

Abstract
The function of many of the uncharacterized open reading frames discovered by genomic sequencing can be determined at the level of expressed gene products, the proteome. However, identifying the cognate gene from minute amounts of protein has been one of the major problems in molecular biology. Using yeast as an example, we demonstrate here that mass spectrometric protein identification is a general solution to this problem given a completely sequenced genome. As a first screen, our strategy uses automated laser desorption ionization mass spectrometry of the peptide mixtures produced by in-gel tryptic digestion of a protein. Up to 90% of proteins are identified by searching sequence data bases by lists of peptide masses obtained with high accuracy. The remaining proteins are identified by partially sequencing several peptides of the unseparated mixture by nanoelectrospray tandem mass spectrometry followed by data base searching with multiple peptide sequence tags. In blind trials, the method led to unambiguous identification in all cases. In the largest individual protein identification project to date, a total of 150 gel spots—many of them at subpicomole amounts—were successfully analyzed, greatly enlarging a yeast two-dimensional gel data base. More than 32 proteins were novel and matched to previously uncharacterized open reading frames in the yeast genome. This study establishes that mass spectrometry provides the required throughput, the certainty of identification, and the general applicability to serve as the method of choice to connect genome and proteome.
Many biological experiments produce patterns of gel-separated stained proteins that then have to be identified to make further experimental progress. Identifications are typically achieved by antibody staining, comparison of two-dimensional (2D) gel positions, or other biochemical techniques. These identifications are often uncertain—for example, due to antibody cross reactivity or unexpected gel mobilities—and generally require prior knowledge of the protein. One then has to resort to protein sequence analysis, which has been laborious and required relatively large quantities of protein. DNA sequencing, in contrast, has made tremendous progress in areas from PCR-based methods to genomic sequencing, with the result that analysis has shifted to the DNA level whenever possible.
Complete genomic sequences of model organisms such as the budding yeast Saccharomyces cerevisiae are now available (1). These have, however, had little direct impact on biochemical experiments directed at the characterization of individual proteins and protein complexes. No function can be assigned to many of the open reading frames (here defined as a 300 bp sequence without stop codon) discovered, nor is it known when or whether they are expressed. The ability to perform large-scale protein identification could help in the elucidation of gene function at the protein level. It is also a precondition for any large-scale study of expressed gene products in general, an approach that has been termed “proteome analysis” (2, 3).
Rapid and generic protein identification requires an efficient method to connect information at the protein level to information at the genome level. Mass spectrometric identification of proteins in sequence data bases is a candidate for such a method. The concept was originally presented by Henzel et al.¶ and later published by a number of independent groups (4–8). Shortcomings of previous approaches for large-scale identification were insufficient sensitivity, the need for extensive sample workup, low confidence in the assignments, and the necessity of including non-mass spectrometric techniques that limited the throughput and sensitivity of the overall procedure (9–14). The development of efficient algorithms (15, 16) for using tandem mass spectrometric data (17, 18) in sequence data base searches has increased confidence in protein assignments, but integration into a straightforward, sensitive, and high throughput strategy has not yet been described.
Here we report an efficient mass spectrometric strategy that is shown to combine high sensitivity, certainty of identification, and high throughput in the analysis of 150 yeast proteins. The data are used in the further construction of a 2D yeast protein map (19) and establishes that mass spectrometry by itself is a very effective and sufficient method for large-scale protein identification.
MATERIALS AND METHODS
Yeast Strains and Gel Preparation.
Yeast strain S288C, the strain whose genome has been sequenced, was used. Growth conditions and gel preparation were as described previously (19). Protein spots were visualized by autoradiography and Coomassie blue staining. As judged by autoradiography, as little as 0.3 pmol could be detected by Coomassie blue staining.
Protein Digestion.
Protein spots were excised from the gel, washed, in-gel reduced, S-alkylated, and in-gel digested with an excess of trypsin (overnight at 37°C) as described (20, 21). A 0.3 μl vol of a total of 15–30 μl digest solution was removed for matrix-assisted laser desorption/ionization (MALDI) mass spectrometric analysis. This aliquot corresponded to about 1–2% of the solution and contained peptides in the range of 1–50 fmol per component. If necessary, gel pieces were further extracted and the resulting peptide mixture was subjected to a single desalting/concentration step before mass spectrometric sequencing (22). In the latter part of the project, sample preparation was performed in a laminar flow hood to avoid contamination by keratins.
MALDI Mass Spectrometry.
Microcrystalline matrix surfaces containing nitrocellulose were made by the fast evaporation technique (23, 24) with modifications as described (21, 25). Acidified droplets (0.6 μl of 2–10% formic acid) were placed on these surfaces, the peptide containing aliquots were added, and the surface was washed twice after drying. MALDI mass spectra of peptide mixtures were obtained on a modified Bruker–Franzen (Bremen, Germany) REFLEX time-of-flight (TOF) mass spectrometer equipped with the SCOUT multiprobe inlet and a gridless delayed extraction ion source and detector bias gating for reduction of the low-molecular weight ion current, which in turn improves peptide ion detection efficiency (26). Instrument operation was as described previously (23, 25). Some of the spectra were acquired in an automatic mode using a fuzzy logic control algorithm to regulate the laser irradiance (Fuzzy Logic Toolbox, Mathworks, Natick, MA). The algorithm evaluated the signal intensity and the resolution of the base peak of each sum spectrum of five laser shots. The laser irradiance was then up- or down-regulated following parallel evaluation of a set of six rules combined with the empirically determined fuzzy membership functions. Mass spectra were calibrated via an AppleScript (Apple Computer, Cupertino, CA) using several matrix-related ion signals (m/z 568.13, 855.10, and 1060.10) and trypsin autodigestion peptide ion signals (m/z 2163.057 and 2289.155). Monoisotopic peptide masses were assigned and used in data base searches.
Nanoelectrospray Mass Spectrometry.
All electrospray experiments were done on an API III triple quadrupole instrument (PE; Sciex, Thornhill, ON, Canada) with an upgraded collision cell (27). The pneumatically assisted electrospray source was replaced by the nanoelectrospray ion source developed in our laboratory and operated as described (22, 28). Nanoelectrospray needles were pulled as described (22). A new needle was used for each analysis to eliminate the risk of cross contamination between different peptide digests.
Data Base Searching Using Mass Spectrometric Data.
A nonredundant protein sequence data base maintained and updated daily at European Molecular Biological Laboratory (EMBL) and the European Bioinformatics Institute (Hinxton, U.K.) was used for all data base searches. This data base that currently contains more than 200,000 entries was searched by the peptidesearch software (5, 29). All searches required less than 20 sec on an Apple Macintosh Power PC 7100/80. No restriction was placed on the species of origin of the protein, on its isoelectric point, and a protein mass range from 0 to 200 kDa was allowed. The nrdb index file (updated weekly) is available at ftp://ftp.ebi.ac.uk/pub/data bases/PeptideSearch and the PeptideSearch software is available via http://www.mann.embl-heidelberg.de/MassSpec/software.html. Data base searching by mass lists and peptide sequence tags (15) is also possible via the World Wide Web at the same home page.
RESULTS
Strategy for Mass Spectrometric Protein Identification.
Our aim was to develop a “layered” strategy that combines the simplicity and very high throughput of peptide mass mapping by MALDI TOF with the certainty of identification by tandem mass spectrometric sequencing, and at the same time reduce sample handling to an absolute minimum. Fig. 1 represents an overview of the strategy for analysis of gel-separated protein spots that was developed in the present study. Excised and destained protein spots were in-gel reduced, alkylated, and proteolytically degraded by trypsin, which was found to be universally applicable because of its excellent properties as a protease. Moreover, trypsin produces peptides of ideal size for mass spectrometry and tryptic peptides contain the basic amino acids Arg and Lys in C-terminal position, which facilitates subsequent mass spectrometric sequencing.
Figure 1.

A strategy based exclusively on mass spectrometry to identify proteins separated by 2D PAGE with certainty, sensitivity, and high throughput.
Small aliquots of the gel supernatant-containing released tryptic peptides were analyzed by automated MALDI TOF with high mass accuracy. The list of measured peptide masses (the peptide mass map) was matched against the list of calculated tryptic peptide masses for each protein or translated open reading frame in a comprehensive sequence data base. Most of the samples were identified by this first screening, resulting in high throughput. If there was insufficient protein or a protein mixture was present, the same sample, of which only 2% had been consumed for MALDI analysis, was further analyzed by nanoelectrospray tandem mass spectrometry. Several of the peptides in the unseparated mixture were partially sequenced resulting in unambiguous identification of the protein.
MALDI Analysis with Delayed Extraction and Automation.
To allow positive identification of a large percentage of proteins, the mass spectrometric performance of MALDI TOF peptide mass mapping had to be increased compared with that previously used in this approach. Recent advances in MALDI MS of peptides (for review, see ref. 30) were implemented, of which sample preparation and the delayed ion extraction technique (31–34) were found to be most important. These improvements allow us to routinely obtain a mass resolution of about 10,000 (full width at half maximum) and a mass accuracy better than 50 ppm over a wide mass range and even higher for a small mass range (25), dramatically increasing search specificity by peptide mass maps.
To further increase throughput, we completely automated MALDI peptide mass mapping and data base searching. The most critical part was the optimal regulation of the laser intensity during automatic acquisition of the spectra. Several ad hoc attempts using feedback control algorithms failed but a real-time fuzzy logic-based algorithm produced MALDI spectra very similar in quality to those of a skilled operator. A systemwide macro language connected the different software components used in the automation.
As an example, Fig. 2 shows the MALDI mass spectrum obtained during the automatic identification of yeast protein ILV5. Average absolute mass accuracy was 25 ppm over the m/z range of 800 to 3600, and 30 peptide peaks matched within 50 ppm of the expected tryptic masses for the protein sequence retrieved by automated data base searching. Two terminal sequence tags (35) could also be assigned. The data base entry found by automatic analysis has a calculated molecular mass of 44.4 kDa and an isoelectric point of 9.46, whereas the apparent positions on the gel were 38.8 kDa and 6.45, respectively. Inspection of the feature table in the Swiss-Prot data base for ILV5 revealed a mitochondrion signal peptide sequence at residues 1–21 and an Arg/Lys rich sequence at residues 3–49. Additionally, the C-terminal peptide was detected in the MALDI mass map, making N-terminal processing of the protein the likely cause of the molecular weight discrepancy. If the start of the mature protein is assumed at position 48 in the sequence, a prominent but unassigned peak at m/z 1853.909 in the peptide mass map matches the N-terminal tryptic peptide within 40 ppm. This truncated protein sequence leads to calculated 2D gel coordinates of 39.2 kDa and a pI of 6.28, which agree with the observed ones. The MALDI mass map covered more than 70% of the sequence of this abundant protein.
Figure 2.
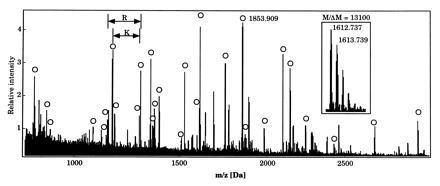
Identification of yeast protein ILV5 by automated MALDI mass spectrometry and automated data base searching. Ion signals whose measured masses match calculated masses of protonated tryptic peptides, (M + H)+, within 50 ppm are indicated with circles. Terminal sequence tags (35) are marked by arrows and with the amino acid producing the ragged end pattern. (Inset) A magnification of one of the tryptic peptide peaks showing isotopically resolved signals differing by 1 Da due to the natural 13C abundance. Sequence coverage is greater than 70 percent. Average absolute mass accuracy is 25 ppm and average resolution is 10,000. Some of the unmarked peaks are matrix related, some are trypsin autolysis products and two matching peptides are outside the mass range shown.
High Sensitivity Nanoelectrospray Tandem MS with Parent Ion Scans.
When proteins could not be identified by MALDI mass mapping, it was previously necessary to at least partially fractionate the peptide mixture by chromatography and perform either tandem mass spectrometry or chemical sequencing on the fractions. We now use nanoelectrospray ionization (22, 28, 36) coupled with a single desalting and concentration step to allow direct analysis of the unseparated peptide mixture at femtomole levels (20, 21).
It turned out that the limit of detection was not determined by the absolute sensitivity of the mass spectrometer. At very low sample levels, peptide ion signals were still present but were obscured by chemical noise. We therefore used parent ion scans of unseparated peptide mixtures (37) to detect these low abundance peptides. Briefly, all precursor (or “parent”) ions producing the m/z 86 fragment are recorded. This fragment is not produced by chemical background but is specific for peptides containing the common amino acids Ile and Leu.
The analysis of the protein sequenced in Fig. 3 marked by an arrow in Fig. 4 illustrates a low level analysis. A faint protein spot was not identified by MALDI peptide mapping since an insufficient number of tryptic peptides was detected. When the sample was analyzed by nanoelectrospray mass spectrometry, no peptide signals apart from trypsin autolysis products were apparent (Fig. 3A). However, a parent ion scan of m/z 86 indicated the masses of several distinct ions. These doubly protonated peptide molecules (Fig. 3B, designated TA–TC) were fragmented in turn. The tandem mass spectrum resulting from TA is typical for peptide amounts of only few femtomoles (Fig. 3C). The spectrum is difficult to interpret completely, but the upper m/z region displays the simple fragmentation pattern typical of doubly charged tryptic peptides. Amino acid residues are easily assigned based on the mass differences between adjacent peaks as indicated in Fig. 3. From this stretch of sequence ions, a peptide sequence tag was constructed by the program peptidesearch. [A peptide sequence tag consists of a short sequence combined with the distance, in mass units, to the N and C termini of the peptide (15).] Independent searches using this tag and the tags obtained from the tandem mass spectra of peaks TB and TC identified the same yeast protein, VMA2. However, its molecular mass of 57.8 kDa, calculated on the basis of the DNA sequence does not agree with the apparent mass of 32.3 kDa. All three peptides (verifying a total of 28 amino acids) mapped to the C-terminal part of the protein, suggesting N-terminal truncation as the reason for the anomalous migration of the protein.
Figure 3.
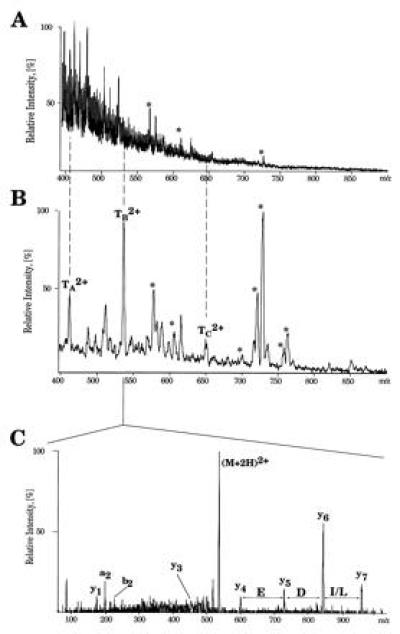
High sensitivity protein identification by nano electrospray mass spectrometry. (A) Mass spectrum (Q1 scan) of a tryptic digest of a spot marked by an arrow on Fig. 4. Ions of trypsin autolysis products are marked (∗). (B) Parent ion scan spectrum for the immonium ions of Leu/Ile (m/z 86) acquired using the same digest. Ions of tryptic peptides are designated TA–TC. (C) Tandem mass spectrum of the doubly charged ion TB2+. The first mass of the fragmentation series, the sequence as determined by the mass differences between fragment peaks and the last mass in the series were entered as (600.2)ED(L/I)(957.4) into peptidesearch together with the measured peptide mass. Yeast protein VMA2 was unambiguously identified by the peptide sequence tag as shown in the spectrum. The letters refer to N-terminal (a and b) and C terminal (y) fragmentation at the amide bonds (38).
Figure 4.
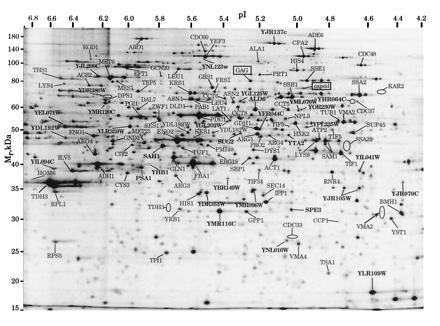
2D PAGE yeast reference map with protein identifications as determined in the current investigation. The protein pattern corresponds to [35S]methionine-labeled polypeptide (19). The arrow indicates the protein sequenced in Fig. 3 and the rectangles indicate proteins of the yeast L-A virus. The elipses mark locations of very weakly staining proteins. Gene names of previously uncharacterized open reading frames are in boldface type.
Certainty of Identification.
To directly test the accuracy of identification, a large set of proteins spots from the yeast 2D gel of the Bordeaux group, which had previously been identified by conventional techniques, were analyzed as a blind trial. For 41 of 46 proteins, previous assignments agreed with the mass spectrometric results.
One protein (pIgel 5.07; Mr,gel 26.9 kDa) had previously been assigned as ANC1 by amino acid analysis and by searching for intron-containing genes. However, eight peptides measured by MALDI MS mapped instead to the sequence of YNL010W with an accuracy of 40 ppm, whereas only four peptides matched another yeast protein, which moreover had a calculated mass of 97 kDa (no peptide mass matched the ANC1 protein). Additionally, three peptides were sequenced by tandem mass spectrometry, each of which uniquely identified YNL010W. We conclude that the previous identification had been incorrect.
In the remaining four cases, the previous analyses had provided only tentative assignments. In each of these cases, mass spectrometry unambiguously identified another protein, the calculated Mr and pI values of which agreed with the apparent ones.
Proteins Identified in this Study.
Overall, 80% of the gel spots marked on Fig. 4 were positively identified by MALDI alone. The high mass accuracy achieved was critical for the positive identification of a large fraction of the spots. In the work reported, the success rate of unambiguous MALDI identification on yeast proteins spots, representing at least one to two picomole starting material, improved from about 50% to more than 90% due to the implementation of new methods (25, 35). A set of 65 protein analyses in the latter part of the project illustrates the power of protein identification by MALDI mass mapping. The peptide mass maps of all proteins were acquired in 6 hr in the automatic mode and 59 of the proteins were identified immediately.
In this large-scale project, nanoelectrospray operation proved to be robust and routine. Even for protein spots close to the limit of detection of about 0.1 to 0.2 pmol of protein material loaded on the gel (20), measurement times were not longer than 1 hr. In the analysis of 49 proteins by nanoelectrospray, unambiguous search results were always obtained. In several cases, initially no match was found in the data base. After more genomic sequence data became available, the same searches then identified those proteins.
The gene names of 134 protein spots are indicated on the reference image of our yeast 2D gel data base (19) in Fig. 4. Another 16 proteins were characterized in targeted gene disruption experiments but only three of these are marked in Fig. 4. As can be seen from the gel picture, proteins with a wide range of molecular weights, isoelectric points, and abundances were identified. A more detailed table of protein assignments can be found on the World Wide Web via http://www.mann.embl-heidelberg.de/.
The 134 protein spots that are marked in the Fig. 4 correspond to 128 different yeast genes; thus, several genes gave rise to more than one protein spot. Of the 134 protein spots in Fig. 4, 107 were within 10% of the expected molecular weight and within 0.5 pI units of the expected isoelectric point. Except for the genes that gave rise to more than one protein spot, the deviating proteins were mainly due to pI differences of proteins found at the basic side of the pH gradient. Altogether, the gel position of 27 proteins differed substantially from the expected one.
Several non-yeast proteins were identified: Two proteins from L-A virus, a double-stranded yeast RNA virus, have been marked in Fig. 4. In the analysis of several of the low abundance spots, human and sheep keratin proteins were identified. Since it is difficult to assess at which stage these contaminating proteins were introduced, these spots were not assigned in the reference data base. In four cases, protein mixtures were encountered. All of them were successfully analyzed, one by MALDI peptide mapping and the rest by nanoelectrospray mass spectrometry.
Interestingly, 32 proteins identified here had not been characterized before. They were previously only known as open reading frames discovered by genomic sequencing and are here analyzed at the protein level. These identifications are listed in Table 1. Homology search suggested the existence of a related protein or at least protein domain for only 23 of the novel proteins identified here.
Table 1.
Identified open reading frames
| Gene name | Suggested function* (% of homology)† |
|---|---|
| ALD6 | Aldehyde dehydrogenase (48) |
| YJL200C | Aconitase (50) |
| YJR137C | Sulfite reductase‡ (13) |
| YJR105W | Ribokinase‡ (5) |
| YMR116C | Guanine nucleotide-binding protein (51) |
| YLR109W | Peroxisomal membrane protein‡ (24) |
| SPE3 | Spermidine synthase (50) |
| YDL182W | Homocitrate synthase‡ (14) |
| PSA1 | NDP-hexose pyrophoshorylase‡ (11) |
| YJR070C | None |
| YFR044C | None |
| YMR096W | None |
| YGL202W | None |
| YIL094C | Isocitrate dehydrogenase‡ (21) |
| YNL010W | None |
| YNL123W | None |
| YPL235W | None |
| YMR120C | Aminoimidazolecarboxamide formyltransferase (61) |
| YLR359W | Adenylosuccinate lyase (61) |
| YDR353W | Thioredoxin reductase (66) |
| YDR380W | Pyruvate decarboxylase‡ (16) |
| YHB1 | Flavohemoprotein‡ (24) |
| YHR064C | HSP70 protein‡ (16) |
| SAH1 | Adenosylhomocysteinase (67) |
| YTA2 | 26S protease regulatory subunit (68) |
| YEL071W | Actin interacting protein 2 (65) |
| YIL041W | None |
| YML070W | Dihydroxyacetone kinase (35) |
| YOR230W | None |
| YGL125W | Methylenetetrahydrofolate reductase‡ (20) |
| YBR149W | GCY protein‡ (24) |
| SUG2 | 26S protease regulatory subunit (34) |
Function as predicted in the Yeast Protein Data Base (www address: http://www.proteome.com/YPDhome.html) and Genequiz (http://www.embl-heidelberg.de/-genequiz/yeast.html).
As a result of blitz search (http://www.ebi.ac.uk/searches/blitz_input.html).
Assigned by homology to a local region by blast (http://expasy.hcuge.ch/cgibin/BLASTEPFL.pl).
Altogether, this investigation resulted in the unambiguous identification of 150 protein spots on our reference 2D gel. Of these, 41 validated existing identifications, 93 are new entries and another 16 were identified in a pilot functional analysis experiment using targeted gene disruption. More than 32 proteins were novel and had not been characterized before.
DISCUSSION
We have shown here that it is now possible to efficiently and with certainty identify large numbers of proteins separated on polyacrylamide gels. This capability should be very useful in many “small-scale” biochemical investigations involving a small numbers of proteins as well as in large-scale projects aiming at the systematic identification of the function of yeast open reading frames.
The strategy developed here has been reduced to a minimum of preparation and analysis steps: Single gels were analyzed, removing the need for gel pooling and concentration gels. No blotting or chromatographic steps to separate the peptide mixtures were used. It is no longer necessary to resort to non-mass spectrometric methods. MALDI with delayed ion extraction, coupled with the sample preparation and automation techniques discussed above, has been shown here to be very powerful for the rapid and unambiguous identification of a large percentage of proteins. In the second screen, peptide sequencing by nanoelectrospray mass spectrometry and parent ion scans were used to increase sensitivity to below the level of chemical noise, which otherwise sets an absolute limit of sensitivity. A “layered” approach to large-scale protein identification has been suggested previously (12), where a first screen would rapidly and economically identify most proteins (for example amino acid analysis), followed by more discriminating but time-consuming analyses (for example, Edman sequencing). Here we have shown that mass spectrometry alone can provide an effective layered approach. The use of two ionization methods, MALDI and electrospray, which are based on completely different physical principles, adds to the analytical flexibility and general applicability of our strategy, which, as shown in Fig. 1, can easily be extended to de novo mass spectrometric sequencing should the protein not be included in a sequence data base (20).
This is the first time that the reliability of identification by mass spectrometry based methods has been rigorously evaluated. All 150 protein identifications were judged to be unambiguous and in no case could conflicting biochemical identifications be sustained. Yeast proteins that were not in sequence data bases at the time of analysis were correctly identified as unknowns. A substantial number of proteins were found at 2D gel coordinates different from the ones expected based on their sequence, suggesting that the 2D gel coordinates should not be used as a search constraint in the identification of gel-separated proteins. In contrast to a previous study on yeast proteins (13), MALDI peptide mapping with high mass accuracy was shown here to lead to unambiguous results rather than to tentative identifications needing further verification. The present investigation represents the largest protein identification project to date, and we have found a substantial number of novel proteins which correspond to previously uncharacterized open reading frames.
Limitations of gel systems such as the one we used here are that they visualize only a subset of expressed yeast proteins (19). Work on restricted pI range gels, alternative staining methods (21) and methods to load membrane proteins may overcome many of these obstacles. Similarly, our purpose here was only the efficient identification of protein spots by mass spectrometry, however, given a somewhat larger protein amount mass spectrometry is increasingly able to map posttranslational modifications as well.
The new protein analysis capabilities have here been used for the rapid enlargement of a 2D gel data base. The analytical preconditions for functional elucidation of yeast genes via gene disruption and observation of the phenotype in 2D protein maps are now met and to determine a large part of the expressed yeast genome i.e. its ‘proteome’ is now rather straightforward and relatively fast. Yeast was used as a model system because it is the first eucariote whose genome has been sequenced completely. We note, however, that all identifications were made in a large sequence data base containing 200,000 entries. Thus, our strategy can be applied unchanged to the 100,000 or so human genes once their coding sequences have been determined. Efficient techniques for large-scale protein characterization could have an impact on molecular biology as great as that of the genomic sequence data bases themselves.
Acknowledgments
We thank our colleagues at EMBL, particularly A. Hyman and D. Tollervey, for fruitful discussions. Work in the Protein and Peptide group at EMBL is partially funded by a generous grant by the German Technology Ministry. Work in the Bordeaux laboratory is partly funded by the European Union pilot project on functional analysis of the yeast genome (B102-CT93-0022). O.N.J. is the recipient of a postdoctoral fellowship from the European Union Biotechnology Program. A.V.P. is supported by a fellowship from Glaxo-Wellcome.
Footnotes
Abbreviations: MALDI, matrix-assisted laser desorption/ionization; TOF, time-of-flight; 2D, two-dimensional; EMBL, European Molecular Biology Laboratory.
Henzel, W. J., Stults, J. T., Watanabe, C., Third symposium of the Protein Society, Seattle, WA, 1989.
References
- 1.Goffeau A, Barrell B G, Bussey H, Davis R W, Dujon B, Feldmann H, Galibert F, Hoheisal J D, Jacq L, Johnston M, Louis E J, Mewes H W, Murakami Y, Philippsen P, Tettelin H, Oliver S B. Science. 1996;274:546–567. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- 2.Wilkins M R, Pasquali C, Appel R D, Ou K, Golaz O, Sanchez J C, Yan J X, Gooley A A, Hughes G, Humphery-Smith I, Williams K L, Hochstrasser D F. Bio/Technology. 1996;14:61–65. doi: 10.1038/nbt0196-61. [DOI] [PubMed] [Google Scholar]
- 3.Kahn P. Science. 1995;270:369–370. doi: 10.1126/science.270.5235.369. [DOI] [PubMed] [Google Scholar]
- 4.Henzel W J, Billeci T M, Stults J T, Wong S C. Proc Natl Acad Sci USA. 1993;90:5011–5015. doi: 10.1073/pnas.90.11.5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mann M, Højrup P, Roepstorff P. Biol Mass Spectrom. 1993;22:338–345. doi: 10.1002/bms.1200220605. [DOI] [PubMed] [Google Scholar]
- 6.Pappin D J C, Højrup P, Bleasby A J. Curr Biol. 1993;3:327–332. doi: 10.1016/0960-9822(93)90195-t. [DOI] [PubMed] [Google Scholar]
- 7.James P, Quadroni M, Carafoli E, Gonnet G. Biophys Biochem Res Commun. 1993;195:58–64. doi: 10.1006/bbrc.1993.2009. [DOI] [PubMed] [Google Scholar]
- 8.Yates J R, Speicher S, Griffin P R, Hunkapiller T. Anal Biochem. 1993;214:397–408. doi: 10.1006/abio.1993.1514. [DOI] [PubMed] [Google Scholar]
- 9.Rasmussen H H, Mørtz E, Mann M, Roepstorff P, Celis J E. Electrophoresis. 1994;15:406–416. doi: 10.1002/elps.1150150159. [DOI] [PubMed] [Google Scholar]
- 10.Clauser K R, Hall S C, Smith D M, Webb J W, Andrews L E, Tran H M, Epstein L B, Burlingame A L. Proc Natl Acad Sci USA. 1995;92:5072–5076. doi: 10.1073/pnas.92.11.5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patterson S D, Thomas D, Bradshaw R A. Electrophoresis. 1996;17:77–891. doi: 10.1002/elps.1150170509. [DOI] [PubMed] [Google Scholar]
- 12.Wheeler C H, Berry S L, Wilkins M R, Corbett J M, Ou K, Gooley A A, Humphery-Smith I, K L, K L W, Dunn M J. Electrophoresis. 1996;17:580–587. doi: 10.1002/elps.1150170329. [DOI] [PubMed] [Google Scholar]
- 13.Sagliocco, F., Guillemot, J. C., Monribot, C., Capdevielle, J., Perrot, M., Ferran, E., Ferrara, P. & Boucherie, H. (1996) Yeast, in press. [DOI] [PubMed]
- 14.Jensen O N, Houthaeve T, Shevchenko A, Cudmore S, Ashford T, Mann M, Griffiths G, Locker J K. J Virol. 1996;70:7485–7497. doi: 10.1128/jvi.70.11.7485-7497.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mann M, Wilm M S. Anal Chem. 1994;66:4390–4399. doi: 10.1021/ac00096a002. [DOI] [PubMed] [Google Scholar]
- 16.Eng J K, McCormack A L, Yates J R. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 17.Biemann K. Anal Chem. 1985;58:1289A–1300A. doi: 10.1021/ac00126a001. [DOI] [PubMed] [Google Scholar]
- 18.Hunt D F, Yates J R, Shabanowitz J, Winston S, Hauer C R. Proc Natl Acad Sci USA. 1986;83:6233–6237. doi: 10.1073/pnas.83.17.6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boucherie H, Dujardin G, Kermorgant M, Monribot C, Slonimski P, Perrot M. Yeast. 1995;11:601–613. doi: 10.1002/yea.320110702. [DOI] [PubMed] [Google Scholar]
- 20.Wilm M, Shevchenko A, Houthaeve T, Breit S, Schweigerer L, Fotsis T, Mann M. Nature (London) 1996;379:466–469. doi: 10.1038/379466a0. [DOI] [PubMed] [Google Scholar]
- 21.Shevchenko A, Wilm M, Vorm O, Mann M. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 22.Wilm M, Mann M. Anal Chem. 1996;66:1–8. doi: 10.1021/ac9509519. [DOI] [PubMed] [Google Scholar]
- 23.Vorm O, Roepstorff P, Mann M. Anal Chem. 1994;66:3281–3287. [Google Scholar]
- 24.Vorm O, Mann M. J Am Soc Mass Spectrom. 1994;5:955–958. doi: 10.1016/1044-0305(94)80013-8. [DOI] [PubMed] [Google Scholar]
- 25.Jensen O N, Podtelejnikov A, Mann M. Rapid Commun Mass Spectrom. 1996;10:1371–1378. doi: 10.1002/(SICI)1097-0231(199608)10:11<1371::AID-RCM682>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 26.Vorm O, Roepstorff P. J Mass Spectrom. 1996;31:351–356. [Google Scholar]
- 27.Thomson B A, Douglas D J, Corr J J, Hager J W, Jolliffe C L. Anal Chem. 1995;67:1696–1704. [Google Scholar]
- 28.Wilm M S, Mann M. Int J Mass Spectrom Ion Proc. 1994;136:167–180. [Google Scholar]
- 29.Mann M. In: Microcharacterization of Proteins. Kellner R, Lottspeich F, Meyer H E, editors. Weinheim: VCH; 1994. pp. 223–245. [Google Scholar]
- 30.Mann M, Talbo G. Curr Opin Biotechnol. 1996;7:11–19. doi: 10.1016/s0958-1669(96)80089-9. [DOI] [PubMed] [Google Scholar]
- 31.Brown R S, Lennon J J. Anal Chem. 1995;67:1998–2003. doi: 10.1021/ac00109a015. [DOI] [PubMed] [Google Scholar]
- 32.King T B, Colby S M, Reilly J P. Int J Mass Spectrom Ion Proc. 1995;145:L1–L7. [Google Scholar]
- 33.Whittal R M, Li L. Anal Chem. 1995;67:1950–1954. doi: 10.1021/ac00109a007. [DOI] [PubMed] [Google Scholar]
- 34.Vestal M L, Juhasz P, Martin S A. Rapid Commun Mass Spectrom. 1995;9:1044–1050. [Google Scholar]
- 35.Jensen O N, Vorm O, Mann M. Electrophoresis. 1996;17:938–944. doi: 10.1002/elps.1150170516. [DOI] [PubMed] [Google Scholar]
- 36.Mann M, Wilm M. Trends Biochem Sci. 1995;20:219–223. doi: 10.1016/s0968-0004(00)89019-2. [DOI] [PubMed] [Google Scholar]
- 37.Wilm M, Neubauer G, Mann M. Anal Chem. 1996;68:527–533. doi: 10.1021/ac950875+. [DOI] [PubMed] [Google Scholar]
- 38.Biemann K. Biomed Environm Mass Spectrom. 1988;16:99–111. doi: 10.1002/bms.1200160119. [DOI] [PubMed] [Google Scholar]