmefene (0.01 mg/kg).
Neuroendocrine Data Analysis
Raw individual prolactin values for each experiment were converted to Δng/ml (i.e., change from baseline) by subtracting mean pre-injection baseline value for each subject. Data were then analyzed with 2- way repeated measures ANOVAs (e.g., time X drug condition) using Sigmastat 3.1, followed by Newman-Keuls post-hoc testing. Values are presented to two decimal places, and the level of significance (α) for all comparisons was set at the 0.05 level.
Drugs
Herkinorin was synthesized in the laboratory of Dr. Prisinzano (Harding et al., 2005), and was dissolved daily in 10%DMSO: 10% Tween 80: sterile water v/v). Salvinorin A (extracted from commercially obtained leaves from Ethnogens.com in Dr. Prisinzano's laboratory) and loperamide HCl (Sigma, St. Louis, MO) were dissolved in 10%ethanol, 10%Tween 80: 80% sterile water (v/v). Quaternary naltrexone methobromide (also known as methylnaltrexone) was kindly supplied by Dr. Chun-Su Yuan (Dept. of Anesthesiology, University of Chicago), and was dissolved daily in sterile water. Nalmefene HCl (Baker-Norton, Miami, FL) was dissolved in sterile water. Drug doses are reported in the forms stated above. All drugs were injected in volumes of 0.05-0.3 ml/kg.
Results
Cumulative dosing: Baseline and vehicle control experiment
Mean baseline prolactin levels in a cumulative vehicle experiment were 6.8 ng/ml (SEM 2.3) in males and 18.8 ng/ml (SEM 6.1) in females. Four consecutive vehicle injections (30-min inter-injection interval, sampling 15 min post-injection), resulted in small decreases in prolactin levels. For example, after the fourth injection, mean prolactin values were -2.6 Δng/ml (SEM 1.4) in males, and -3.3 Δng/ml (SEM 9.9) in females. Mean Δng/ml values over four consecutive cycles with vehicle administration are presented in Fig. 1.
Figure 1.
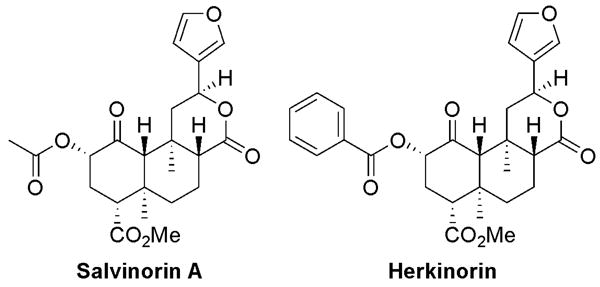
Chemical structures of the parent compound salvinorin A (left), and its μ-selective analog, herkinorin (right).
Cumulative dosing studies
Herkinorin
Up to the largest dose that could be studied (0.32 mg/kg), herkinorin only caused a small increase in prolactin levels in males (mean 14.4 Δng/ml [SEM 7.7]). However, in females, a greater maximum effect was detected at the largest dose (mean 175.8 [SEM 47.4]) (Figure 1). See below for statistical analysis of these cumulative dose-effect curve data.
Salvinorin A
In a comparative study, salvinorin A was studied in a similar cumulative dosing procedure in males vs. females. Dose ranges were adjusted based on prior information of the greater effectiveness of a salvinorin A dose in males vs. females (Butelman et al., 2007). Salvinorin A (0.001-0.032 mg/kg) caused robust dose-dependent effects in males (maximum mean 135.7 Δng/ml [SEM 29.4], at 0.032 mg/kg). Salvinorin A (0.00032-0.01 mg/kg) caused even more robust effect in females (maximum mean 314.9 [SEM 90.6], at 0.01 mg/kg) (Fig. 1). See below for statistical analysis of these cumulative dose-effect curve data.
Loperamide
Loperamide caused dose-dependent prolactin increases in males (103 Δng/ml [48.4] at 0.32 mg/kg). Loperamide also caused robust, dose-dependent prolactin increases in females (maximum mean 202.1 Δng/ml [SEM 23.7] at 0.32 mg/kg) (Fig. 1). See below for statistical analysis of these cumulative dose-effect curve data.
Analysis of dose-effect curve data for all compounds
For males and females, 2-way repeated measures ANOVAs were carried out for drug (vehicle, herkinorin, salvinorin A or loperamide) vs. dose (as used in this 4-cycle procedure), using Δng/ml values (i.e., after subtraction of individual pre-injection baseline). The aim of this analysis was to describe the observed effects in the dose-effect curve studies, while taking into account individual baseline differences, and the slight decrease that is observed through repeated vehicle administration in this setting (see above).
In males, this drug X dose ANOVA resulted in main effects of drug (F[3,9]=9.94; p<0.003), dose (F[3,9]=9.89; p<0.003) and their interaction (F[9,27]=6.23; p<0.001). Herkinorin was not different from its cycle-matched vehicle control, in any of the doses tested. Salvinorin A was significantly different from vehicle at the two largest doses studied (0.01 and 0.032 mg/kg; q=5.60 and 10.08, respectively [all p<0.05]). Loperamide was significantly different from vehicle only at largest dose studied (0.32 mg/kg; q=7.70; p<0.05).
In females, the drug X dose ANOVA also resulted in main effects of drug (F[3,9]=8.58; p<0.005), dose (F[3,9]=22.02; p<0.001) and their interaction (F[9,27]=7.74; p<0.001). Herkinorin was significantly different from vehicle at the largest dose studied (0.32 mg/kg; q=7.18; p<0.05). Salvinorin A was significantly different from vehicle at the two largest doses studied (0.0032 and 0.01 mg; q=4.61 and 12.76, respectively [all p<0.05]). Loperamide was different from vehicle only at the largest dose studied (0.32 mg/kg; q=8.24; p<0.05).
Timecourse studies: Baseline and vehicle control experiment
In the vehicle control timecourse experiment, males exhibited mean baseline pre-injection prolactin values of 8.7 ng/ml (SEM 1.1). Females had similar mean pre-injection values 8.3 ng/ml (SEM 1.9). Bolus administration of the vehicle used in herkinorin studies (1:1:8 DMSO: Tween 80 : sterile water v/v) resulted in slight gradual decreases in prolactin levels over the course of the experiment, reaching -4 Δng/ml (SEM 1) for males and -3.2 Δng/ml (SEM 0.6) for females, at the last (120 min) time point (Fig. 2).
Figure 2.

Cumulative dose- effect curves for i.v. salvinorin A, loperamide and herkinorin in gonadally intact males and females (upper and lower panels, respectively). Abscissae: Cumulative dose in mg/kg. Ordinates: Effects on prolactin levels compared to individual pre-injection baseline (Δng/ml, ±SEM). Dashed line is the mean value for repeated vehicle injection in these subjects (4 consecutive dosing cycles, with a 30 min inter-injection interval). *Significantly different from cycle-matched vehicle control [p<0.05]. *# Both loperamide and herkinorin were significantly different from cycle-matched vehicle control [p<0.05]. See text for details.
Timecourse effects of herkinorin
Up to the largest dose that could be administered (0.32 mg/kg, i.v.), herkinorin caused a modest but significant time-dependent increase in serum prolactin levels in males (peak mean levels of 19.1 Δng/ml [SEM 7.3]) (Fig. 2; note ordinate axis break). A rapid onset was observed in this effect, with peak effects observed at the earliest sample point (5 min after administration). A 2-way repeated measures ANOVA (condition [vehicle or herkinorin] vs. post-injection time) revealed a main effect of condition (F[1,3]=12.2; p<0.04), time (F[5,15]=6.0; p<0.003) and their interaction (F[5,15]=4.0; p<0.02). A Newman-Keuls test confirmed that herkinorin caused an increase in prolactin levels compared to vehicle at 5, 15 and 30 min after administration (q values were 5.73, 5.41 and 5.0 respectively [all p<0.05]), but not at later time points (i.e., 60-120 min).
By contrast, the same herkinorin dose in females caused a greater maximum effect 5 min after administration (peak maximum 306.6 Δng/ml [SEM 91]) (Fig. 2; note ordinate axis break). A 2-way repeated measures ANOVA (condition [vehicle or herkinorin] vs. post-injection time) revealed a main effect of condition (F[1,3]=15.34; p<0.03), time (F[5,15]=10.74; p<0.001) and their interaction (F[5,15]=10.48; p<0.001). A Newman-Keuls test confirmed that herkinorin caused an increase in prolactin levels compared to vehicle at 5, 15, and 30 min after administration (q values were 9.56, 6.55 and 4.03, respectively [all p<0.05]), but not at later time points (i.e., 60-120 min).
Antagonism Experiments
Due to the small magnitude of herkinorin's effect in males, subsequent antagonism experiments were only carried out in females, against the herkinorin 0.32 mg/kg dose. Nalmefene was administered in two separate pretreatment experiments (0.01 or 0.1 mg/kg). In either case, nalmefene alone (measured 20 min after administration) did not cause a change in prolactin levels (not shown). Nalmefene, at two pretreatment doses (0.01 mg/kg and 0.1 mg/kg) produced a partial and complete blockade of herkinorin's effect, respectively. A 2-way repeated measures ANOVA (condition [vehicle alone, herkinorin alone, herkinorin after nalmefene 0.01 or 0.01 mg/kg pretreatment] X post-injection time) revealed a significant main effect of condition (F[3,9]=11.98; p<0.002), post-injection time (F[5,15]=10.48; p<0.001) and their interaction (F[15,45]=8.16; p<0.001) (Fig. 3, left panel). Newman-Keuls tests revealed that there was a significant of nalmefene (0.01 mg/kg) pretreatment vs. herkinorin alone at 5, 15 and 30 min after administration (q values were 9.38, 5.95 and 3.94, respectively [all p<0.05]). The larger nalmefene pretreatment dose (0.1 mg/kg) also caused a significant difference vs. herkinorin alone at 5, 15 and 30 min (q values were 12.51, 8.61 and 5.37, respectively [all p<0.05]).
Figure 3.

Timecourse of herkinorin (0.32 mg/kg, i.v.) in males and females, compared to i.v. vehicle, 5-120 min after injection. Abscissa: Time from bolus i.v. injection (min). Ordinate: Effects on prolactin levels compared to individual pre-injection baseline (Δng/ml, ±SEM). Please note break in ordinate axis, to illustrate difference between effects in males vs. females. *Significantly different from respective vehicle time point; see text for details.
In a comparison experiment, the μ-selective agonist fentanyl (0.01 mg/kg) produced prolactin-releasing effects of a similar magnitude to those of herkinorin, but was fully blocked by the smaller dose of nalmefene (0.01 mg/kg) (Fig. 3, right panel). A 2-way repeated measures ANOVA (condition [vehicle alone, fentanyl alone, fentanyl after nalmefene 0.01 mg/kg pretreatment] X post-injection time) revealed a significant main effect of post-injection time (F[5,15]=3.89; p<0.02) and condition X post-injection time interaction F[10,30]=3.06; p<0.009). Newman-Keuls tests revealed that nalmefene (0.01 mg/kg) pretreatment resulted in significant differences vs. fentanyl alone at 5 and 15 min (q values were 3.87 and 5.68 [all p<0.05]).
The effects of herkinorin (0.32 mg/kg) were also studied after pretreatment with the peripherally selective antagonist, quaternary naltrexone (1 mg/kg). Quaternary naltrexone alone did not have an effect on prolactin levels (20 min after administration; not shown). Quaternary naltrexone caused a partial reduction in herkinorin's effect (Fig. 4). A 2-way repeated measures ANOVA (condition [herkinorin alone, vs. herkinorin after quaternary naltrexone pretreatment] X post-injection time) revealed a significant main effect of post-injection time (F[5,15]=8.13; p<0.001) and condition X post-injection time interaction F[5,15]=5.87; p<0.003). Newman-Keuls comparisons confirmed that quaternary naltrexone pretreatment caused a blockade in herkinorin's effect at 5 and 15 min (q=6.89 and 4.0, respectively [all p<0.05]). Larger quaternary naltrexone doses were not probed herein due to supply limitations.
Figure 4.

Antagonism of the effects of herkinorin (0.32 mg/kg) in females by nalmefene (0.01 or 0.1 mg/kg; left panel) or of fentanyl (0.01 mg/kg) by nalmefene (0.01 mg/kg; right panel). Nalmefene alone, measured 20 min after administration, had no effect on prolactin levels (not shown). See Fig. 2 for other details. *# either dose of nalmefene was significantly different from herkinorin alone [p<0.05]; see text for details.
Discussion
Herkinorin is the first compound derived from the salvinorin A diterpenoid scaffold to have μ- over κ- selectivity (Harding et al., 2005). The present neuroendocrine biomarker assay (prolactin release) is responsive to both μ- and κ- agonists in humans and non-human primates and humans, and is therefore a translationally viable assay to study the in vivo pharmacology of this novel compound (Hoehe et al., 1988; Bart et al., 2003).
In cumulative dosing studies in males (0.01-0.32 mg/kg, i.v.), herkinorin only caused a small, and non-statistically significant effect, up to the largest dose that could be studied under the present solubility conditions. For comparison, the parent compound salvinorin A displayed greater potency and efficacy than herkinorin in males. The peripherally selective μ-agonist loperamide also displayed greater effectiveness than herkinorin in males.
Herkinorin's effect in females was more robust. The parent compound, salvinorin A, was approximately 30-fold more potent than herkinorin in females. The peripherally selective μ-agonist loperamide was approximately equipotent and equieffective to herkinorin in this determination in females.
The relative ineffectiveness of herkinorin in males could be potentially interpreted as a sign of partial agonist effects in this assay, compared to loperamide and salvinorin A (both of these latter ligands have high efficacy and selectivity at their respective receptor targets, the μ- and κ- receptors, respectively) (DeHaven-Hudkins et al., 1999; Roth et al., 2002). However, herkinorin also has relatively high efficacy at both μ- and κ- receptors in vitro (with greater relative potency at μ over κ) (Harding et al., 2005). Furthermore, recent unpublished pilot studies with herkinorin (0.32 mg/kg) as a pretreatment to either salvinorin A (0.032 mg/kg) or loperamide (0.32 mg/kg) were not consistent with partial agonist effects by herkinorin (i.e., no herkinorin-induced blockade of salvinorin A or loperamide was observed). Thus, based on these findings, it is more likely that the present lack of herkinorin effectiveness in males is due to practical limits to reach a sufficiently high dose in vivo, as limited by solubility.
In order to further investigate the agonist effects of herkinorin, timecourse studies at the largest herkinorin dose were carried out (0.32 mg/kg, measured 5-120 min after i.v. bolus). These findings were generally consistent with the cumulative dose-effect curve study. In the timecourse studies, a small but significant effect was observed in males, whereas a robust effect was observed in females. Herkinorin displayed a fast onset after i.v. administration (peak values were observed at 5-15 min). Intriguingly, while the effectiveness of herkinorin was greater in females than in males, the duration of action of herkinorin was similar in subjects of either sex. Specifically, herkinorin was significantly different from vehicle only up to 30 min after administration in either females or males. These are the first studies to compare the effects of herkinorin in male and female subjects of any species, to our knowledge. Similarly to the parent compound, salvinorin A, herkinorin displayed robustly increased effectiveness in females vs. males. These studies are consistent with available research in humans, showing that opioid agonists can cause more robust effects on this neuroendocrine endpoint in females than in males (Kreek et al., 1999).
The demonstration that a compound can cause prolactin release is not sufficient to implicate opioid receptors, since several types of compounds in addition to opioids also have this effect (including dopaminergic antagonists or serotonergic agonists; see for example (e.g., Aloi et al., 1984; Nordstrom and Farde, 1998). Consequently, antagonism studies were designed to determine whether opioid (μ- and or κ- receptor) mechanisms were involved in the effects of herkinorin. Antagonism studies such as apparent pA2 or pKB analyses were not undertaken herein, because herkinorin's solubility under these conditions precluded investigation of surmountability. Also, antagonism studies were only undertaken in females, due to the small magnitude of herkinorin's observed effect in males.
Nalmefene has a relative μ- over κ- selectivity as an antagonist in non-human primates (France and Gerak, 1994; Butelman et al., 2002), and does not in itself cause prolactin release in non-human primates (as found in the present study and prior references; Butelman et al., 1999; Mello et al., 2000). Interestingly, nalmefene exhibits partial κ-agonist effects in cloned human κ-receptors in the GTPγS assay, and does cause detectable prolactin release in humans (Bart et al., 2005). In the present studies, nalmefene 0.01 mg/kg produced a partial reduction (by approximately 75%, from a mean peak of 306.6 Δng/ml to 79.8 Δng/ml) in the effects of herkinorin in females. The larger dose of nalmefene (0.1 mg/kg) was able to produce essentially complete blockade of this effect (by approximately 98%, to a mean peak of 4.4 Δng/ml). Given prior data on doses of nalmefene active against μ- and κ- opioid ligands in rhesus monkeys (France and Gerak, 1994; Butelman et al., 2002; Butelman et al., 2007), this suggests that the effects of herkinorin are mediated principally by μ-receptors, and that κ-receptors may also be partially involved. These data are consistent with the in vitro profile of herkinorin, which shows relative (approximately 8-fold) binding selectivity for μ- over κ- receptors (Harding et al., 2005). As a direct confirmation in this setting, the smaller dose of nalmefene (0.01 mg/kg) was indeed sufficient to cause complete blockade of the effects of the selective μ-agonist fentanyl (see also Butelman et al., 2008). This confirms that an agonist thought to act solely through μ-receptors in this assay is fully blocked by the smaller nalmefene dose under these experimental conditions. Antagonism experiments with more selective κ-opioid antagonists (e.g., nor-BNI) were not undertaken due to the potential for ultra-long duration of such antagonists on neuroendocrine endpoints (at least several weeks) (Pascoe et al., 2008).
As recently shown and confirmed herein (e.g., with loperamide), μ-receptors located outside the blood-brain barrier (probably in the hypothalamus) can mediate prolactin-releasing effects in primates (Merchenthaler, 1991; Zheng et al., 2005; Butelman et al., 2008). For comparison, we recently reported that fentanyl may produce this effect by acting on μ-receptor populations inside as well as outside the blood-brain barrier (Butelman et al., 2008). In this study, the peripherally selective opioid antagonist quaternary naltrexone (methylnaltrexone; 1 mg/kg) (Valentino et al., 1983; Yuan et al., 2002) caused a partial blockade of herkinorin's peak effect (by approximately 70%, to a mean peak of 88.3 Δng/ml). This confirms that herkinorin may cause the present effect by acting at least partially on opioid receptors outside the blood brain barrier (Zheng et al., 2005). Based on prior studies (Butelman et al., 2004) and recent pilot data, we have also found that quaternary naltrexone is not able to fully block the effects of κ-agonists in this endpoint. Thus, it may not be excluded that the part of the effect of herkinorin that was insensitive to quaternary naltrexone may have been mediated by κ-receptors, rather than by central μ-receptors (Butelman et al., 2008). Such a conclusion would in fact be consistent with the present nalmefene antagonism data (see above).
To our knowledge, this is the first direct dose-effect curve comparison of prolactin-releasing effects of μ- and κ- opioids in gonadally intact male and female primates. The present study confirms that κ-opioids (e.g., salvinorin) cause prolactin-releasing effects of both greater potency and greater apparent efficacy in females than in males (Kreek et al., 1999). Sex differences for a peripherally selective μ-opioid (loperamide) were more modest, and observable mostly as a difference in maximum effect. Overall, the present dose-effect curve determinations support the use of this translationally viable neuroendocrine biomarker for the study of novel μ- and κ- opioid analogs in primates. These findings support use of the biomarker in human clinical populations, as a means to determine responsiveness of either μ- or κ- receptor pools, and their association to disease status.
This is also the first evaluation of herkinorin, the first μ-selective ligand from the salvinorin scaffold (Harding et al., 2005), in non-human primates, and the first evaluation of its neuroendocrine effects in any species. This study shows that herkinorin exhibits moderate potency in vivo, and that its effectiveness in this neuroendocrine biomarker is greater in females than in males. Antagonism studies confirm that herkinorin can produce μ-agonist effects in vivo, although it may have moderate μ- over κ- selectivity in this respect. Lastly, studies with quaternary naltrexone suggest that herkinorin may produce these effects by acting at least partially on opioid receptors located outside the blood-brain barrier. Additional studies to improve the pharmacokinetic properties and μ-selectivity of the herkinorin template are ongoing.
Fig. 5.

Antagonism of the effects of herkinorin (0.32 mg/kg) in females by quaternary naltrexone (1 mg/kg, s.c.). Quaternary naltrexone alone, measured 20 min after administration, had not effect on prolactin levels (not shown). *Significantly different from herkinorin alone [p<0.05]. See Fig. 2 and text for other details.
Acknowledgments
The technical assistance of Mr. Marek Mandau for parts of these studies is gratefully acknowledged.
These studies were funded by NIH-NIDA grants DA017369 (ERB), DA018151 (TEP) and DA05130 (MJK).
Abbreviations
- Δng/ml
change in prolactin levels from individual pre-injection baseline
- PT
pretreatment
Footnotes
Studies were reviewed by the Rockefeller University Animal Care and Use Committee, in accordance with the Guide for the Care and Use of Animals (National Academy Press; Washington DC, 1996).
References
- Aloi JA, Insel TR, Mueller EA, Murphy DL. Neuroendocrine and behavioral effects of m-chlorophenylpiperazine administration in rhesus monkeys. Life Sci. 1984;34:1325–1331. doi: 10.1016/0024-3205(84)90003-1. [DOI] [PubMed] [Google Scholar]
- Bart G, Borg L, Schluger JH, Green M, Ho A, Kreek MJ. Suppressed prolactin response to dynorphin A(1-13) in methadone maintained versus control subjects. J Pharmacol Exp Ther. 2003;306:581–587. doi: 10.1124/jpet.103.050682. [DOI] [PubMed] [Google Scholar]
- Bart G, Schluger JH, Borg L, Ho A, Kreek MJ. Nalmefene induced elevation in serum prolactin in normal human volunteers: Partial kappa-opioid agonist activity? Neuropsychopharmacology. 2005;30:2254–2262. doi: 10.1038/sj.npp.1300811. [DOI] [PubMed] [Google Scholar]
- Bowen CA, Negus SS, Kelly M, Mello NK. The effects of heroin on prolactin levels in male rhesus monkeys: use of cumulative dosing procedures. Psychoneuroendocrinology. 2002;27:319–336. doi: 10.1016/s0306-4530(01)00053-1. [DOI] [PubMed] [Google Scholar]
- Brown NA, Bethea CL. Cloning of decidual prolactin from rhesus macaque. Biol Reproduction. 1994;50:543–552. doi: 10.1095/biolreprod50.3.543. [DOI] [PubMed] [Google Scholar]
- Butelman ER, Ball JW, Kreek MJ. Comparison of the discriminative and neuroendocrine effects of centrally-penetrating kappa-opioid agonists in rhesus monkeys. Psychopharmacology. 2002;164:115–120. doi: 10.1007/s00213-002-1195-y. [DOI] [PubMed] [Google Scholar]
- Butelman ER, Ball JW, Kreek MJ. Peripheral selectivity and apparent efficacy of dynorphins: Comparison to non-peptidic kappa-opioid agonists in rhesus monkeys. Psychoneuroendocrinology. 2004;29:307–326. doi: 10.1016/s0306-4530(03)00030-1. [DOI] [PubMed] [Google Scholar]
- Butelman ER, Harris TJ, Kreek M. Apparent efficacy of kappa-opioid receptor ligands on serum prolactin levels in rhesus monkeys. Eur J Pharmacol. 1999;383:305–309. doi: 10.1016/s0014-2999(99)00640-8. [DOI] [PubMed] [Google Scholar]
- Butelman ER, Mandau M, Tidgewell K, Prisinzano TE, Kreek MJ. Effects of salvinorin A, a kappa-opioid hallucinogen, on a neuroendocrine biomarker assay in non-human primates with high kappa-receptor homology to humans. J Pharmacol Exp Ther. 2007;320:300–306. doi: 10.1124/jpet.106.112417. [DOI] [PubMed] [Google Scholar]
- Butelman ER, Reed B, Chait BT, Mandau M, Yuferov V, Kreek MJ. Limited effects of beta-endorphin compared to loperamide or fentanyl in a neuroendocrine biomarker assay in non-human primates. Psychoneuroendocrinology. 2008;33:292–304. doi: 10.1016/j.psyneuen.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeHaven-Hudkins DL, Burgos LC, Cassel JA, Daubert JD, DeHaven RN, Mansson E, Nagasaka H, Yu G, Yaksh TL. Loperamide (ADL-2-1294), an opioid antihyperalgesic agent with peripheral selectivity. J Pharmacol Exp Ther. 1999;289:494–502. [PubMed] [Google Scholar]
- France CP, Gerak LR. Behavioral effects of 6-methylene naltrexone (nalmefene) in rhesus monkeys. J Pharmacol Exp Ther. 1994;270:992–999. [PubMed] [Google Scholar]
- Groer CE, Tidgewell K, Moyer RA, Harding WW, Rothman RB, Prisinzano TE, Bohn LM. An opioid agonist that does not induce mu-opioid receptor - arrestin interactions or receptor internalization. Mol Pharmacol. 2007;71:549–557. doi: 10.1124/mol.106.028258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding WW, Tidgewell K, Byrd N, Cobb H, Dersch CM, Butelman ER, Rothman RB, Prisinzano TE. Neoclerodane diterpenes as a novel scaffold for mu opioid receptor ligands. J Med Chem. 2005;48:4765–4771. doi: 10.1021/jm048963m. [DOI] [PubMed] [Google Scholar]
- Hoehe M, Duka T, Doenicke A. Human studies on the mu opiate receptor agonist fentanyl: neuroendocrine and behavioral responses. Psychoneuroendocrinology. 1988;13:397–408. doi: 10.1016/0306-4530(88)90046-7. [DOI] [PubMed] [Google Scholar]
- Kreek MJ, Schluger J, Borg L, Gunduz M, Ho A. Dynorphin A1-13 causes elevation of serum levels of prolactin through an opioid receptor mechanism in humans: gender differences and implications for modulation of dopaminergic tone in the treatment of addictions. J Pharmacol Exp Ther. 1999;288:260–269. [PubMed] [Google Scholar]
- Mello NK, Mendelson JH, Kelly M. Acute effects of nalmefene on LH, prolactin, and testosterone in male rhesus monkeys. Pharmacol Biochem Behav. 2000;66:275–283. doi: 10.1016/s0091-3057(00)00190-8. [DOI] [PubMed] [Google Scholar]
- Merchenthaler I. Neurons with access to the general circulation in the central nervous system of the rat: a retrograde tracing study with fluoro-gold. Neuroscience. 1991;44:655–662. doi: 10.1016/0306-4522(91)90085-3. [DOI] [PubMed] [Google Scholar]
- Nordstrom AL, Farde L. Plasma prolactin and central D2 receptor occupancy in antipsychotic drug-treated patients. J Clin Psychopharmacol. 1998;18:305–310. doi: 10.1097/00004714-199808000-00010. [DOI] [PubMed] [Google Scholar]
- Pascoe JE, Williams KL, Mukhopadhyay P, Rice KC, Woods JH, Ko MC. Effects of mu-, kappa, and delta- opioid receptor agonists on the function of hypothalamic-pituitary adrenal axis in monkeys. Psychoneuroendocrinology. 2008 doi: 10.1016/j.psyneuen.2008.01.006. online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecins-Thompson M, Brown NA, Kohama SG, Bethea CL. Ovarian steroid regulation of tryptophan hydroxylase mRNA expression in rhesus macaques. J Neurosci. 1996;16:7021–7029. doi: 10.1523/JNEUROSCI.16-21-07021.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prisinzano TE, Rothman RB. Salvinorin A analogs as probes in opioid pharmacology. Chemical Reviews. 2008 doi: 10.1021/cr0782269. online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Salvinorin A: A potent naturally occuring nonnitrogenous kappa opioid selective agonist. Proc Natl Acad Sci U S A. 2002;99:11934–11939. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ur E, Wright DM, Bouloux PM, Grossman A. The effects of spiradoline (U-62066E), a kappa-opioid receptor agonist, on neuroendocrine function in man. Br J Pharmacol. 1997;120:781–784. doi: 10.1038/sj.bjp.0700971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentino RJ, Katz JL, Medzihradsky F, Woods JH. Receptor binding, antagonist, and withdrawal precipitating properties of opiate antagonists. Life Sci. 1983;32:2887–2896. doi: 10.1016/0024-3205(83)90325-9. [DOI] [PubMed] [Google Scholar]
- Xu H, Partilla JS, Wang X, Rutherford JM, Tidgewell K, Prisinzano TE, Bohn LM, Rothman RB. A comparison of noninternalizing (herkinorin) and internalizing (DAMGO) mu-opioid agonists on cellular markers related to opioid tolerance and dependence. Synapse. 2007;61:166–175. doi: 10.1002/syn.20356. [DOI] [PubMed] [Google Scholar]
- Yuan CS, Wei G, Foss JF, O'Connor M, Karrison T, Osinski J. Effects of subcutaneous methylnaltrexone on morphine-induced peripherally mediated side effects: a double-blind randomized placebo-controlled trial. J Pharmacol Exp Ther. 2002;300:118–123. doi: 10.1124/jpet.300.1.118. [DOI] [PubMed] [Google Scholar]
- Zheng SX, Bosch MA, Ronnekleiv OK. mu-opioid receptor mRNA expression in identified hypothalamic neurons. J Comp Neurol. 2005;487:332–344. doi: 10.1002/cne.20557. [DOI] [PubMed] [Google Scholar]