

Abstract
QS-21 is one of the most promising new adjuvants for immune response potentiation and dose-sparing in vaccine therapy given its exceedingly high level of potency and its favorable toxicity profile. Melanoma, breast cancer, small cell lung cancer, prostate cancer, HIV-1, and malaria are among the numerous maladies targeted in more than 80 recent and ongoing vaccine therapy clinical trials involving QS-21 as a critical adjuvant component for immune response augmentation. QS-21 is a natural product immunostimulatory adjuvant, eliciting both T-cell- and antibody-mediated immune responses with microgram doses. Herein is reported the synthesis of QS-21Aapi in a highly modular strategy, applying novel glycosylation methodologies to a convergent construction of the potent saponin immunostimulant. The chemical synthesis of QS-21 offers unique opportunities to probe its mode of biological action through the preparation of otherwise unattainable nonnatural saponin analogues.
Introduction
The development of therapeutic vaccines for the treatment of diseases has rapidly grown beyond the established strategy of using lifeless or attenuated microorganisms to include the use of macromolecules and/or small molecules to elicit immune response. For example, the use of subunit vaccine antigens encompasses selected disease-associated glycoproteins, recombinant proteins, synthetic peptides, and even nonimmunogenic complex carbohydrates, which can be rendered immunogenic when conjugated to an appropriate immunocarrier protein. However, subunit antigen vaccines are inherently less immunogenic than those employing attenuated microorganisms. Consequently many antigen formulations require co-administration with an adjuvant or immunostimulating complex (ISCOM),1 a substance that is itself not necessarily immunogenic but functions in concert with the antigen to enhance/prolong immune response.
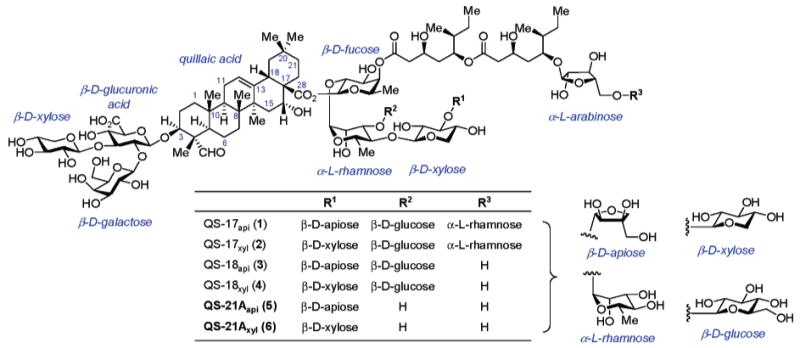
It had been known for decades that semi-purified extracts of the South American tree, Quillaja saponaria Molina, exhibit remarkable adjuvant activity.2 In 1991, Kensil et al. reported the purification, isolation, and partial characterization of selected minor constituents of the bark extract from Quillaja saponaria.3 Among the more adjuvant-active components of these extracts are several complex triterpene saponins (Chart 1), including QS-17api/xyl (1 and 2), QS-18api/xyl (3 and 4), and QS-21Aapi/xyl (5 and 6).3 Of these constituents, both QS-21Aapi (5) and QS-21Axyl (6) have emerged as being among the most promising new adjuvants for immune response potentiation and dose-sparing in vaccine therapy, given their exceedingly high level of potency and favorable toxicity profile.4 QS-21A (the 21st fraction from RP-HPLC) was identified through extensive chemical degradation and spectroscopic studies5 to be a mixture of two principal isomeric triterpene glycoside saponins, QS-21Aapi (5) and QS-21Axyl (6), each incorporating a central quillaic acid triterpene core, flanked on either side by complex oligosaccharides. The trisaccharide moiety attached to the C3-position of quillaic acid is composed of d-glucuronic acid, d-galactose, and d-xylose, while the tetrasaccharide substructure attached at the C28 carboxylate of the triterpene comprises a linear array of d-fucose, followed by l-rhamnose and d-xylose linked to one of two isomeric sugars, d-apiose or d-xylose, for QS-21Aapi (5) or QS-21Axyl (6), respectively. The remaining component, a structurally elaborate l-arabinose-terminated fatty acyl chain attached to the 4-position of the fucose residue, completes the structural makeup of these amphiphilic triterpene saponins.
Chart 1.
Clinical trials employing QS-21A adjuvant have involved, inter alia, formulations of ganglioside–keyhole limpet hemocyanin (KLH) conjugates for melanoma,6 globo-H oligosaccharide–KLH conjugates for prostate7 and breast8 cancers, MUC1 peptide–KLH conjugates for breast cancer,9 and N-propionylated polysialic acid–KLH conjugates for small-cell lung cancer.10 In addition, several murine and human studies have demonstrated promising adjuvant effects of QS-21A in vaccine formulations with recombinant gp120 against HIV-111 and with synthetic Plasmodium falciparum peptides against malaria.12 Both animal and human studies on the adjuvant activity of QS-21A indicate that it elicits both Th1- and Th2-type cytokines and amplifies both the T-cell- and B-cell-mediated immune responses in microgram doses. Thus, QS-21A is an immunostimulatory adjuvant, possibly functioning to facilitate the uptake of the antigen into antigen-presenting cells.4
However, QS-21A is essentially nonrenewable in that it is only present as a minor constituent in the bark of Quillaja saponaria.3 The development of a chemical synthesis of QS-21A would not only expand its availability but also offer opportunities to access otherwise unattainable nonnatural saponin analogues to probe its mode of biological action. Reported synthetic efforts directed at QS-21A are sparse, consisting of the preparation of the fully acetylated trisaccharide and tetrasaccharide components.13 Herein is reported a detailed account of the synthesis of QS-21Aapi (5).14
Results and Discussion
Construction of the triterpene saponin with maximum convergence (Chart 2) relies on the acquisition of the triterpene (7), the branched trisaccharide (8), the linear tetrasaccharide (10), and fatty acyl chain (9) as the principal substructure quadrants. With this strategy, the synthesis of the oligosaccharide portions of the natural product through various glycosylation methods appears to be, at least on the surface, a straightforward task in that all of the glycosidic linkages are of the 1,2-trans configuration, and are thereby susceptible to traditional neighboring group participation effects.15 However, the vast majority of neighboring group participatory functionalities consist of acyl protective groups such as esters, which have recently been shown to be useful in the synthesis of the oligosaccharide fragments of QS-21.13b Unfortunately, such intermediates are unlikely to be directly viable in a synthesis of QS-21A without extensive protective group exchange due to functional group incompatibilities during late-stage protecting group (i.e., ester) removal. Indeed, the reported hydrolytic lability of the acyl chain16 necessitates the selection of differentiated oligosaccharide protective groups that can be removed without disrupting this sensitive ester functionality in the final stages of the synthesis.
Chart 2.

Synthesis of the Branched Trisaccharide and Its Conjugation to Quillaic Acid
The quillaic acid subunit (11, Scheme 1) is a 30-carbon terpene of the Δ12-oleanane family. Semi-purified bark of the Quillaja saponaria tree is commercially available and has been analyzed to contain 25% of various terpenes, with the major terpene constituent being the desired quillaic acid. Quillaic acid has previously been isolated from the Quillaja bark via acid hydrolysis followed by extraction and recrystallization.17 Acid-mediated hydrolysis of the oligosaccharide components from the crude commercial Quillaja bark was performed in refluxing aqueous 1.0 N HCl followed by continuous extraction with ether, providing gram quantities of 11, following silica gel chromatography. The C28-carboxyl group was then selectively protected as its methyl ester 12 (Cs2-CO3, MeI, 68%) to provide a suitable model glycosyl acceptor for coupling with a preformed trisaccharide fragment of QS-21A.
Scheme 1a.

a Reagents and conditions: (a) MeI, Cs2CO3, DMF (68%).
The branched trisaccharide subunit of QS-21A (8, Chart 2) is composed of a central d-glucuronate residue (GlcUA) and two peripheral monosaccharides, d-galactopyranose (Gal) and d-xylopyranose (Xyl). The presence of the C2-O-branched substructure within the glucuronate fragment of the branched trisaccharide reveals an opportunity to apply our sulfonium-mediated oxidative glycosylation procedure18 with a glucuronal substrate such as 15 (Scheme 2). The synthesis of the glucuronal 15 commenced with glucurono-6,3-lactone (13) according to the four-step procedure of Nishimura19 providing the methyl 3,4-di-O-acetyl-d-glucuronal 14 in 72% overall yield. Installation of orthogonal protective groups was then accomplished by methanolysis of the acetate esters within 14 followed by site-selective sequential TBS protection of the C3-hydroxyl (TBSCl, imidazole) and acetylation at the C4-hydroxyl (Ac2O, DMAP) to provide the glucuronal 15 in 59% yield (three steps).
Scheme 2a.
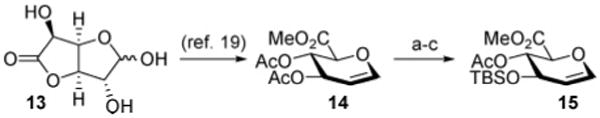
a Reagents and conditions: (a) NaOMe, MeOH (81%); (b) TBSCl, imidazole, DMF (76%); (c) Ac2O, DMAP, CH2Cl2 (96%).
Access to glucuronal 15 permitted application of our oxidative glycosylation employing triflic anhydride and diphenyl sulfoxide (Scheme 3).18 Initial model investigations with 2-propanol as a glycosyl acceptor led to the efficient formation of the C2-hydroxy-β-glucuronate 17 (85%). Good yields as well as exclusive β-selectivities were also observed when allyl alcohol, o-nitrobenzyl alcohol and p-methoxybenzyl alcohol were employed as acceptors, (i.e., 18–20), each of which could function as a potentially useful anomeric protective group. It is worth noting that 1H NMR studies conducted on the oxidative glycosylation with glucuronal 15 revealed that the α-1,2-anhydroglucuronate 16 was indeed formed in >90% as the principal carbohydrate species prior to introduction of the glycosyl acceptor. Finally, attempts at oxidative glycosylation of quillaic acid methyl ester (12, HO-QA-Me) were less successful, leading to a low yield of the GlcUA–O-QA-Me conjugate 21 (31%), presumably a result of the increased steric bulk associated with the neopentyl-like structure of this hydroxyl glycosyl acceptor. As a consequence, the glucuronate 20, incorporating the anomeric p-methoxybenzyl group, was advanced in the synthesis of an appropriate oligosaccharide fragment suitable for glycosylation of the triterpene core.
Scheme 3a.

a Reagents and conditions: (a) Ph2SO, Tf2O, TBP; MeOH, Et3N; ROH, ZnCl2.
With the availability of C2-hydroxyglucuronate 20, efforts were directed toward the synthesis of the galactose β-(1→2) glucuronate glycosidic linkage (Table 1) within the trisaccharide subunit of QS-21. Activation of the 2,3,4,6-tetra-O-benzylgalactopyranose (22) with Ph2SO and Tf2O (entry 1),20 followed by addition of glucuronate 20, provided the disaccharide 24 (85%), although with complete selectivity for the undesired α-anomer. With the aim of effecting anomeric control through neighboring-group participatory effects, 2,3,4,6-O-tetrabenzoyl-d-galactopyranose (23) was employed as the donor. This proved only moderately effective as sulfoxide-mediated dehydrative glycosylation of 20 with 23 to form 25 (entries 2 and 3), in either PhMe/CH2Cl2 (3:1) or CHCl3 as solvent, led to 3:2 and 1:3 (α:β) anomeric selectivities, respectively. The lack of high β-selectivity in these glycosylation reactions is likely due to competing anchimeric influences from the Lewis basic carbonyl functionalities of both the C2-α-benzoate (favoring β-anomeric selectivity) and the C4-β-benzoate (favoring α-anomeric selectivity) within the donor 23.21
Table 1a.
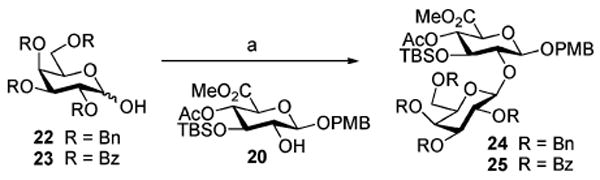 | ||||
|---|---|---|---|---|
| entry | donor | solvent | yield (%) | (α/β) |
| 1 | 22 | CH2Cl2 | 85 (24) | α only |
| 2 | 23 | PhMe/CH2Cl2 (3:1) | 99 (25) | 3:2 |
| 3 | 23 | CHCl3 | 96 (25) | 1:3 |
Reagents and conditions: (a) Ph2SO, Tf2O, TBP; add 20.
However, exclusive β-glycoside formation could be achieved through a simple protective group modification of the galactopyranose donor, such as 3,4,6-tetra-O-benzyl-2-O-benzoyl-d-galactopyranose (27, Scheme 4), wherein the C4-hydroxyl is capped with a nonparticipatory benzyl ether. This donor could be easily accessed via our recently disclosed hypervalent iodine-mediated 1,2-bis(acyloxylation)22 of tribenzyl galactal 26 employing PhI(OBz)2 and BF3·OEt2 to provide benzoyl 3,4,6-tri-O-benzyl-2-O-benzoyl-β-d-galactopyranoside. Subsequent deprotection of the resulting anomeric benzoate (NH3, MeOH) yielded the desired galactose hemiacetal 27 in 60% yield over two steps, whereas conventional methodologies would have required a protracted multistep sequence to access 27. As expected, use of the newly synthesized galactose hemiacetal donor 27 in the glycosylation of the C2-hydroxy glucuronate acceptor 20 afforded exclusively the β-disaccharide 28 (80%).
Scheme 4a.
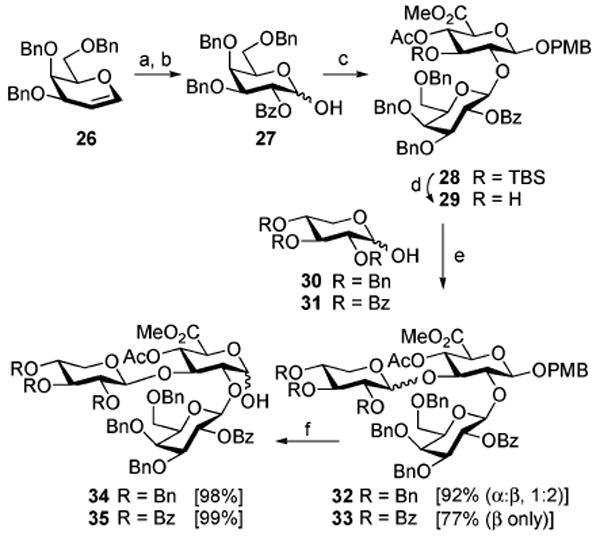
a Reagents and conditions: (a) PhI(OBz)2, BF3·OEt2, CH2Cl2 (83%); (b) NH3, MeOH, THF (72%); (c) Ph2SO, Tf2O, TBP; add 20 (80%); (d) HF·pyr, THF (99%); (e) 31 or 32, Ph2SO, Tf2O, TBP; then 29; (f) TFA, H2O, CHCl3.
Completion of the synthesis of the trisaccharide fragment involved removal of the TBS group within 28 with HF·pyridine, unveiling alcohol 29 (99%) with no evidence of C4-acetyl migration. Glycosylation of disaccharide 29 with 2,3,4-tri-O-benzyl-d-xylose (30) afforded trisaccharide 32 (92%) with an anomeric ratio of 1:2 (α:β). In an attempt to increase the anomeric ratio to favor exclusively the β-xyloside, 2,3,4-tri-O-benzoyl-d-xylose (31) was investigated as an alternate donor. Indeed, with 31 and disaccharide acceptor 29, the corresponding β-anomer of trisaccharide 33 was obtained exclusively (77%). Subsequent removal of the anomeric PMB acetal in 32 and 33 (TFA) provided the trisaccharide hemiacetals 34 and 35, respectively, in near quantitative yields. Although the β-anomeric selectivity with the use of 2,3,4-tri-O-benzyl-d-xylose (30) to form 32 was lower than that with 2,3,4-tri-O-benzoyl-d-xylose (31) to form 33, the isolated yield of the β-anomer of 32 versus that of 33 were not significantly disparate. Given the likely difficulties associated with multiple simultaneous saponification events required for future protective group exchanges of the ester functionalities in the synthesis, the trisaccharide 32β was isolated and advanced to 34 for glycosylation of the triterpene core.
Identification of feasible glycosylation conditions for the coupling of the branched trisaccharide 34 with the triterpene fragment turned out to be one of the most challenging tasks during the synthesis of QS-21A. This difficulty arises from pronounced steric hindrance in forming the glucuronate anomeric bond as this is proximal to a sterically demanding array of both C2- and C3-carbohydrate appendages. Unfortunately, several attempts at sulfoxide-mediated glycosylations with a variety of selectively protected trisaccharide hemiacetals with the triterpene 12 met with limited success. The most efficient dehydrative glycosylation procedure arose from enhancement of the nucleophilicity of the triterpene glycosyl acceptor in the form of its 3-O-stannyl ether (37, entry 1, Table 2)23 to form 36 (79%), albeit with exclusive undesired α-anomeric selectivity. Moreover, extensive investigations into the use of other classes of glycosyl donors such as anomeric phosphites, fluorides, and sulfides were all unproductive.24 However, the most promising method in this screen of glycosylation protocols involved the use of the trichloroacetimidate donor, pioneered by Schmidt.25 Thus, conversion of anomeric trisaccharide hemiacetal 34 to the α-trichloroacetimidate donor 38 was accomplished (CCl3CN, DBU) in 95% yield. The use of TBSOTf (entry 2, Table 2) in the glycosylation of HO-QA-Me (12) with imidate 38 proceeded with good efficiency to provide glycoconjugate 36 (78%, comparable to entry 1), although not surprisingly with the thermodynamically favored undesired α-stereochemistry. On the other hand, the use of BF3·OEt2 catalyst (entry 3, Table 2) provided a small but significant quantity of the desired β-glycoside 36β (33% in the highest yielding trial). Notably, the unproductive formation of the glycosyl fluoride derivative 39 (≥18%) during the course of this reaction could not be avoided in our hands, despite extensive purification and drying of both the coupling substrates and the acid catalyst. Nevertheless, the procedure in this last entry at least indicated the feasibility of accessing the desired β-glycoconjugate, albeit with suboptimal efficiency. Therefore, probing appropriate modifications of the trichloroacetimidate glycosylation protocol should lead to a viable means with which to secure the trisaccharide–triterpene β-glycosidic linkage in QS-21 (vide infra).
Table 2a.
 | |||||
|---|---|---|---|---|---|
| entry | donor | acceptor | reagent | 36 (%) | 39 (%) |
| 1 | 34 | (Bu3SnO-QA-Me) | Ph2SO, Tf2O | 79 (α) | – |
| 2 | 38 | 12 | TBSOTf | 78 (α) | – |
| 3 | 38 | 12 | BF3·OEt2 | 33 (β) | 18 (α) |
Reagents and conditions: (a) CCl3CN, DBU, CH2Cl2 (96%); (b) 12 or 37, “reagents,” CH2Cl2.
Synthesis of the Tetrasaccharide Fragment of QS-21Aapi
The linear tetrasaccharide component of QS-21A is composed of the four constituent monosaccharides, l-rhamnose, d-xylose, d-apiose, and d-fucose. Each of the selectively protected forms of these monosaccharides was prepared according to standard carbohydrate functional group interconversion processes (Scheme 5). The selectively protected apiose derivative 41 (Scheme 5A) was synthesized from 2,3-di-O-isopropylidene-d-apiose (40)26 via selective silylation of the C4′-hydroxyl (74%). Triisopropylsilyl 2,3-di-O-isopropylidene-β-l-rhamnopyranoside (43, Scheme 5B) was accessed from 4-O-acetyl-2,3-di-O-isopropylidene-β-d-rhamnopyranose (42)27 via anomeric silylation (TIP-SCl, imidazole) followed by acetate hydrolysis (K2CO3, MeOH). Preparation of 2,4-di-O-benzyl-d-xylose (45, Scheme 5C) was achieved via a short sequence involving anomeric acetal exchange of d-xylose (44) with allyl alcohol (77%), one-pot dibenzylation of the C2- and C4-hydroxyl groups (BnCl, NaH) with moderate selectivity (45%),28 and finally allyl acetal removal (t-BuOK, then HCl) to provide 45 (71%). The initial route to preparation of the differentially protected fucose moiety 51 (Scheme 5D) involved conversion of d-fucose (46) to tri-O-acetylfucosyl bromide 47 (Ac2O, Et3N; HBr, AcOH), which then served to glycosylate o-nitrobenzyl alcohol (49%, two steps). The esters within the resulting fucoside were then hydrolyzed (K2CO3, MeOH; 90%) to form triol 48,29 allowing for monobenzylation of the C3-hydroxyl with moderate selectivity via its transient stannylene acetal derivative to generate 49. Subsequent sequential C2-O-TBS protection and C4-O-acetylation, followed by acid hydrolysis of the TBS ether provided o-nitrobenzyl 4-O-acetyl-3-O-benzyl-β-d-fucopyranoside (51). While this initial attempt at securing a suitable differentially protected d-fucose building block followed a somewhat suboptimal protracted sequence, it did serve us well in the evaluation of initial model studies for the synthesis of a protected form of QS-21Aapi.
Scheme 5a.
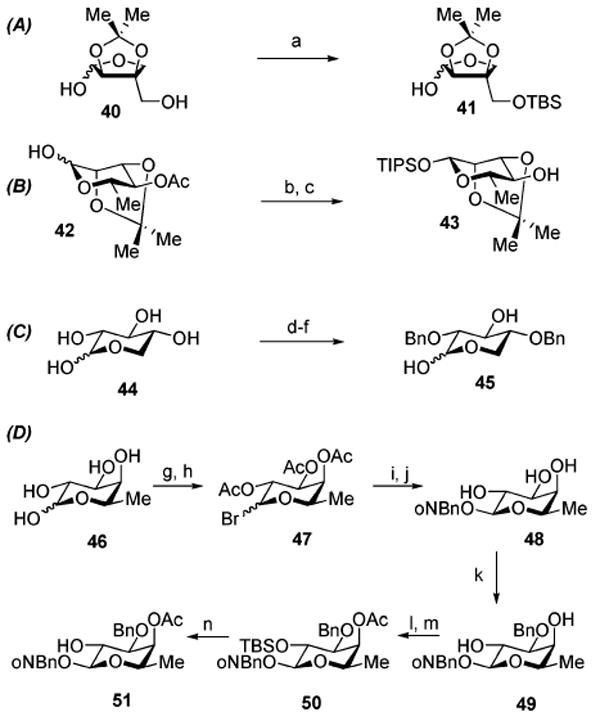
a Reagents and conditions: (a) TBSCl, imid, DMF (74%); (b) TIPSCl, AgNO3, imid, DMF (48% β (and 40% α)); (c) K2CO3, MeOH (98%); (d) allyl-OH, MeCOCl (77%, 3:1, α: β); (e) BnCl, NaH, 95 °C (45%); (f) KO-t-Bu, DMSO, 100 °C; then HCl, acetone, reflux (71%); (g) Ac2O, Et3N, CH2Cl2 (84%); (h) HBr, AcOH, CH2Cl2; (i) o-NO2-BnOH, Ag2O, CH2Cl2 (58%, 2 steps); (j) K2CO3, MeOH (90%); (k) Bu2SnO, BnBr, Bu4NBr, PhCH3 (40%); (l) TBSCl, imid, DMF (96%); (m) Ac2O, DMAP, CH2Cl2 (84%); (n) p-TsOH, MeOH (72%).
The assembly of the linear tetrasaccharide fragment of QS-21Aapi commenced with a novel chemoselective application of our sulfoxide-mediated dehydrative glycosylation20 in which a 1,3-diol glycosyl donor is employed in the coupling (Scheme 6). In this sequence, 2,4-di-O-benzyl-d-xylopyranose (45) was activated with excess Ph2SO (5.6 equiv) and Tf2O (2.8 equiv), followed by the introduction of triisopropylsilyl 2,3-di-O-isopropylidene-β-l-rhamnopyranose (43) to afford the 1→4-β-linked disaccharide 52 (55%) as a single constitutional and stereoisomer.30 The resulting disaccharide 52, with its free hydroxyl group on the xylose ring, was immediately used as the nucleophilic glycosyl acceptor in a direct glycosylation with apiose 41, providing the trisaccharide 53 (88%, α only). Subsequent selective anomeric desilylation of 53 (TBAF) furnished the trisaccharide hemiacetal 54 (98%), which served to glycosylate the C2-hydroxyl of fucose 51 (Ph2SO, Tf2O) to provide the fully protected linear tetrasaccharide 55 (85%). With de-acylation of the fucosyl-C4-hydroxyl group in 55, the tetrasaccharide 56 was produced (91%), ready to be coupled to the acyl side chain.
Scheme 6a.
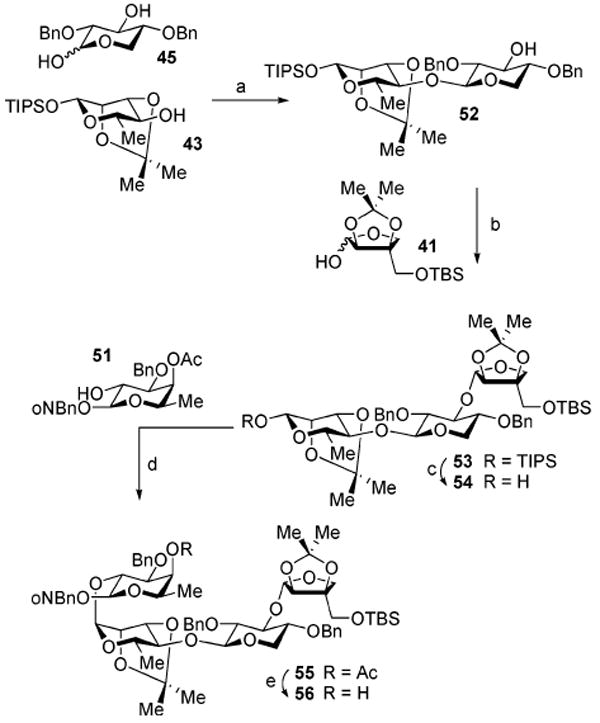
a Reagents and conditions: (a) Ph2SO, Tf2O, TBP, CH2Cl2; 43 (55%); (b) 41, Ph2SO, Tf2O, TBP, CH2Cl2; add 52 (88%); (c) TBAF, THF (98%); (d) Ph2SO, Tf2O, TBP, CH2Cl2; add 51 (85%); (e) K2CO3, MeOH (91%).
Synthesis of the Acyl Chain of QS-21
The asymmetric synthesis of the dimeric fatty acyl chain of QS-21A began (Scheme 7) with treatment of commercially available isobutyl-acetoacetate (57) with sodium hydride followed by n-BuLi to provide the corresponding dianion, which immediately was exposed to BOMCl to provide the β-ketoester 58 (70%). Asymmetric reduction of the ketone in 58 with Noyori's (R)-BINAP–RuBr2 catalyst31 at elevated pressure (750 psi) provided the desired (R)-enantiomer 59 in near quantitative yield and excellent enantioselectivity (>98:2 er). The resulting alcohol 59 was protected as its TBS ether (>99%), which underwent ester reduction (DIBAL-H) to afford aldehyde 60 (93%). Diastereoselective Brown crotylation32 of aldehyde 60 with (Z)-crotyl(diisopinocamphenyl)borane provided a mixture of the diastereomers 61 and 62 (83%) in a diastereomeric ratio of 1:2 favoring the desired (S,S,S)-diastereomer 62.33 Despite the poor diastereoselectivity in this crotylation due to a stereochemical “mismatch” in this pair of reagent substrates, the major isomer 62 was separated and advanced in the synthesis of the acyl chain.
Scheme 7a.
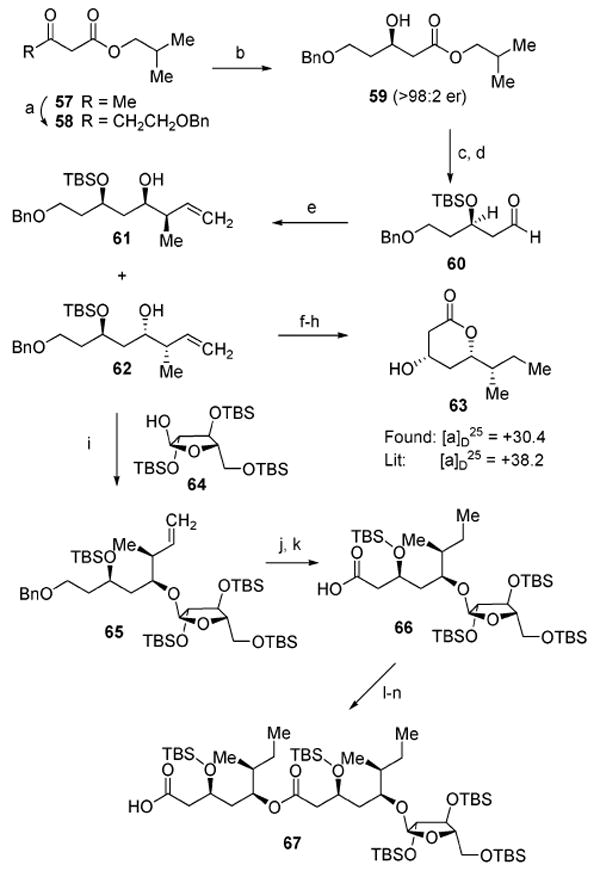
a Reagents and conditions: (a) NaH, THF; n-BuLi; BOMCl (70%); (b) (R)-BINAP·RuBr2·Et3N (cat.), H2, MeOH (>99%); (c) TBSCl, imid, DMF (>99%); (d) DIBAL-H, PhMe, −78 °C (93%); (e) (+)-(ipc)2B(OMe), Z-MeCH=CHCH2Li (83%, 1:2, 61:62); (f) H2, Pd/C, MeOH (73%); (g) TPAP, NMO, CH2Cl2 (85%); (h) TBAF, THF (51%); (i) 64, Ph2SO, Tf2O, TBP, PhMe/CH2Cl2 (1:1); 62 (90%, 2:1, α: β); (j) H2, Pd/C, MeOH (94%); (k) RuCl3·H2O, NaIO4, H2O, MeCN (88%); (l) 2,4,6-Cl3C6H2COCl, Et3N, DMAP, CH2Cl2; add 62 (96%); (m) H2, Pd/C, MeOH (73%); (n) RuCl3·H2O, NaIO4, H2O, MeCN (96%).
At this point, it was prudent to verify that the homoallylic alcohol 62 indeed possessed the resident stereocenters whose absolute configurations conformed to that of the natural product. This involved its derivatization to the lactone 63, which had been previously synthesized and verified as containing the correct absolute configuration (S,S,S) of the QS-21 acyl chain. Thus, hydrogenation/hydrogenolysis of 62 (H2, Pd/C) provided the corresponding saturated diol (73%), which was then subjected to TPAP oxidation (85%) and TBS removal (51%) to provide lactone 63. Comparison of data of this compound to the reported data of the naturally derived lactone revealed identical 1H NMR resonances and comparable optical rotation values.5b
With access to the desired diastereomer of the homoallylic alcohol 62, efforts were directed at glycosylation of its hydroxyl group with the arabinofuranose 64. Following extensive investigations, the best result obtained involved sulfoxide-mediated dehydrative glycosylation (Ph2SO, Tf2O) performed in a 1:1 (v:v) solvent mixture of PhMe and CH2Cl2, providing the desired α-anomer 65 in 60% yield along with the undesired β-anomer in 30% yield. Completion of the synthesis of the dimeric acyl chain was then accomplished with a series of operations including: (1) simultaneous alkene hydrogenation and benzyl ether hydrogenolysis (94%); (2) oxidation of the primary alcohol to the carboxylic acid 66 with RuCl3 and NaIO4 (88%); (3) esterification of the homoallylic alcohol 62 with the activated Yamaguchi anhydride34 derivative of 66 (96%); and (4) sequential hydrogenation/hydrogenolysis of the alkene/benzyl ether (73%) and alcohol oxidation to provide the carboxylic acid 67 (96%).
Synthesis of Protected QS-21Aapi
With the availability of all fully protected quadrants of QS-21Aapi, efforts focused on their late-stage convergent assembly. The initial task involved the preparation of the trisaccharide–triterpene substructure following a modified protocol for trichloroacetimidate glycosylation. It was clear from our earlier model studies for trichloroacetimidate glycosylation of protected quillaic acid (see Table 2, entry 2) that the key issue in maximizing the coupling yield with trisaccharide 38 was to minimize the unwanted formation of glycosyl fluoride 39. To circumvent this problem of glycosyl fluoride formation, tris(pentafluorophenyl)borane (B(C6F5)3)35 was selected as a potentially useful catalyst in this context. B(C6F5)3, lacking the reactive B–F bond, has been used as a catalyst in a variety of transformations, including Mukaiyama aldol,36 Sakurai–Hosomi allylation,37 and epoxide rearrangement,38 exhibiting a reactivity profile similar to that of BF3·OEt2.
Thus, the suitably protected quillaic acid C28-O-allyl derivative 68 (Scheme 8) was prepared by allylation of the cesium carboxylate of 11, and 68 was then subjected to glycosylation with the α-trichloroacetimidate 38 employing 3 mol % of tris-(pentafluorophenyl)borane ((C6F5)3B) as the catalyst. Under these conditions, the desired glycoconjugate 69 was produced in 59% (α: β, 1:7) (15% recovered 34) with no evidence of formation of unwanted glycosyl fluoride that plagued previous glycosylations employing BF3·OEt2 as catalyst. Subsequent exchange of the ester protecting groups, initially required to secure anomeric selectivity in the preceding coupling reactions, to groups which are labile to either mild acid or hydrogenolysis in the final deprotection steps in the synthesis was then performed. These interconversions include: (1) sequential treatment of 69 with sodium hydroxide (NaOH) and Cs2CO3 for ester group hydrolysis; (2) protection of the glucuronic acid carboxylate group as the benzyl ester using benzyl bromide (BnBr) and KHCO3 (92%, two steps); and (3) removal of the allyl ester on C28 in the triterpene (70%) to afford triol-acid 70.
Scheme 8a.
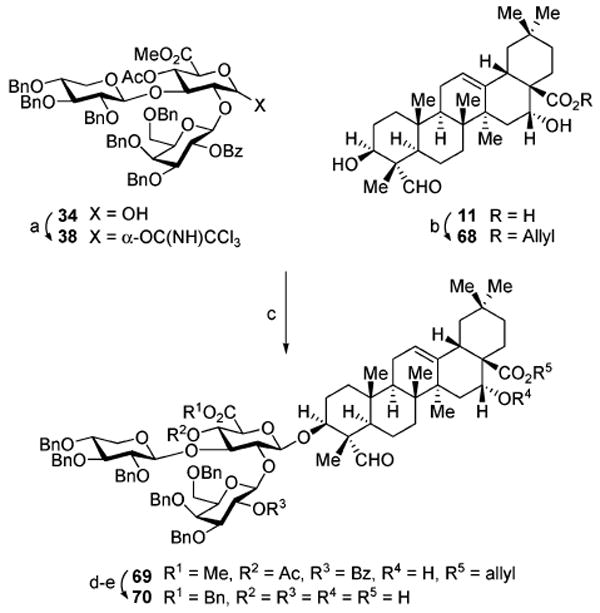
a Reagents and conditions: (a) CCl3CN, DBU, CH2Cl2 (95%); (b) Cs2CO3, allyl-Br, DMF (70%); (c) (B(C6F5)3), CH2Cl2 (59%, 1:7, α: β; plus 15% 34 and 21% 68); (d) NaOH, 1,4-dioxane; then Cs2CO3, H2O, MeOH; (e) KHCO3, BnBr, DMF (92%, 2 steps); (e) HCO2H, Pd(OAc)2), Et3N, PPh3, 1,4-dioxane (70%).
In assembling the tetrasaccharide-acyl chain hemisphere of the immunostimulant (Scheme 9), acylation of the fucose C4-hydroxyl on the linear tetrasaccharide 56 was effected by way of the Yamaguchi anhydride derivative of 67 to form 71 (75%). Exchange of the o-nitrobenzyl acetal to the anomeric trichloroacetimidate was achieved in moderate yield (50%, two steps). Trichloroacetimidate 71 and the trisaccharide–triterpene conjugate 70 were then subjected to BF3·OEt2 activation, resulting in the formation of the β-glycosyl ester 72 (72%), which could be separated from minor side products derived from multiple glycosylations of the hydroxyl groups in 70. During the initial stages of planning the synthesis, care was taken to include only acid- and hydrogenolysis-labile protective groups in the final stages of the synthesis to facilitate global deprotection without basic hydrolysis of the noted labile of ester functionality of the acyl chain. While this particular goal was attained, critical problematic deprotection issues surfaced with the attempted acid-mediated removal of the isopropylidene acetal on the apiose fragment. Despite earlier model studies which suggested otherwise, this ketal could not be removed without acid hydrolysis of specific glycosidic linkages within the saponin construct. Indeed, after extensive experimentation on the deprotection of 72, only a trace quantity of QS-21Aapi among a variety of glycoconjugate fragments could be detected by LC–MS under various hydrogenolysis–acid hydrolysis conditions.
Scheme 9a.
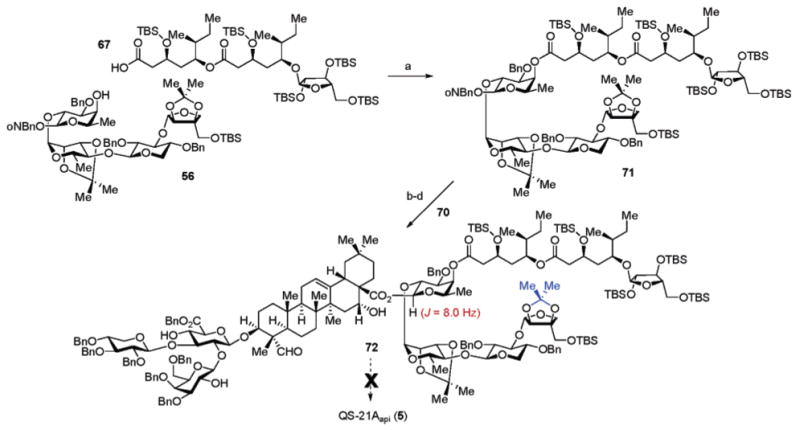
a Reagents and conditions: (a) 67, 2,4,6-Cl3C6H2COCl, Et3N, DMAP, CH2Cl2; add 56 (75%); (b) hv (350 nm), THF; (c) CCl3CN, DBU, CH2Cl2 (50%, 2 steps); (d) 70, BF3·OEt2, CH2Cl2 (72%).
Preparation of a New Acylated Tetrasaccharide Fragment: Synthesis of QS-21Aapi
The unsuccessful attempts at global deprotection of 72, coupled with some suboptimal stereoselective transformations involved in the preparation of the acyl chain, led us to overhaul the synthesis of the entire acylated tetrasaccharide fragment of the natural product. One of the key bottlenecks in acquiring adequate quantities of forefront synthetic intermediates involved the rather protracted preparation of the selectively protected d-fucose moiety within QS-21A (Scheme 5D, vide supra). Attempts to streamline this process involved a preparation of 75 (Scheme 10) commencing with the formation of allyl fucopyranoside (88%) from d-fucose (46). Regioselective benzylation (CsF, BnBr) of the intermediate allyl fucoside via its stannylene acetal intermediate afforded the diol 73 (67%), whose remaining equatorial hydroxyl group was silylated (TBSCl, imidazole) as the TBS ether (94%). Following acetylation of the axial C4-hydroxyl (99%), anomeric allyl acetal removal (Et2Zn, P(PPh3)4) and subsequent desilylation (TBAF) afforded the corresponding 1,2-diol (59%, two steps), which could be resilylated at the anomeric position to afford the β-anomeric TIPS fucoside 75 (49% plus 30% 1,2-di-O-TIPS derivative available for recycling).
Scheme 10a.

a Reagents and conditions: (a) allyl-OH, AcCl (88%); (b) Bu2SnO, PhMe; CsF, BnBr, DMF (67%); (c) TBSCl, imid, DMF (94%); (d) Ac2O, Et3N, DMAP, CH2Cl2 (99%); (e) Pd(PPh3)4, Et2Zn, THF; (f) TBAF, THF (59%, 2 steps); (g) TIPSCl, imid, DMAP, DMF (49%).
With 75 available in sufficient quantity, preparation of a new suitably protected tetrasaccharide fragment that would obviate the problematic late-stage hydrolysis of the apiose-2,3-di-O-isopropylidene was initiated. Thus, the previously prepared xylose–rhamnose disaccharide 52 (Scheme 6, vide supra), with its free hydroxyl group on the xylose ring, was employed (Scheme 11) as the nucleophilic glycosyl acceptor in a TESOTf-catalyzed glycosylation39 with acetyl 2,3-di-O-acetyl-5-O-benzylapiofuranose (76) to provide the β-apiose anomer of the fully protected trisaccharide (51%). Being mindful of the base-hydrolytic instability of QS-21A, the acetate protecting groups, which controlled anomeric selectivity in the glycosylation, were exchanged for the benzylidene acetal (K2CO3, MeOH; then C6H5CH(OMe)2, p-TsOH). Subsequent removal of the anomeric TIPS group (TBAF) on the rhamnose residue afforded the trisaccharide hemiacetal 77 in 95% over three steps.40 Dehydrative glycosylation (Ph2SO, Tf2O) of triisopropyl 4-O-acetyl-3-O-benzyl-β-d-fucopyranose (75) with the trisaccharide hemiacetal 77 provided the fully protected tetrasaccharide fragment 78 (54%).
Scheme 11a.

a Reagents and conditions: (a) 76, TESOTf, CH2Cl2 (51%); (b) K2CO3, MeOH; (c) C6H5CH(OMe)2, p-TsOH; (d) TBAF, THF (95%, 3 steps); (e) Ph2SO, Tf2O, TBP, CH2Cl2; then 75 (54%).
The synthesis of the acyl chain of QS-21A was also overhauled in light of the “mismatched” diastereoselective crotylation reaction in our initial synthesis (Scheme 7) of this fragment. The modified sequence (Scheme 12) began with asymmetric diastereoselective crotylation32 of 3-O-TBS-propionaldehyde (79) with (+)-(ipc)2B(OMe) and (Z)-MeCH=CHCH2Li to afford the homoallylic alcohol 80 as a single diastereomer (89%, >99:1 dr, 98:2 er). Protection of the hydroxyl group as the benzyl ether, followed by sequential desilylation (TBAF) and Swern oxidation (DMSO, (COCl)2) of the resulting primary alcohol provided the β-alkoxy aldehyde 81 in 81% yield over three steps.
Scheme 12a.

a Reagents and conditions: (a) (+)-(ipc)2B(OMe), Z-MeCH=CHCH2Li (89%, >99:1 dr, 98:2 er); (b) BnBr, NaHMDS, THF, DMF (91%); (c) TBAF, THF (94%); (d) DMSO, (COCl)2, Et3N, CH2Cl2 (95%); (e) 82, LDA, THF; aldehyde 81, MgBr2, −115 °C, (4:1 dr); (f) NaOMe, MeOH (89%, 2 steps); (g) TBSCl, imid, DMF (82%); (h) H2, 10% Pd/C, MeOH (92%); (i) 64, Ph2SO, Tf2O, TBP, CH2Cl2; then 84 (72%); (j) Ba(OH)2·8H2O, MeOH (77%); (k) 2,4,6-C6H2Cl3COCl, Et3N, PhMe; then 84, DMAP (>99%); (l) Ba(OH)2·8H2O, MeOH (83%).
Diastereoselective aldol reaction of the aldehyde 81 with the enolate derived from (R)-2-acetoxy-1,1,2-triphenylethanol (82, plus 2.4 equiv LDA)41 afforded the corresponding β-hydroxy ester (4:1 dr), whose chiral auxiliary was removed by methanolysis to provide 83 (89%, two steps). The methyl ester 83 was then subjected to a series of functional group interconversions, including: (1) protection of the β-hydroxyl group as its TBS ether and (2) reduction with H2 (Pd/C) to effect simultaneous benzyl ether hydrogenolysis and alkene hydrogenation, providing the δ-hydroxy ester 84 in 75% over two steps. Dehydrative glycosylation (Ph2SO, Tf2O) of the hydroxyl group in 84 with 2,3,5-tri-O-TBS-l-arabinofuranose (64) afforded the corresponding α-glycoconjugate (72%), which then provided the carboxylic acid 66 after saponification of its methyl ester (77%) with Ba(OH)2·8H2O. The corresponding carboxylic acid in 66 was then activated as the mixed 2,4,6-trichlorobenzoyl anhydride, which engaged in quantitative acylation of the previously synthesized hydroxy-ester 84. Subsequent hydrolysis of the methyl ester with Ba(OH)2·8H2O, taking care to avoid unwanted hydrolysis of the internal ester group, was accomplished to yield the fully intact glycosylated fatty acyl chain subunit 67 (83%, two steps).
The final convergent stages of the synthesis involved initial acetate saponification in tetrasaccharide 79 to form 85, followed by esterification (90%, two steps) of the fucose C4-hydroxyl in 85 with the mixed 2,4,6-trichlorobenzoyl anhydride of 67, generated in situ from its treatment with 2,4,6-trichlorobenzoyl chloride (2,4,6-C6H2Cl3COCl). Removal of the anomeric TIPS group in 86 (TBAF) provided the hemiacetal 87 (81%), whose anomeric hydroxyl was converted (CCl3CN, DBU) to the corresponding α-trichloroacetimidate 88 (56%, plus 40% recovered hemiacetal 87) to serve as the glycosyl donor in triterpenoid glycosylation.
Generation of the corresponding trisaccharide–triterpene glycosyl acceptor for coupling with 88 also incorporated minor modifications relative to the earlier synthetic route to 72 (Scheme 9, vide supra). To avoid the potential for multiple glycosylations onto a partially protected glycosyl acceptor such as 70 (Scheme 8, vide supra), a fully protected version of the trisaccharide–triterpene-C28-carboxylic acid (89, Scheme 13) was prepared from 69, involving the steps of (1) treatment of 69 with sodium hydroxide (NaOH) and Cs2CO3 for ester group hydrolysis; (2) protection of the glucuronic acid carboxylate group as the benzyl ester using benzyl bromide (BnBr) and KHCO3; (3) protection of the remaining hydroxyl groups as the TES ethers; and (4) removal of the allyl ester on C28 in the triterpene, all of which proceeded in 75% yield over the four steps to afford 89.
Scheme 13a.
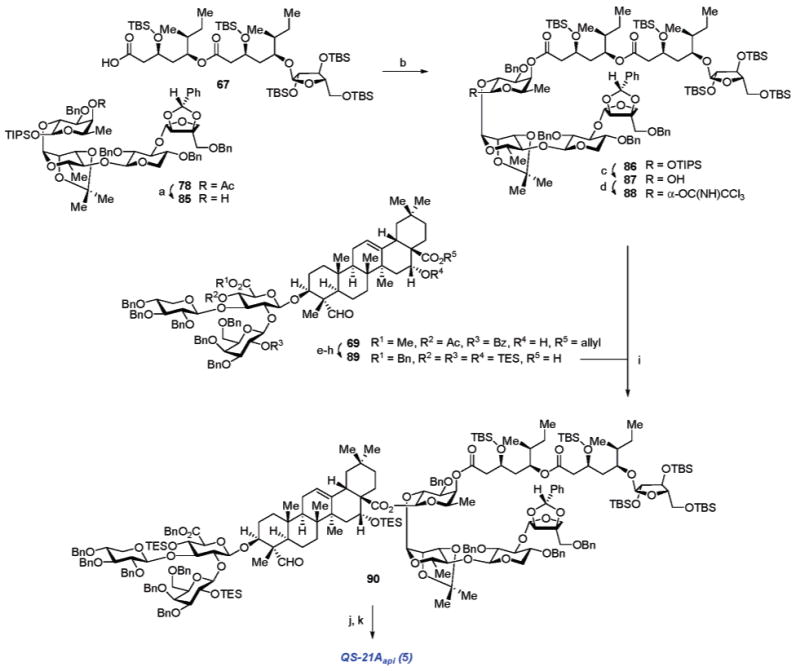
a Reagents and conditions: (a) K2CO3, MeOH; (b) 67, 2,4,6-C6H2Cl3COCl, Et3N, PhMe; 85, DMAP (90%, 2 steps); (c) TBAF, THF (81%); (d) CCl3CN, DBU, CH2Cl2 (56% α, plus 40% recovered 87); (e) NaOH, 1,4-dioxane; then Cs2CO3, H2O, MeOH; (f) KHCO3, BnBr, DMF (92%, 2 steps); (g) TESOTf, 2,6-lutidine, CH2Cl2; (h) HCO2H, Pd(OAc)2, Et3N, PPh3, 1,4-dioxane (81%, 2 steps); (i) BF3·OEt2, CH2Cl2 (70%); (j) TFA, H2O, CH2Cl2, 0 °C; (k) 150 psi H2, Pd/C, THF, MeOH (75%, 2 steps).
The final coupling in the synthesis involves the glycosylation of the trisaccharide–triterpene 70 with 89, employing BF3·OEt2 catalyst to afford the fully protected QS-21Aapi intermediate 90 (70%). The remaining critical operations to complete the synthesis relied on the proper early selection of protecting groups. Thus, mild acid hydrolysis of the rhamnose isopropylidene ketal and all of the silicon (TBS and TES) ethers was accomplished with a 4:1 (v:v) mixture of trifluoroacetic acid (TFA) and water, without compromising the glycosidic linkages or the sensitive ester linkages on the acyl chain of the natural product. Subsequent hydrogenolysis of all benzylic protecting groups with H2 and Pd/C occurred efficiently without reduction of the trisubstituted alkene in the triterpene, providing synthetic QS-21Aapi (5) in 75% yield.42
Conclusion
With the completion of the first synthesis of QS-21Aapi (5), the initial reported structure of the adjuvant has been verified, and availability of this therapeutically important immunostimulant has been expanded to synthetic sources. The potency of QS-21A and its favorable toxicity profile over a broad spectrum of vaccine formulations have established it as one of the most promising new adjuvants for immune response potentiation and dose-sparing. Although reasonable hypotheses highlight the potential of QS-21A to influence cell membrane permeability or to induce cytokine response in local tissue as its means of immunostimulation,4 the mechanism of action of QS-21A has yet to be ascertained. The highly modular synthetic approach to 5 should allow for facile generation of designed structural analogues for determination of its putative minimal pharmacophore requirements as well as investigations into its mechanism of immunostimulatory activity.
Supplementary Material
Experimental details for the preparation and analytical characterization of synthetic intermediates. This information is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This research was supported by the NIH-NIGMS (GM58833), the Alfred P. Sloan Foundation, and Research Corporation. Y.J.K. thanks Lubrizol and Pharmacia (Pfizer) for predoctoral fellowships. We thank Ms. Michelle Adams for proofreading of this manuscript.
References
- 1.(a) Barr IG, Mitchell GF. Immunol Cell Biol. 1996;74:8. doi: 10.1038/icb.1996.2. [DOI] [PubMed] [Google Scholar]; (b) Sjölander A, Cox JC. Adv Drug Delivery Rev. 1998;34:321. doi: 10.1016/s0169-409x(98)00046-5. [DOI] [PubMed] [Google Scholar]
- 2.Dalsgaard K. Arch Gesamte Virusforsch. 1974;44:243. [PubMed] [Google Scholar]
- 3.Kensil CR, Patel U, Lennick M, Marciani D. J Immunol. 1991;148:431–437. [PubMed] [Google Scholar]
- 4.Kensil CR. Crit Rev Ther Drug Carrier Syst. 1996;13:1–55. [PubMed] [Google Scholar]
- 5.(a) Jacobsen NE, Fairbrother WJ, Kensil CR, Lim A, Wheeler DA, Powell MF. Carbohydr Res. 1996;280:1. doi: 10.1016/0008-6215(95)00278-2. [DOI] [PubMed] [Google Scholar]; (b) Zhu X, Yu B, Hui Y, Higuchi R, Kusano T, Miyamoto T. Tetrahedron Lett. 2000;41:717. [Google Scholar]
- 6.Ragupathi G, Livingston PO, Hood C, Gathuru J, Krown SE, Chapman PB, Wolchok JD, Williams LJ, Oldfield RC, Hwu WJ. Clin Cancer Res. 2003;9:5214. [PubMed] [Google Scholar]
- 7.Slovin SF, Ragupathi G, Adluri S, Ungers G, Terry K, Kim S, Spassova M, Bornmann WG, Fazzari M, Dantis L, Olkiewicz K, Lloyd KO, Livingston PO, Danishefsky SJ, Scher HI. Proc Natl Acad Sci USA. 1999;96:5710. doi: 10.1073/pnas.96.10.5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gilewski T, Ragupathi G, Bhuta S, Williams LJ, Musselli C, Zhang XF, Bencsath KP, Panageas KS, Chin J, Hudis CA, Norton L, Houghton AN, Livingston PO, Danishefsky SJ. Proc Natl Acad Sci USA. 2001;98:3270. doi: 10.1073/pnas.051626298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim SK, Ragupathi G, Cappello S, Kagan E, Livingston PO. Vaccine. 2001;19:530. doi: 10.1016/s0264-410x(00)00195-x. [DOI] [PubMed] [Google Scholar]
- 10.Krug LM, Ragupathi G, Ng KK, Hood C, Jennings HJ, Guo Z, Kris MG, Miller V, Pizzo B, Tyson L, Baez V, Livingston PO. Clin Cancer Res. 2004;10:916. doi: 10.1158/1078-0432.ccr-03-0101. [DOI] [PubMed] [Google Scholar]
- 11.(a) Newman MJ, Wu JY, Gardner BH, Anderson CA, Kensil CR, Recchia J, Coughlin RT, Powell MF. Vaccine. 1997;15:1001. doi: 10.1016/s0264-410x(96)00293-9. [DOI] [PubMed] [Google Scholar]; (b) Evans TG, McElrath MJ, Matthews T, Montefiori D, Weinhold K, Wolff M, Keefer MC, Kallas EG, Corey L, Gorse GJ, Belshe R, Graham BS, Spearman PW, Schwartz D, Mulligan MJ, Goepfert P, Fast P, Berman P, Powell M, Francis D. Vaccine. 2001;19:2080. doi: 10.1016/s0264-410x(00)00415-1. [DOI] [PubMed] [Google Scholar]
- 12.Carcaboso AM, Hernández RM, Igartua M, Rosas JE, Patarroyo ME, Pedraz JL. Vaccine. 2004;22:1423. doi: 10.1016/j.vaccine.2003.10.020. [DOI] [PubMed] [Google Scholar]
- 13.(a) Kim YJ, Gin DY. Org Lett. 2001;3:1801–1804. doi: 10.1021/ol015651u. [DOI] [PubMed] [Google Scholar]; (b) Zhu X, Yu B, Hui Y, Schmidt RR. Eur J Org Chem. 2004:965. [Google Scholar]
- 14.Wang P, Kim YJ, Navarro-Villalobos M, Rohde BD, Gin DY. J Am Chem Soc. 2005;127:3256–3257. doi: 10.1021/ja0422007. [DOI] [PubMed] [Google Scholar]
- 15.Nukada T, Berces A, Zgierski MZ, Whitfield DM. J Am Chem Soc. 1998;120:13291–13295. [Google Scholar]
- 16.Cleland JL, Kensil CR, Lim A, Jacobsen NE, Basa L, Spellman M, Wheeler DA, Wu JY, Powell MF. J Pharm Sci. 1996;85:22. doi: 10.1021/js9503136. [DOI] [PubMed] [Google Scholar]
- 17.Elliot DF, Kon GAR. J Chem Soc. 1939:1130–1135. [Google Scholar]
- 18.(a) Di Bussolo V, Kim YJ, Gin DY. J Am Chem Soc. 1998;120:13515–13516. [Google Scholar]; (b) Honda E, Gin DY. J Am Chem Soc. 2002;124:7343–7352. doi: 10.1021/ja025639c. [DOI] [PubMed] [Google Scholar]
- 19.Nishimura SI, Nomura S, Yamada K. Chem Commun. 1998:617. [Google Scholar]
- 20.(a) Garcia BA, Poole JL, Gin DY. J Am Chem Soc. 1997;119:7597–7598. [Google Scholar]; (b) Garcia BA, Gin DY. J Am Chem Soc. 2000;122:4269–4279. [Google Scholar]
- 21.Zinner H, Thielebeule W. Chem Ber. 1960;93:2791. [Google Scholar]
- 22.Shi L, Kim YJ, Gin DY. J Am Chem Soc. 2001;123:6939–6940. doi: 10.1021/ja015991a. [DOI] [PubMed] [Google Scholar]
- 23.David S, Hanessian S. Tetrahedron. 1985;41:643. [Google Scholar]
- 24.Toshima K, Tatsuta K. Chem Rev. 1993;93:1503–1531. [Google Scholar]
- 25.Schmidt RR, Kinzy W. Adv Carbohydr Chem Biochem. 1994;50:21. doi: 10.1016/s0065-2318(08)60150-x. [DOI] [PubMed] [Google Scholar]
- 26.Ho PT. Can J Chem. 1979;57:381. [Google Scholar]
- 27.Nguyen HM, Poole JL, Gin DY. Angew Chem, Int Ed. 2001;40:414. doi: 10.1002/1521-3773(20010119)40:2<414::AID-ANIE414>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 28.Fukase K, Hase S, Ikenaka T, Kusumoto S. Bull Chem Soc Jpn. 1992;65:436. [Google Scholar]
- 29.Nicolaou KC, Hummel CW, Nakada M, Shibayama K, Pitsinos EN, Saimoto H, Mizuno Y, Baldenium KU, Smith AL. J Am Chem Soc. 1993;115:7625–7635. [Google Scholar]
- 30.The specific reason for the high β-selectivity in the glycosylation of 43 in the absence of the C2-participatory group in 45 is unclear, although it is likely that this outcome is highly substrate dependent.
- 31.Noyori R, Ohkuma T, Kitamura M, Takaya H, Sayo N, Kumobayashi H, Akutagawa S. J Am Chem Soc. 1987;109:5856–5858. [Google Scholar]
- 32.Brown HC, Bhat KS. J Am Chem Soc. 1986;108:5919–5931. doi: 10.1021/ja00279a042. [DOI] [PubMed] [Google Scholar]
- 33.(a) White JD, Hong J, Robarge LA. Tetrahedron Lett. 1999;40:1463. [Google Scholar]; (b) Blakemore PR, Browder CC, Hong J, Lincoln CM, Nagornyy PA, Robarge LA, Wardrop DJ, White JD. J Org Chem. 2005;70:5449–5460. doi: 10.1021/jo0503862. [DOI] [PubMed] [Google Scholar]
- 34.Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull Chem Soc Jpn. 1979;52:1989. [Google Scholar]
- 35.Ishihara K, Yamamoto H. Eur J Org Chem. 1999:527. [Google Scholar]
- 36.Ishihara K, Hanaki H, Yamamoto H. Synlett. 1993:577. [Google Scholar]
- 37.Ishihara K, Hanaki H, Funahashi M, Miyata M, Yamamoto H. Bull Chem Soc Jpn. 1995;68:1721–1730. [Google Scholar]
- 38.Ishihara K, Hanaki H, Yamamoto H. Synlett. 1995:721–722. [Google Scholar]
- 39.Roush WR, Bennett CE, Roberts SE. J Org Chem. 2001;66:6389–6393. doi: 10.1021/jo015756a. [DOI] [PubMed] [Google Scholar]
- 40.The strategy of using 2,3-di-O-benzylidine-5-O-benzylapiofuranose instead of 76 as a the glycosyl donor for direct coupling with 52 was less attractive, given that such a donor required its preparation from 76 in a low-yielding sequence. Given that the conversion of 52 + 76 → → 77 is nearly quantitative over three steps, the alternate approach would have been considerably less efficient.
- 41.Braun M, Waldmuller D. Synthesis. 1989:856. [Google Scholar]
- 42.RP-HPLC, 1H NMR, and MS data of synthetic 5 were identical to those of natural 5, obtained by RP-HPLC of Quil A (Accurate Chemical & Scientific Corp.), a Quillaja extract known to contain traces of 5.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental details for the preparation and analytical characterization of synthetic intermediates. This information is available free of charge via the Internet at http://pubs.acs.org.