

Oculocutaneous albinism (OCA) comprises a group of autosomal recessive disorders with a clinical spectrum that ranges from the most severe OCA type 1A (OCA1A, OMIM 203100), with a complete lack of melanin production throughout life, to milder forms (OCA1B, OMIM 606952; OCA2, OMIM 203200; OCA3, OMIM 203290; and OCA4, OMIM 606574) [Grønskov, 2007]. At least four human genes are implicated; TYR, P, TYRP1and MATP. There are also syndromic forms of OCA, among which is Hermansky-Pudlak syndrome (HPS, OMIM 203300), an autosomal recessive disease characterized by OCA, bleeding tendency due to platelet storage-pool deficiency, and variable manifestations due to lysosomal ceroid lipofuscinosis in multiple organs. [Wei, 2006; Huizing and Gahl, 2002; Gahl et al., 1998; Li et al., 2003]. The clinical manifestations of different HPS types are variable and at least 8 human types have been described associated with mutations in 4 ubiquitously expressed protein complexes genes; adaptor protein-3 (AP-3) and biogenesis of lysosome-related organelles complex 1 through -3 (BLOC-1,-2 and -3) [Dell’Angelica, 2004].
Here we present a patient with oculocutaneous albinism type 1, recurrent rhabdomyolysis and bleeding diathesis. Our patient (Fig. 1A) was a 14.5 year old girl from Corfu island (Greece), born at term to healthy, nonconsanguineous parents. She had oculocutaneous albinism, horizontal nystagmus, photophobia and severely decreased visual acuity. Her motor and mental development were reported to be normal with no musculoskeletal abnormalities. She had a history of Henoch-Schönlein purpura, post-streptococcal glomerulonephritis and bleeding tendency with heavy menstruation. She had no history of respiratory problems, frequent bacterial infections, neutropenia or any evidence of immunodeficiency. Perinatal history was non-contributory and there was no family history of recurrent rhabdomyolysis or OCA. Since age 10 years she has developed recurrent episodes of rhabdomyolysis and myoglobinuria, which occurred during febrile illness or after strenuous exercise, associated with progressively worsening generalized myalgia, severe proximal muscle weakness and severe abdominal pain. During these episodes she had significant elevation in serum creatine kinase (max 140,000 IU/L), aspartate and alanine transaminase, lactate dehydrogenase, aldolase and urine myoglobin casts. Between episodes, her creatine kinase was 2.5 times the upper normal limit. Her third episode occurred at the age of 14.5 years, with complete recovery within 10 days, with aggressive hydration only. She denied exposure to toxins, drugs, anesthetics, recreational drugs, inhalation of irritant substances, any serious trauma, allergies, bites or unusual food intake. Within the following 2.5 years she frequently complained of muscle cramps that resolved with rest, and severe lower abdominal pain, responding to analgesics.
Figure 1.
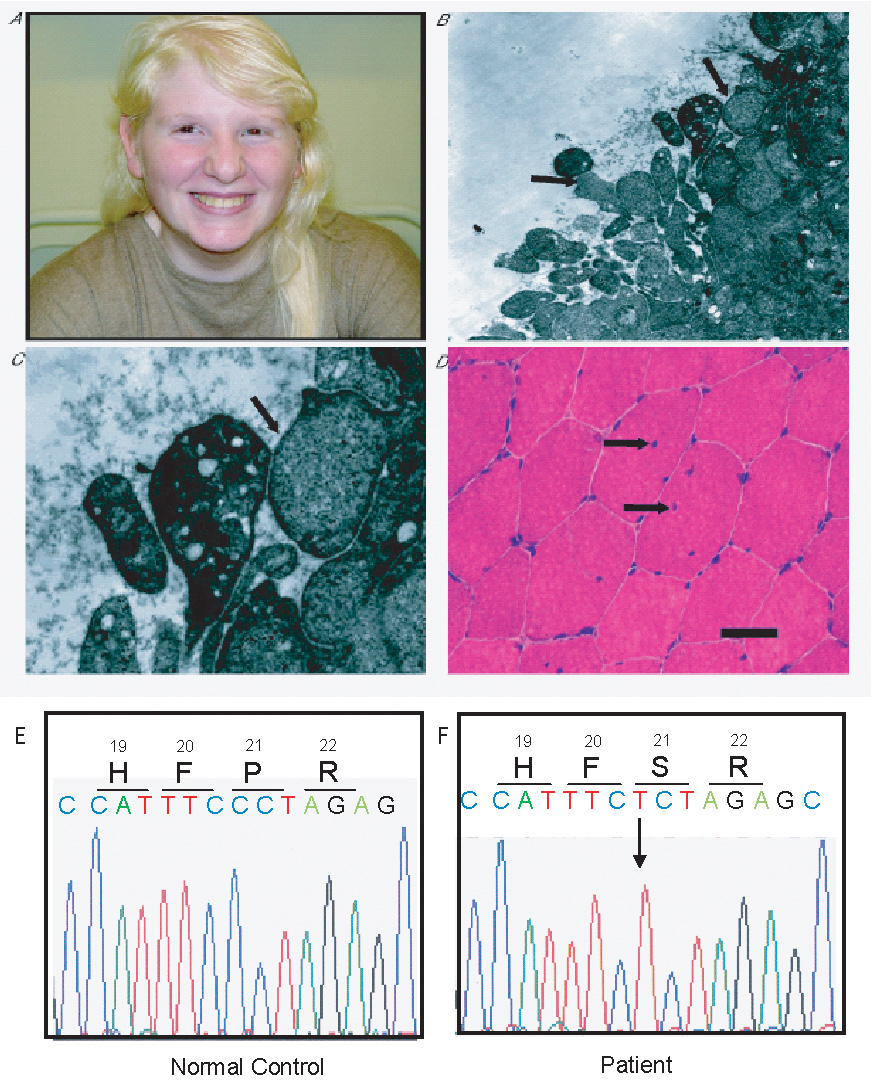
A: Image of our patient with oculocutaneous albinism (white skin and hair; eyebrows with slight brown pigmentation; few melanocytic nevi). B: Platelets electron micrograph (magnification X7000), with absent dense bodies and uniform cytoplasm, without membranous inclusions in the majority of the platelets (long arrows). C: Enlarged image of platelets subpopulation corresponding to right long arrow of Fig 1A. (magnification X17000;Ultra thin sections of dehydrated platelet pellets were stained with uranyl acetate and lead citrate and observed under a Zeiss EM900 transmission electron microscope). D: Transverse muscle biopsy section from the quadriceps muscle showing 3 muscle fibers with 1 or more internal nuclei; in 1 of these fibers a splitting line can be seen. (Black bar: 50 micron. Hematoxylin-Phloxine stain). E–F: Sequencing results for tyrosinase (TYR) gene of DNA samples obtained from a normal individual (E) and our patient (F). The arrow indicates the position of the base substitution c.61C>T identified in our patient, corresponding to the p.P21S amino-acid substitution, that has been associated with OCA type 1 in Caucasians.
Results of workup for infectious and endocrine causes of recurrent rhabdomyolysis were normal. Blood gases, serum glucose, lactate, ammonia, amino acids, acyl-carnitines and urine organic acids were normal. Electromyography, electroneurography, brain MRI/ MRI spectroscopy and forearm-ischemia exercise test (McArdle test) were normal, with no ketonuria during the test. Mild non-ketotic hypoglycemia developed after an 18 hours fasting-tolerance test. Fatty acid oxidation studies in skin fibroblasts, including palmitate loading test, carnitine palmitoyl-transferases, short, middle, long and very-long chain acyl-CoA dehydrogenases, and oxidative phosphorylation complex I–IV enzymatic activity were also normal. Pyrosequencing analysis of mitochondrial DNA did not document any of the known point mutations (MELAS m.3243A>G, MERRF m.8344A>G and Leigh/NARP m. 8993T>G/C).
Due to the history of bleeding tendency, she was further investigated for possible HPS. Electron microscopy of platelets, showed platelet storage-pool deficiency, with two aberrant subpopulations; one with complete absence of dense-bodies and the other with very few dense bodies (Fig.1B,C). Platelet dense-bodies absence is considered pathognomonic for HPS. Nevertheless, normal platelets were also present and a platelet aggregation test was normal. Results of coagulation studies, clotting factors, complement component C4, platelet counts and bone marrow aspirate were all normal. Sequencing analysis for the 5 most frequently mutated HPS-related genes-HPS1 and HPS4 (BLOC-3); HPS3, HPS5 and HPS6 (BLOC-2); DTNBP1/HPS7 and BLOC1S3/HPS8 (BLOC-1)-did not show any mutations in their exons or their surrounding intron/exons boundaries [Wei, 2006; Dell’Angelica, 2004; Huizing and Gahl, 2002].
On physical examination she had an elevated body mass index (BMI:31), symmetrically decreased muscle strength (4/5), slight dysmetria, minimal tremor in the coordination test of upper limbs, OCA, horizontal nystagmus and decreased visual acuity (Fig. 1A); otherwise, she had no hearing deficit, skeletal abnormalities, ophthalmoplegia, blepharoptosis or any other anomalies.
On muscle biopsy an increased percentage (30%) of muscle fibers was hypertrophic; 15% of the muscle fibers had 1 or more internal nuclei (normally up to 3%) (Fig. 1D). Moreover, fiber splitting correlating with muscle fiber hypertrophy was also observed. Staining for oxidative enzyme activity showed only a few muscle cores. No lipid accumulation or glycogen storage was detected. Moreover, muscle phosphofructokinase and phosphorylase kinase activity was normal. On electron-microscopy only one abnormal mitochondrial structure was seen; no abnormalities were detected in the Golgi structure and the lysosomes. No biochemical evidence was found for mitochondrial disease; substrate oxidation and adenosine triphosphate (ATP) production from pyruvate oxidation were normal and no alteration was found in the oxidative phosphorylation (OXPHOS) enzyme activities I–V and the activity of the pyruvate dehydrogenase complex. As the muscle biopsy histologic findings of fiber hypertrophy and presence of internal nuclei can be found in malignant hyperthermia syndrome (MHS, OMIM 145600) and central core disease (CCD, OMIM 11700) [Jungbluth, 2007], further sequencing was performed for the ryanodine receptor-1 (RYR1) gene, which has been associated with these two conditions. No mutations were identified in any of the 15 RYR1 sequenced exons (exons 2,6,9,11,12,14,15,17,39,40,45,46, 100–102, including the 3 mutational “hot spots” in the N-terminal, central and C-terminal residues) [Quane et al., 1993]. Moreover, screening for microdeletions, with array-comparative genomic hybridization (array-CGH) assays (Affymetrix, 250k SNP-array platform) did not uncover any aberrations such as loss or gain of genetic material. However, three large homozygous regions were identified; a 19 Mb region on chromosome 11q13.5–q21 (76.0–95.0), a 6.5 Mb on chromosome 13q21.1–q21.2 (54.5–61.0) and a 1.6 Mb on chromosome 13q33.1–q33.2 (103.5–105.1). Sequencing analysis of the tyrosinase (TYR) gene, located in 11q14–q21, identified a homozygous missense mutation P21S (c. 61C>T) (Fig. 1E–F). This mutation has previously been reported in Caucasian patients with OCA1 [King et al., 2003]. Sequencing of the P gene (OCA2) revealed no mutations. Although there was no reported parental consanguinity, the presence of large homozygous regions suggests that this possibility could not be completely excluded as both parents originated from the same island (Corfu).
Here we presented a patient with a previously unreported combination of oculocutaneous albinism type-1, platelet storage-pool deficiency and recurrent rhabdomyolysis. Based on the muscle biopsy histologic findings and the patient’s clinical history, we considered that our patient had an MHS-susceptibility phenotype. MHS-susceptible persons can develop severe rhabdomyolysis post anesthesia, but also post severe exercise in hot conditions and post infections [Rosenberg et al., 2007]. This was probably the case with our patient. MHS has common genetic background and overlapping manifestations with CCD [OMIM 117000; Jungbluth, 2007]; however, the histopathology in our case did not confirm CCD. Moreover, although muscle fiber hypertrophy can occur post rhabdomyolysis, the muscle biopsy in our patient was performed more than 1 year post rhabdomyolysis, making this alternative less likely.
MHS usually is inherited as an autosomal dominant disease [OMIM 145600], but there are cases of autosomal recessive inheritance and rare sporadic cases [Manzurt et al., 1998]. Almost half of MHS cases have been linked to at least 25 mutations in the RYR-1 gene [Rosenberg et al., 2007; Quane et al., 1993]. RYR-1 is one of the genomically most complex genes in humans, with 15.5 kilobases (106 exons), located on 19q12–13.2 [Phillips et al., 1996], encoding a large sarcoplasmic reticulum 563 kDa protein [MacLennan and Phillips, 1992]. Our sequencing analysis did not detect any RYR-1 mutation. However, it is possible that mutations in other non-sequenced susceptibility loci may have been implicated [Rosenberg et al., 2007].
Platelet storage-pool deficiency has not been previously reported to be associated with recurrent rhabdomyolysis or any type of muscle dysfunction. Moreover, none of the reported cases of genetically confirmed HPS patients had recurrent rhabdomyolysis or any known mutation for OCA type 1–4 [Garrison et al., 2004]. HPS patients with BLOC 2–3 defects display a complete absence of platelet dense bodies [Huizing and Gahl, 2002]. Only two HPS patients with BLOC-1 defects are described; one with DTNBP1 (HPS7) mutations [Li et al., 2003] and one with BLOC1S3 (HPS8) mutations [Morgan et al., 2006]. Mouse models for almost every subunit of BLOC-1 exist, but none of these mice has apparent neurological/muscle involvement [Li et al., 2004]. Of interest is that DTNBP1/HPS7 codes for dysbindin, which binds dystrobrevins. Dysbindin, when assembled into the BLOC-1 complex cannot bind dystrobrevins [Nazarian et al., 2006]; however, mutations may alter it's binding characteristics, allowing for dystrobrevin binding and thus decreasing the dystrobrevin pool available for physiologic muscle function. Dysbindin mutations can also alter its binding to myospryn, and thus also affect muscle function [Benson et al., 2004]. Nevertheless, sequencing in our patient for 7 HPS-related genes, including DTNBP1, did not reveal any mutations. Furthermore, it is unlikely that our patient has a defect in AP-3 [Wei, 2006; Huizing et al., 2002] as among the few reported patients all had a history of frequent infections and none had rhabdomyolysis.
In conclusion, this is the first report of a patient with OCA1 presenting with recurrent rhabdomyolysis due to an MHS-susceptibility phenotype and bleeding diathesis due to platelet storage-pool deficiency. As the clinical spectrum of OCA1 is very limited to ocular defects and melanin deficiency in the skin, it is unlikely that these findings represent an extended clinical spectrum of the disease. If this co-occurrence is not a coincidence, it might represent a new distinct genetic syndrome with platelet storage-pool deficiency and severe muscle involvement in a patient with OCA1 disease.
ACKNOWLEDGMENTS
This work was supported by the Intramural Research program of the National Human Genome Research Institute (NHGRI), National Institutes of Health, Bethesda, Maryland, USA
REFERENCES
- Benson MA, Tinsley CL, Blake DJ. Myospryn is a novel binding partner for dysbindin in muscle. J Biol Chem. 2004;279:10450–10458. doi: 10.1074/jbc.M312664200. [DOI] [PubMed] [Google Scholar]
- Dell‘Angelica EC. The building BLOC(k)s of lysosomes and related organelles. Curr Opin Cell Biol. 2004;16:458–464. doi: 10.1016/j.ceb.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Gahl WA, Brantly M, Kaiser-Kupfer MI, Iwata F, Hazelwood S, Shotelersunk V, Duffy LF, Kuehl EM, Troendle J, Bernadini I. Genetic defects and clinical characteristics of patients with a form of oculocutaneous albinism (Hermansky-Pudlak syndrome) New Engl J Med. 1998;338:1258–1264. doi: 10.1056/NEJM199804303381803. [DOI] [PubMed] [Google Scholar]
- Garrison NA, Yi Z, Cohen-Barak O, Huizing M, Hartnell LM, Gahl WA, Brilliant MH. P gene mutations in patients with oculocutaneous albinism and findings suggestive of Hermansky-Pudlak syndrome. J Med Genet. 2004;41:e86. doi: 10.1136/jmg.2003.014902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grønskov K, Ek J, Brøndum-Nielsen K. Oculocutaneous albinism. Orphanet J Rare Dis. 2004;2:43–50. doi: 10.1186/1750-1172-2-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huizing M, Gahl WA. Disorders of vesicles of lysosomal lineage: the Hermansky-Pudlak syndromes. Curr Mol Med. 2002;2:451–467. doi: 10.2174/1566524023362357. [DOI] [PubMed] [Google Scholar]
- Huizing M, Scher CD, Strovel E, Fitzpatrick DL, Hartnell LM, Anikster Y, Gahl WA. Nonsense mutations in ADTB3A cause complete deficiency of the beta3A subunit of adaptor complex-3 and severe Hermansky-Pudlak syndrome type 2. Pediatr Res. 2002;51:150–158. doi: 10.1203/00006450-200202000-00006. [DOI] [PubMed] [Google Scholar]
- Jungbluth H. Central core disease. Orphanet J Rare Dis. 2007;2:25–33. doi: 10.1186/1750-1172-2-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RA, Pietsch J, Fryer JP, Savage S, Brott MJ, Russell-Eggitt I, Summers CG, Oetting WS. Tyrosinase gene mutations in oculocutaneous albinism 1 (OCA1): definition of the phenotype. Hum Genet. 2003;113:502–513. doi: 10.1007/s00439-003-0998-1. [DOI] [PubMed] [Google Scholar]
- Li W, Zhang Q, Oiso N, Novak EK, Gautam R, O'Brien EP, Tinsley CL, Blake DJ, Spritz RA, Copeland NG, Jenkins NA, Amato D, Roe BA, Starcevic M, Dell'Angelica EC, Elliott RW, Mishra V, Kingsmore SF, Paylor RE, Swank RT. Hermansky-Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1) Nat Genet. 2003;35:84–89. doi: 10.1038/ng1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Rusiniak ME, Chintala S, Gautam R, Novak EK, Swank RT. Murine Hermansky Pudlak syndrome genes: regulators of lysosome-related organelles. Bioessays. 2004;26:616–628. doi: 10.1002/bies.20042. [DOI] [PubMed] [Google Scholar]
- MacLennan DH, Phillips MS. Malignant hyperthermia. Science. 1992;256:789–794. doi: 10.1126/science.1589759. [DOI] [PubMed] [Google Scholar]
- Manzur AY, Sewry CA, Ziprin J, Dubowitz V, Muntoni F. A severe clinical and pathological variant of central core disease with possible autosomal recessive inheritance. Neuromuscul Disord. 1998;8:467–473. doi: 10.1016/s0960-8966(98)00064-9. [DOI] [PubMed] [Google Scholar]
- Morgan NV, Pasha S, Johnson CA, Ainsworth JR, Eady RA, Dawood B, McKeown C, Trembath RC, Wilde J, Watson SP, Maher ER. A germline mutation in BLOC1S3/reduced pigmentation causes a novel variant of Hermansky-Pudlak syndrome (HPS8) Am J Hum Genet. 2006;78:160–166. doi: 10.1086/499338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarian R, Starcevic M, Spencer MJ, Dell'Angelica EC. Reinvestigation of the dysbindin subunit of BLOC-1 (biogenesis of lysosome-related organelles complex-1) as a dystrobrevin-binding protein. Biochem J. 2006;395:587–598. doi: 10.1042/BJ20051965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Online Mendelian Inheritance in Man. OOMIM (TM) Baltimore, MD: Johns Hopkins University; OMIM. World Wide Web URL: http://www.ncbi.nlm.nih.gov/oOMIM/ [Google Scholar]
- Phillips MS, Fujii J, Khanna VK, DeLeon S, Yokobata J, de Jong PJ, MacLennan DH. The structural organization of the human skeletal muscle ryanodine receptor (RYR1) gene. Genomics. 1996;34:24–41. doi: 10.1006/geno.1996.0238. [DOI] [PubMed] [Google Scholar]
- Quane KA, Healy JM, Keating KE, Manning BM, Couch FJ, Palmucci LM, Doriguzzi C, Fagerlund TH, Berg K, Ording H, Bendixen D, Mortier W, Linz U, Muller CR, McCarthy TV. Mutations in the ryanodine receptor gene in central core disease and malignant hyperthermia. Nat Genet. 1993;5:51–55. doi: 10.1038/ng0993-51. [DOI] [PubMed] [Google Scholar]
- Rosenberg H, Davis M, James D, Pollock N, Stowell K. Malignant hyperthermia. Orphanet J Rare Dis. 2007;2:21–34. doi: 10.1186/1750-1172-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei ML. Hermansky-Pudlak syndrome: a disease of protein trafficking and organelle function. Pigment Cell Res. 2006;19:19–42. doi: 10.1111/j.1600-0749.2005.00289.x. [DOI] [PubMed] [Google Scholar]