

Abstract
While the etiology of Parkinson’s disease (PD) remains unknown, there is overwhelming evidence that neuroinflammation plays a critical role in the progressive loss of dopamine (DA) neurons. Because nearly all persons suffering from PD receive l-DOPA, it is surprising that inflammation has not been examined as a potential contributor to the abnormal involuntary movements (AIMs) that occur as a consequence of chronic l-DOPA treatment. As an initial test of this hypothesis, we examined the effects of exogenously administered corticosterone (CORT), an endogenous anti-inflammatory agent, on the expression and development of l-DOPA-induced dyskinesia (LID) in unilateral DA-depleted rats. To do this, male Sprague-Dawley rats received unilateral medial forebrain bundle 6-hydroxydopamine lesions. Three weeks later, l-DOPA primed rats received acute injections of CORT (0–3.75 mg/kg) prior to l-DOPA to assess the expression of LID. A second group of rats was used to examine the development of LID in l-DOPA naïve rats co-treated with CORT and l-DOPA for 2 weeks. AIMs and rotations were recorded. Exogenous CORT dose-dependently attenuated both the expression and development of AIMs without affecting rotations. Real-time RT-PCR of striatal tissue implicated a role for IL-1β in these effects as its expression was increased on the lesioned side in rats treated with l-DOPA (within the DA-depleted striatum) and attenuated with CORT. In the final experiment, IL-1 receptor antagonist (IL-1ra) was microinjected into the striatum of l-DOPA-primed rats to assess the impact of IL-1 signaling on LID. Intrastriatal IL-1ra reduced the expression of LID without affecting rotations. These findings indicate a novel role for neuroinflammation in the expression of LID, and may implicate the use of anti-inflammatory agents as a potential adjunctive therapy for the treatment of LID.
Keywords: interleukin-1, corticosterone, l-DOPA, dyskinesia, abnormal involuntary movements, striatum
INTRODUCTION
Parkinson’s disease (PD) is a progressive neurodegenerative disease characterized by resting tremor, poverty of movement, postural instability, and rigidity (Dauer & Przedborski, 2003). The pathological hallmark of PD is the presence of proteinaceous inclusions called Lewy bodies and the preferential death of dopamine (DA) neurons within the substantia nigra pars compacta (SNpc). Although the causative agents underlying PD development remain speculative, there is accumulating experimental and clinical evidence that neuroinflammation contributes significantly (McGeer & McGeer, 2004; Whitton, 2007). For example, inflammatory factors such as tumor necrosis factor-α (TNF-α) and interleukin-1β IL-1β are increased within the substantia nigra pars compacta (SNpc) and striatum of post-mortem PD brains ( Mogi et al., 1994; Hirsch et al.,1998) and promote SNpc DA cell death in animals (Viviani et al., 2004; Ferrari et al., 2006). Moreover, anti-inflammatory agents such as minocycline and dexamethasone attenuated nigral cell loss induced by the DA neurotoxin 1-methyl 4-phenyl 1,2,3,6-tetrahydropyridine (MPTP) and the endotoxin lipopolysaccharide (LPS), respectively (Du et al., 2001; Arimoto & Bing, 2003).
For symptomatic treatment, nearly all PD patients receive DA replacement therapy in the form of L-3,4-dihydroxyphenylalanine (l-DOPA). While initially efficacious, prolonged l-DOPA treatment often leads to abnormal and excessive involuntary movements referred to as l-DOPA-induced dyskinesia; LID (Stocchi et al., 1997; Ahlskog & Muenter, 2001). Although the precise mechanism of LID is not fully understood, it is clear that pre- and post-synaptic elements within the striatum contribute (Picconi et al., 2005; Cenci, 2007). For example, LID is associated with supraphysiological increases in striatal DA (Buck & Ferger, 2007) and glutamate (Picconi et al., 2002; Robelet et al., 2004) leading to overactive striatal output through the extracellular regulated kinase (ERK) signaling pathway (Santini et al., 2007) that is exemplified by enhanced preprodynorphin (PPD) expression (Cenci, 2002; Tel et al., 2002). While it is well-documented that DA and glutamate (GLUT) receptor stimulation induce LID and their respective antagonism reduces LID (Bibbiani et al., 2005; Taylor et al., 2005; Mela et al., 2007), excessive extracellular striatal DA and GLUT may also create a transient pro-inflammatory environment (Farber et al., 2005). This aberrant extracellular milieu may exacerbate ongoing neuroinflammatory processes and lead to an exaggerated or sensitized pro-inflammatory response in a condition in which damage has already occurred, such as PD (Gao et al., 2003; Cunningham et al., 2005). Thus, the development and expression of LID may include or be accompanied by an abnormal (sensitized) inflammatory response.
The present study examined the potential relationship between inflammation and LID utilizing the endogenous anti-inflammatory agent corticosterone (CORT) in a rodent model of LID. Acute endogenous CORT has been shown to suppress the immune response at multiple levels via stimulation of glucocorticoid receptors (GR; Guyre et al., 1984). Because GR are found within all basal ganglia nuclei (Elenkov & Chrousos, 2002) it was hypothesized that acute CORT may reduce LID-induced inflammation and as a result alleviate the expression of LID. However, chronic CORT has also been shown to facilitate neurodegeneration and worsen motor behaviors (Sapolsky, 1985; Sapolsky et al., 1985a; Behl et al., 1997; Metz et al., 2005). Thus, we also examined the impact of adjunctive CORT on LID development. To test this, we employed the abnormal involuntary movements (AIMs) model of LID (Lundblad et al., 2002; Eskow et al., 2007) in 6-hydroxydopamine (6-OHDA)-lesioned rats. The present results indicate that CORT administration reduced both the expression and development of AIMs in a dose-dependent manner. We subsequently employed real-time PCR to examine transcriptional changes as a result of l-DOPA and CORT treatment. Examination of striatal mRNA revealed that IL-β, a pro-inflammatory cytokine, is upregulated in l-DOPA-treated rats and attenuated by CORT. In further support of these findings, intrastriatal injection of IL-1 receptor antagonist (IL-1ra) site-specifically reduced the expression of AIMs. Collectively, these novel results implicate an important functional relationship between inflammation and LID and in particular a role for striatal IL-1β.
EXPERIMENTAL PROCEDURES
ANIMALS
Adult male Sprague-Dawley rats were used (225–250 g upon arrival; Taconic Farms, Hudson, NY, USA). Animals were housed in plastic cages (22 cm high, 45 cm deep and 23 cm wide) and had free access to standard lab chow (Rodent Diet 5001; Lab Diet, Brentwood, MO, USA) and water. The colony room was maintained on a 12/12 hr light/dark cycle (lights on at 0700 hrs) at a temperature of 22–23° C. Animals were maintained in accordance with the guidelines of the Institutional Animal Care and Use Committee of Binghamton.
SURGERY
6-HYDROXYDOPAMINE LESION AND CANNULATION SURGERIES
One week after arrival, all rats (N=73) received unilateral 6-hydroxydopamine (6-OHDA; Sigma, St. Louis, MO, USA) lesions of the left medial forebrain bundle to destroy DA neurons. Desipramine HCl (25 mg/kg, ip; Sigma) was given 30 min prior to 6-OHDA injection to protect norepinephrine (NE) neurons. Rats were anesthetized with inhalant isoflurane (2–3%; Sigma) in oxygen (2.5 L/min), then placed in a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA, USA). The coordinates for 6-OHDA injections were AP: −1.8 mm, ML: +2.0 mm, DV: −8.6 mm relative to bregma with the incisor bar positioned 3.3 mm below the interaural line (Paxinos & Watson, 1998). Using a 10 µl Hamilton syringe attached to a 26 gauge needle, 6-OHDA (12 µg) dissolved in 0.9% NaCl + 0.1% ascorbic acid was infused through a small burr hole in the skull at a rate of 2 µl/min for a total volume of 4 µl. The needle was withdrawn 1 min later. For a subset of rats (n=40), 22 gauge guide cannula (Plastics One, Roanoke, VA, USA) were placed bilaterally into the central striatum, AP: +0.4 mm, ML: ±2.9 mm, DV: −3.6 mm (Paxinos & Watson, 1998) immediately following 6-OHDA lesion. Cannulae were fixed in place using liquid and powder dental acrylic (Plastics One Roanoke, VA, USA). At the completion of surgery, guide cannulae were fitted with 28 gauge inner stylets (Plastics One) to maintain patency. Following surgery, all rats were placed in clean cages on warming pads to recover from the surgery, after which they were returned to group-housing (2 rats/cage for lesion only, single housing for cannulated rats). Soft chow was provided as needed to facilitate recovery during the first week after surgery. All rats were allowed to recover for 3 weeks before testing commenced.
EXPERIMENTAL DESIGN
EXPERIMENT 1: DOSE RESPONSE TO EXOGENOUS CORTICOSTERONE
A between subjects design was utilized to determine plasma CORT levels in non-lesioned rats produced by peripheral CORT injections that fall within the physiological range for what is normally evoked by stressors across a wide range of intensities (Kalman & Spencer, 2002). Rats (n=6–7 per group) were injected subcutaneously (sc) with vehicle, 1.25 mg/kg, 2.5 mg/kg, or 3.75 mg/kg of CORT (Sigma, St. Louis, MO, USA) dissolved in EtOH (16%), propylene glycol (44%), and phosphate buffer saline (40%; see Kalman & Spencer, 2002). Blood was sampled at 30, 60, 120, and 240 min following CORT injection. Repeated blood samples (~150 ml) were collected using the tail-clip method by gently stroking the tail as described previously (Deak et al., 2005; Barnum et al., 2007). All blood was collected within 2 min to ensure the samples were not tainted by stress of the sampling procedure. Total plasma CORT was determined using radioimmunoassay (see detailed methods below).
EXPERIMENT 2: IMPACT OF ACUTE ADMINISTRATION OF CORT ON THE EXPRESSION OF AIMS
The second experiment used a within subjects design to examine whether the expression of LID changed as a result of acute CORT administration. To test this, rats (n=13) were primed with 3,4-dihydroxyphenylacetic acid methyl ester (l-DOPA; 4 mg/kg, ip; Sigma) + dl-Serine 2-(2,3,4-trihydroxybenzyl) hydrazide hydrochloride (benserazide; 15 mg/kg, ip; Sigma) (Taylor et al., 2005; Putterman et al., 2007) for 7 days until all rats demonstrated consistent abnormal involuntary movements (ALO AIMs ≥ 10; see detailed methods below). All rats that reached criterion by day 5 received each dose of CORT (1.25, 2.5, 3.75 mg/kg, sc) and vehicle 30 min prior to l-DOPA beginning on day 8 and ALO AIMs and rotations were quantified every 20 min for 2 hrs every 3–4 days thereafter. Treatments were counterbalanced to control for order effects. Following the completion of the study, rats were killed and striata were processed for monoamine analysis with High Performance Liquid Chromatography (HPLC) as described below.
EXPERIMENT 3: THE EFFECTS OF CHRONIC CORT ON THE DEVELOPMENT OF AIMS
The third experiment employed a between subjects design to examine the development of LID in l-DOPA-naïve rats (n=8–9 per group). Two weeks post-lesion and a week prior to experimentation, rats were subjected to the forepaw adjusting steps (FAS) test to determine motor disability (see Eskow et al., 2007 for details) which strongly correlates with degree of DA depletion (Olsson et al., 1995; Chang et al., 1999). Based on these results, rats were assigned to equally disabled groups. Beginning on day 1, rats (n=8–9 per group) were treated with either vehicle, low (1.25 mg/kg), or high (3.75 mg/kg) dose of CORT 30 min prior to l-DOPA (4 mg/kg) + benserazide (15 mg/kg) for 9 consecutive days. From day 10–15, all rats received l-DOPA + benserazide only. Immediately following l-DOPA administration on days 1, 3, 5, 8, and 15, ALO AIMs and rotations were quantified every 20th min for 2 hrs. Following the completion of the AIMs test on day 15, rats were killed and striata were processed for HPLC analysis.
EXPERIMENT 4: STRIATAL mRNA EXPRESSION IN RATS CO-TREATED WITH L-DOPA AND CORT
Our initial transcriptional analyses focused on three sets of factors: (i) mRNA for PPE, PPD, and PPT given the extant literature supporting an association between activity of these neuropeptides and the expression of LID (Cenci 2007); (ii) mRNA for D1 and D2 receptors as a preliminary test of whether exogenous CORT influenced DA function; and (iii) mRNA for several key pro-inflammatory cytokines (IL-1, TNF-α & IL-6) to establish a potential mechanism(s) underlying the anti-dyskinetic properties of exogenous CORT because corticosteroid treatment has pronounced anti-inflammatory properties (Munck et al., 1984). To test this, rats (from experiment 2) were assigned to one of two equally dyskinetic groups (n=6–7 per group) and were injected (i.p.) with either CORT (3.75 mg/kg) or vehicle 30 min prior to l-DOPA (4 mg/kg) + benserazide (15 mg/kg) treatment and then killed by rapid decapitation 2 hrs later. Striata were dissected and processed for HPLC and real-time reverse-transcription polymerase chain reaction (RT-PCR) analysis as described below. As described below, relative gene expression was quantified using the 2−ΔΔCT method according to Pfaffl (2001) and Livak and Schmittgen (2001) using the non-lesioned, vehicle-treated striata as the ultimate control.
EXPERIMENT 5: THE ROLE OF IL-1 IN THE EXPRESSION OF AIMS
To further delineate a potential mechanism by which CORT reduces LID, we examined whether AIMs could be reduced by intrastriatal injection of IL-1 receptor antagonist (IL-1ra; Amgen, Thousand Oaks, CA). To test this, intrastriatally cannulated rats (n=9–12 per group) were primed with l-DOPA (4 mg/kg) + benserazide (15 mg/kg) for 7 days until all rats demonstrated consistent abnormal involuntary movements (AIMs). If criterion was achieved (ALO AIMs ≥ 10), rats were matched for AIMs and assigned to one of three groups in a between subjects design. Three days after the last day of priming, rats received an acute bilateral intrastriatal infusion of 1.34-µl (0.5 µl/min) of 10-µg IL-1ra, 100-µg IL-1ra, or vehicle (sterile saline) immediately prior to l-DOPA after which ALO AIMs and rotations were quantified over 2 hrs. Following these tests, rats were killed and striata were processed for HPLC and verification of cannula placement.
ABNORMAL INVOLUNTARY MOVEMENTS (AIMs)
Rats were monitored for AIMs using a procedure described previously (Dupre et al., 2007; Eskow et al., 2007) and similar to that initially depicted by Lundblad and colleagues (2002). On test days (0900–1400 hrs), rats were individually placed in plastic trays (60 cm × 75 cm) 5 min prior to pretreatments. Following l-DOPA injection, a trained observer blind to treatment condition assessed each rat for exhibition of axial, limb, orolingual, AIMs. Each new rater was trained for a minimum of 3 sessions and then correlated with a well-trained instructor. A correlation of ≥ 90% with the instructor is required before new raters can score AIMs. Inter-rater reliability for AIMs in the current studies was ≥ 95%. In addition, contralateral rotations, defined as complete 360 degree turns away from the lesioned side of the brain, were tallied. No ipsilateral rotations, defined as complete 360 degree turns toward the lesioned side of the brain, were observed during testing at the doses tested. Dystonic posturing of the neck and torso, involving positioning of the neck and torso in a twisted manner directed toward the side of the body contralateral to the lesion, were referred to as “axial” AIMs. “Forelimb” AIMs were defined as rapid, purposeless movements of the forelimb located on the side of the body contralateral to the lesion. “Orolingual” AIMs were composed of repetitive openings and closings of the jaw and tongue protrusions. The movements are considered abnormal since they occur at times when the rats are not chewing or gnawing on food or other objects. Every 20th min for 2 hrs, rats were observed for 2 consecutive min. Rats were rated for AIMs during the 1st min and rotational behavior in the 2nd min. During the AIMs observation periods (beginning 20, 40, 60, 80, 100, and 120 min post-injection), a severity score of 0–4 was assigned for each AIMs category: 0= not present, 1= present for less than 50% of the observation period (i.e., 1–29 sec), 2= present for more than 50% or more of the observation period (i.e., 30–59 sec), 3 = present for the entire observation period (i.e., 60 sec) and interrupted by a loud stimulus (a tap on the wire cage lid), or 4 = present for the entire observation period but not interrupted by a loud stimulus. For each AIMs category, the scores were summed for the entire 2 hr period. Thus, the theoretical maximum score for each type of AIM was 24 (4 × 6 periods) although observed scores were never this severe. For statistical analysis, three of the AIMs subcategories (axial, forelimb, and orolingual; ALO AIMs) and rotations were summed for the entire 2-h period.
TISSUE PROCESSING
Tissue was harvested after rapid decapitation and striata were quickly removed on a cold plate, flash frozen, and stored at −70°C until time of assay. For each rat, left and right striata (between + 0.6 − 0.6 from bregma) were bissected for HPLC and/or RT-PCR analysis depending upon the experiment. Striata processed for HPLC were homogenized in 0.1M Perchloric Acid using a motorized pestle and centrifuged at 4°C at 14,000 rpm for 30 min. Supernatant was transferred to a new tube and stored at −80°C until time of assay. The residual pellet was re-suspended in phosphate buffer saline (PBS) and assessed for total protein using the method of (Bradford, 1976). For RT-PCR, tissue was processed using Qiagen’s RNeasy mini protocol for isolation of total RNA from animal tissues (RNeasy Mini Handbook, 3rd editon, 2001) with slight modifications. Briefly, on the day of assay, frozen striata were quickly placed into a 1.5 ml eppendorf containing 350 µl Buffer RLT + β-mercaptoethanol (Sigma). Tissue was homogenized using a motorized pestle and passed through Qiagen QIAshredder columns to shear residual genomic DNA and ensure thorough homogenization of samples. Equal volume of 70% ethanol was added to the supernatant and purified through RNeasy mini columns. Columns were washed with buffer and eluted with 30 µL of RNase-free water (65°C). First strand cDNA synthesis was performed according to manufacturer’s instructions with 8 µL of total RNA using oligo DT primer according to manufacturer protocols (First-Strand cDNA Synthesis Kit, Amersham Biosciences) and stored at −20°C.
HISTOLOGY
To verify cannula placement, a coronal dissection was made posterior to the cannulation and the anterior section of the brain was flash frozen in 2-Methylbutane (EMD Chemicals Inc., Gibbstown, NJ, USA). Striata were sectioned at 20 µm (coronal) on a cryostat, stained with cresyl violet, and examined under light microscopy. All rats were found to have injector placements within the boundries of the striatum (see Figure 6).
Figure 6.
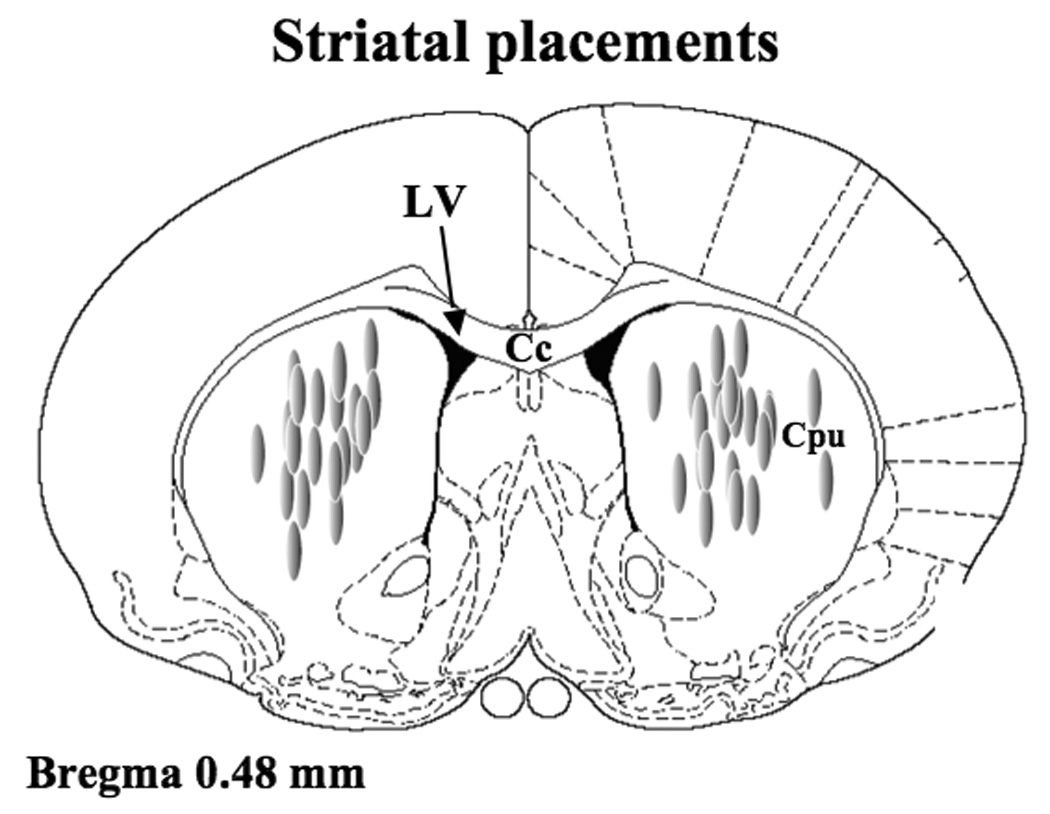
Schematic representation of coronal sections of the rat brain taken from Paxinos and Watson (1998). Shaded ovals depict the location of striatal microinfusion sites in rats microinjected with IL-1ra. Anatomical structures: Cc corpus collosum; Cpu caudate putamen; LV lateral ventricle.
HPLC
Reverse-phase HPLC coupled to electrochemical detection was performed on striatal tissue according to the protocol of Kilpatrick and colleagues (1986). The ESA system (Chelmsford, MA, USA) included an autoinjector (Model 542), an ESA solvent delivery system (1582), an external pulse dampener (ESA), a ESA Guard-Pak column, and a MD-150 column (ESA). Samples were homogenized in ice-cold perchloric acid (0.1 M), 1% ethanol, and 0.02% EDTA and spun for 30 min at 16,100 g with the temperature maintained at 4° C. Aliquots of supernatant were then analyzed for abundance of DA and 3,4-dihydroxyphenylacetic acid (DOPAC). Samples were separated using a mobile phase composed of 90 mM sodium dihydrogen phosphate (monobasic, anhydrous), 0.05 mM EDTA, 1.7 mM octane sulfonic acid, and 10% acetonitrile, adjusted to pH 3.0 with o-phosphoric acid. A coulometric detector configured with 3 electrodes (Coulochem III, ESA) measured content of monoamines and metabolites. An ESA model 5020 guard cell (+300 mV) was positioned prior to the autoinjector. The analytical cell (ESA model 501lA; first electrode at −100 mV, second electrode at +250 mV) was located immediately after the column. The second analytical electrode emitted signals that were recorded and analyzed by EZChrom Elite software via a Scientific Software, Inc. (SS420χ) module. The final oxidation current values were compared to known standard concentrations (10−6–10−9) and adjusted to total striatal protein content using the method of Bradford (1976) and expressed as nanogram (ng) of monoamine or metabolite per milligram (mg) tissue protein (mean ± 1 SEM).
RADIOIMMUNOASSAY FOR CORT
Plasma CORT was measured by radioimmunoassay using rabbit antiserum (antibody B3-163, Endocrine Sciences, Tarzana, CA, USA) as previously described (Deak et al., 2005; Barnum et al., 2007). This antiserum was employed due to its low cross reactivity with other glucocorticoids and their metabolites. Assay sensitivity was 0.5 µg/dl (assay volume = 20 µl plasma). The intra-assay coefficient of variation was 8%.
REAL-TIME RT-PCR
PCR product was amplified using the IQ SYBR Green Supermix kit (BioRad). Briefly, a reaction mastermix (total volume 20 µL) consisting of 10 µL SYBR Green Supermix, 1 µL primer (final concentration 250 nM), 1 µL cDNA template, and 8 µL RNase-free water was run in duplicate in a 96 well plate (BioRad) according to the manufacturers instructions and captured in real-time using the iQ5 Real-Time PCR detection system (BioRad). Following a 3 min hot start (95°C), samples underwent 30 sec of denaturation (95°C), 30 sec of annealing (60°C), and 30 sec of extension (72°C) for 40 cycles. An additional denaturation (95°C, 1 min) and annealing cycle (55°C, 1 min) were conducted to ensure proper product alignment prior to melt curve analysis. For melt curve analysis, samples underwent 0.5°C changes every 15 sec ranging from 55°C – 95°C. A single peak expressed as the negative first derivative of the change in fluorescence as a function of temperature indicated the presence of a single amplicon. Primer sequences are presented in Table 1. Relative gene expression was quantified using the 2−ΔΔCT method as described previously using calcium modulating cyclophilin ligand (CAML) as a reference gene (Livak & Schmittgen, 2001; Pfaffl, 2001).
TABLE 1.
Specific gene sequences were obtained from Genbank at the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/) and copied into Primer3 for primer design (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). Primer specificity was verified using the Basic Local Alignment Search Tool (http://www.ncbi.nlm.nih.gov/blast/), and ordered from Integrated DNA Technologies (Coralville, IA). In some cases, primers were taken from the literature. Whenever possible primers were designed to span an intron.
| Name | Oligo | Sequence | Product size | Accession Number |
|---|---|---|---|---|
| CAML | Forward | 5’-ggacgacggaagagtttgac-3’ | 247 bp | AF302085 |
| Reverse | 5’-tccatggaccggtttatcac-3’ | |||
| DR1 | Forward | 5’-tccactctcctgggcaatac-3’ | 240 bp | NM_012546.1 |
| Reverse | 5’-tcacgcagaggttcagaatg-3’ | |||
| DR2 | Forward | 5’-aaaatctgggagacctgcaa-3’ | 317 bp | X53278 |
| Reverse | 5’-tctgcggctcatcgtcttaag-3’ | |||
| IL-1 | Forward | 5’-aggacccaagcaccttcttt-3’ | 152 bp | NM_031512 |
| Reverse | 5’-agacagcacgaggcattttt-3’ | |||
| IL-6 | Forward | 5’-ccggagaggagacttcacag-3’ | 134 bp | NM_012589 |
| Reverse | 5’-cagaattgccattgcacaac-3’ | |||
| PPD | Forward | 5’-gggttcgctggattcaaata-3’ | 197 bp | NM_019374 |
| Reverse | 5’-tgtgtggagagggacactca-3’ | |||
| PPE | Forward | 5’-aaaatctgggagacctgcaa-3’ | 243 bp | K02807.1 |
| Reverse | 5’-catgaaaccgccatacctct-3’ | |||
| PPT | Forward | 5’-agcctcagcagttctttgga-3’ | 255 bp | NM_012666 |
| Reverse | 5’-cggacacagatggagatgaa-3’ | |||
| TNF-α | Forward | 5’-gtctgtgcctcagcctcttc-3’ | 113 bp | X66539 |
| Reverse | 5’-cccatttgggaacttctcct-3’ |
CAML - calcium modulating cyclophilin ligand; DR1 – dopamine receptor 1; DR2 – dopamine receptor 2; IL-1 & IL-6 – interleukin-1/6; PPD – preprodynorphin; PPE – preproenkephalin; PPT – preprotachykinin; TNF-α – tumor necrosis factor-α.
Data Analyses
Monoamine and metabolite levels in the striatum were analyzed using paired t-tests (comparing intact versus lesioned striata). Treatment effects for ALO AIMs were analyzed by employing non-parametric between subjects Kruskal–Wallis or within subjects Friedman tests. Rotations and plasma CORT levels were analyzed using a 2-way and repeated measures analysis of variance (ANOVAs), respectively. Significant differences between treatments were determined by Wilcoxon post hoc comparisons for ALO AIMs, and planned comparison post hoc tests for ALO AIMs and rotations in experiment 5. Relative gene expression (expressed as means ± 1 SEM from control) was analyzed using two-way ANOVA followed by planned comparison post hoc tests. All data are expressed as means ± 1 SEM. Analyses were performed with the use of Statistica software ’98 (Statsoft Inc., Tulsa, OK, USA). Alpha was set at P< 0.05.
RESULTS
MONOAMINE & METABOLITE LEVELS
The effects of the 6-OHDA lesion on concentrations of monoamine and metabolite levels and turnover ratios (metabolite/monoamine) in the intact (right) versus lesioned (left) striata were examined using HPLC across all experiments (see Table 2). Unilateral 6-OHDA injection into the medial forebrain bundle produced significant reductions in striatal DOPAC and DA levels (P<0.05), 88% and 96% respectively, compared to intact striatum. The denervated side also showed an increased DOPAC/DA turnover rate (295%) compared to control (P <0.05).
TABLE 2. Effects of unilateral medial forebrain bundle (MFB) 6-OHDA lesions on concentration of 3,4-dihydroxyphenylacetic acid (DOPAC), dopamine (DA), and their metabolites/monoamine ratios in the striatum.
Values are nanogram monoamine or metabolite per milligram protein or ratios of metabolite to monoamine (mean ± 1 SEM) and percentage of control group. Differences between group means were determined by paired t-tests
| DOPAC (ng/mg) | DA (ng/mg) | DOPAC/DA | |
|---|---|---|---|
| Experiment 2: Expression of LID following acute CORT injection | |||
| Intact (right) | 94.6 ± 15.5 | 376.2 ± 63.8 | 0.27 ± 0.1 |
| Lesioned (left) | 5.6 ± 1.1* | 11.9 ± 3.3* | 0.63 ± 0.1* |
| (lesioned/intact, %) | 6% | 3% | 237% |
| Experiment 3: Development of LID with chronic CORT injection | |||
| Intact (right) | 38.1 ± 13.1 | 221.7 ± 57.9 | 0.26 ± 0.1 |
| Lesioned (left) | 5.8 ± 2.0* | 9.3 ± 4.9* | 0.78 ± 0.1* |
| (lesioned/intact, %) | 15% | 4% | 299% |
| Experiment 5: Role of interleukin-1 (IL-1RA) in the expression of LID | |||
| Intact (right) | 32.1 ± 5.8 | 55.4 ± 4.4 | 0.76 ± 0.1 |
| Lesioned (left) | 4.6 ± 0.7* | 2.5 ± 0.4* | 2.66 ± 0.4* |
| (lesioned/intact, %) | 14% | 5% | 350% |
(p < 0.05 compared to the intact side).
EXPERIMENT 1: DOSE RESPONSE TO EXOGENOUS CORTICOSTERONE
A 2-way ANOVA was used to determine potential differences in plasma CORT following doses of exogenous CORT injection over time (see Figure 1). Plasma levels of CORT differed depending on the CORT dose administered [F(3,21)=78.6,P<0.05] and the time-point examined [F(3,63)=191.7,P<0.05]. A time-point × dose interaction was also noted [F(9,63)=13.2,P<0.05]. Post hoc analyses revealed a dose-dependent increase in plasma CORT levels at the 30 and 60 min time-points as all doses were significantly different from each other (P<0.05) with the exception of the 1.25 mg/kg vs. 2.5 mg/kg at 60 min. A similar trend was observed at the 2 hr time-point, as all doses were significantly different from each other (P<0.05) with the exception of the 2.50 mg/kg and 3.75 mg/kg dose of CORT. At the 4 hr time-point, only vehicle and 1.25 mg/kg vs. 3.75 mg/kg were significantly different from each other (P<0.05).
Figure 1.
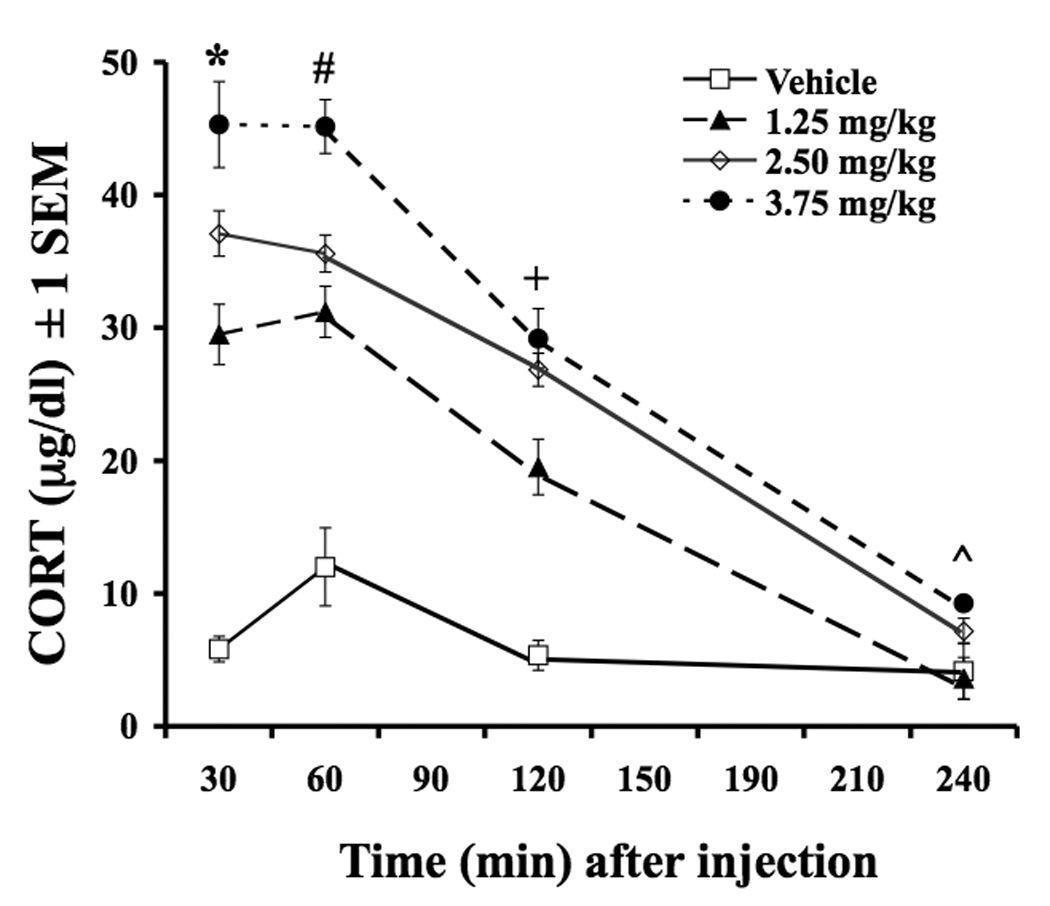
Effects of peripheral CORT injections on plasma CORT levels. Rats were injected with Vehicle (16% EtOH, 44% propylene glycol, 40% phosphate buffer saline) or CORT (1.25 mg/kg, 2.50 mg/kg, 3.75 mg/kg) and tail blood was sampled at 30, 60, 120, and 240 min later. Figure 1: * All doses are different from each other at 30 min time-point (p<0.05). # All doses are different from each other except 1.25 mg/kg vs. 2.50 mg/kg at 60 min time-point (p<0.05). + All doses are different from each other except 2.50 mg/kg vs. 3.75 mg/kg at 120 min time-point (p<0.05). ^ Only 1.25 mg/kg vs. 3.75 mg/kg are different at 240 min time-point (p<0.05). Data are expressed as mean ± 1 SEM.
EXPERIMENT 2: IMPACT OF ACUTE ADMINISTRATION OF CORT ON AIMS EXPRESSION
The same doses of CORT used in experiment 1 were tested in l-DOPA-primed rats to determine their effects on ALO AIMs and rotations. As shown in Figure 2, CORT dose-dependently reduced the expression of ALO AIMs (χ32=23.12,P<0.05) as post hoc analyses demonstrated that all doses of CORT reduced ALO AIMs compared to vehicle treated rats (P<0.05). A similar pattern of effects in rotations was also observed although this failed to achieve statistical significance [F(3,47)=2.2, P<0.05].
Figure 2.
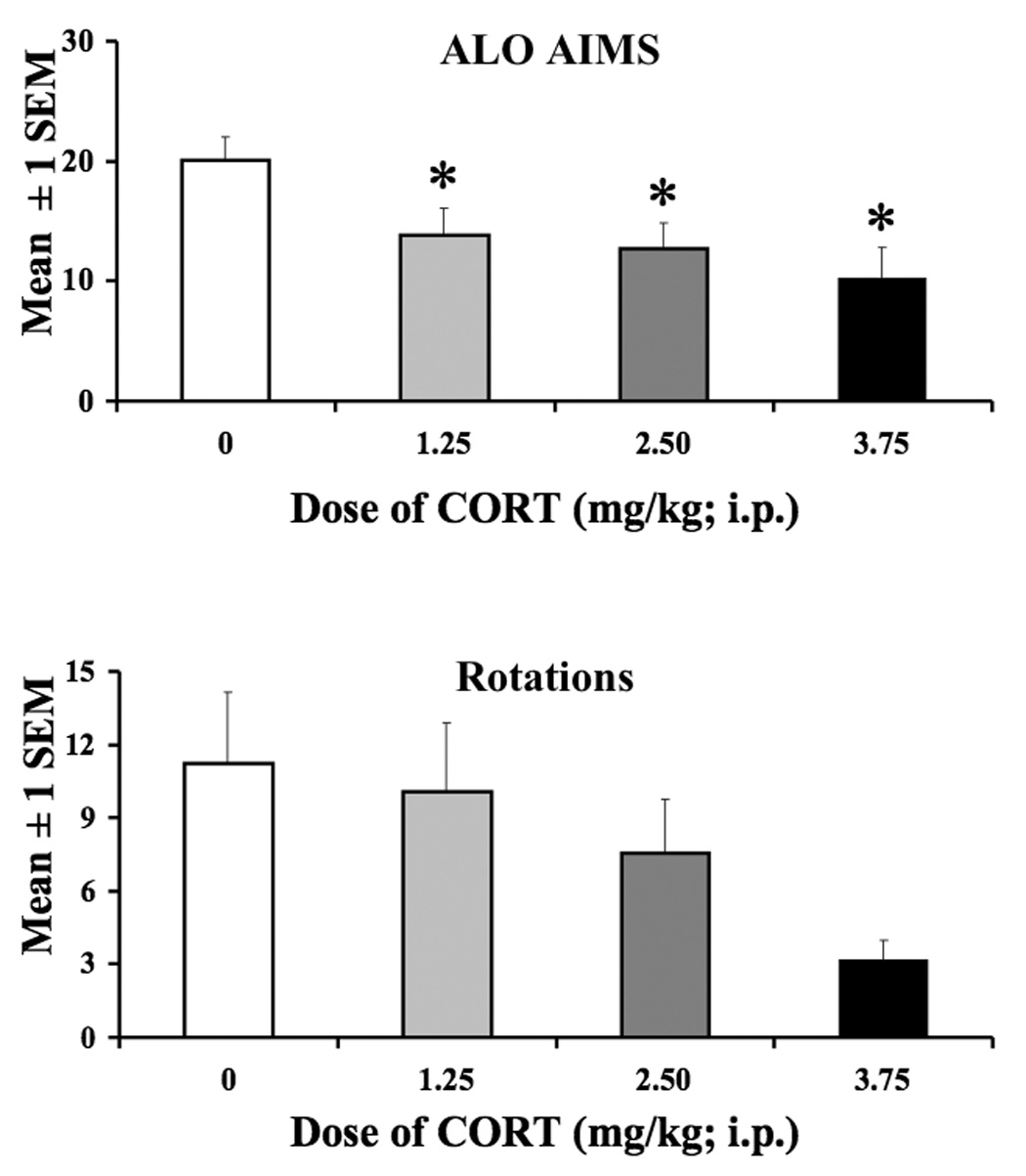
The effects of CORT on the expression of ALO AIMs and rotations. A within subjects design was used to examine the effects of CORT (1.25 mg/kg, 2.5 mg/kg, or 3.75 mg/kg) and Vehicle (16% EtOH, 44% propylene glycol, 40% phosphate buffer saline) prior to l-DOPA treatment on AIMs and rotations in L-DOPA-primed rats. Each dose of CORT reduced ALO AIMs compared to Vehicle-treated rats. * Significantly different from Vehicle, p<0.05. Data are expressed as the mean ± 1 SEM.
EXPERIMENT 3: THE EFFECTS OF CHRONIC CORT ON THE DEVELOPMENT OF AIMS
The third experiment examined the development of AIMs in l-DOPA-naïve rats. As shown in Figure 3, analysis of the development of ALO AIMS revealed significant treatment effects at day 1 (χ22=5.8,P< 0.05), day 3 (χ22=4.0,P<0.05), and day 5 (χ22=4.0,P<0.05). Post hoc analyses revealed that ALO AIMs were reduced in rats receiving 3.75 mg/kg of CORT (vs. vehicle) on day 1, and rats receiving 1.25 & 3.75 mg/kg of CORT on days 3 & 5 (P<0.05) compared to vehicle. No changes in rotations were noted [F(8,60)=1.2,P>0.05].
Figure 3.
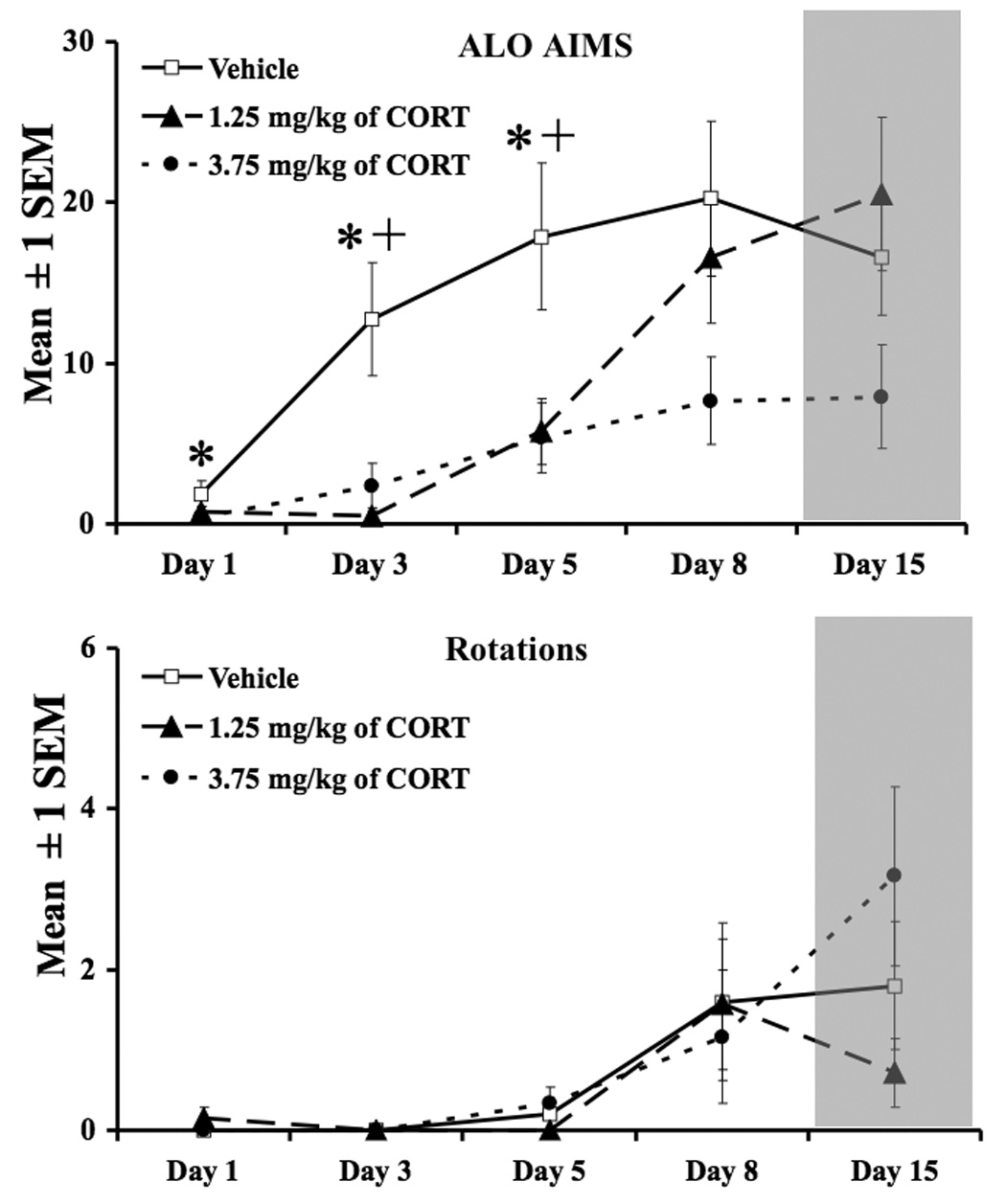
The development of ALO AIMs and rotations in rats chronically treated with CORT and l-DOPA. From days 1–9, rats received either CORT (1.25 mg/kg or 3.75 mg/kg) and l-DOPA (4 mg/kg + 15 mg/kg of benserazide) or Vehicle (16% EtOH, 44% propylene glycol, 40% phosphate buffer saline) and l-DOPA. From days 10 –15, rats only received l-DOPA (shaded region). The development of ALO AIMs was delayed in rats co-administered CORT and l-DOPA on days 1, 3, and 5. LID developed once CORT treatment ceased (see day 15). All symbols denote p<0.05. (* Vehicle vs. 3.75 mg/kg; + vehicle vs. 1.25 mg/kg). Data are presented as mean ± 1 SEM.
EXPERIMENT 4: STRIATAL mRNA EXPRESSION IN RATS CO-TREATED WITH L-DOPA AND CORT
RT-PCR was employed to test whether CORT pre-treatment altered the expression of inflammatory factors and neuropeptides within the striatum of l-DOPA-primed rats (see Figure 4). Analyses revealed main effects of CORT treatment on measures of IL-1 [F(1,24)=9.08,P<0.05], PPE [F(1,24)=6.13,P<0.05], and PPD [F(1,24)=7.30,P<0.05]. Main effects of lesion were also shown on IL-1 [F(1,24)=4.80,P<0.05], PPE [F(1,24)=28.11,P<0.05], and PPT [F(1,24)=8.60,P<0.05]. Significant interactions were demonstrated on measures of IL-1 [F(1,24)=4.8,P<0.05], PPE [F(1,24)=8.94,P<0.05], and PPD [F(1,24)=5.66,P<0.05]. Planned comparisons on highest order effects revealed that 6-OHDA lesion reduced striatal PPT mRNA and CORT suppressed l-DOPA-induced increases in IL-1, PPE, and PPD mRNA expression within the lesioned striatum (P<0.05). No changes in TNF-α, IL-6, D1R, or D2R mRNA were observed.
Figure 4.
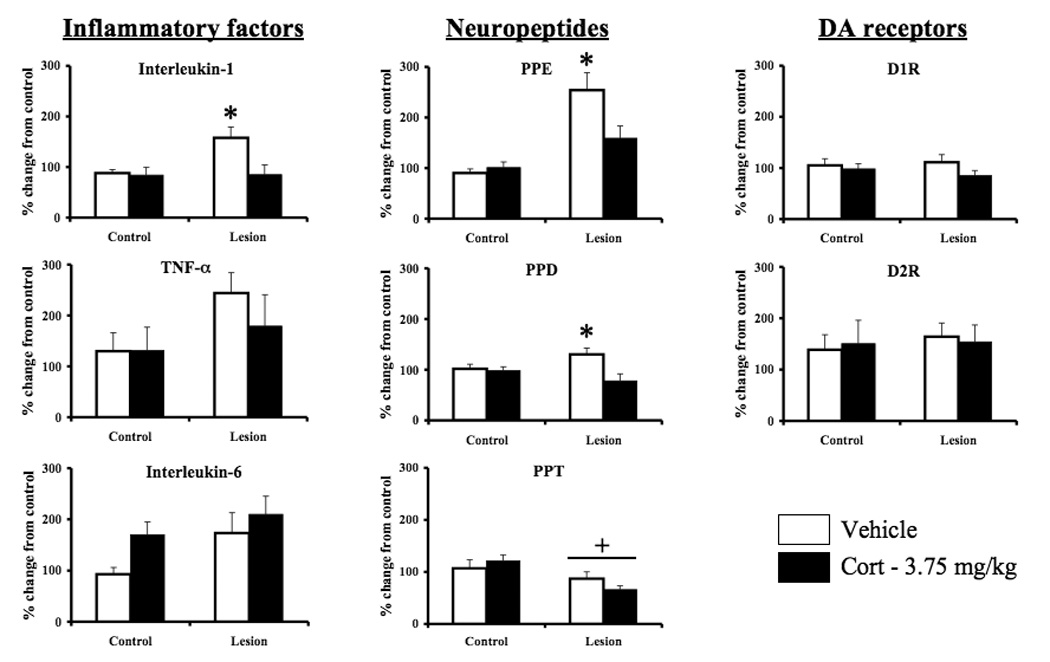
Striatal mRNA expression of pro-inflammatory cytokines, neuropeptides, and DA receptors in rats co-treated with CORT (3.75 mg/kg) and l-DOPA (4 mg/kg + 15 mg/kg of benserazide). L-DOPA primed rats received an injection of CORT prior to l-DOPA treatment and were examined for changes in mRNA 2 hrs later. Pre-treatment with CORT reduced l-DOPA-induced increases in IL-1β, PPE, and PPD mRNA within the lesioned striatum. No changes in DA receptors were observed as a result of treatment. For IL-1β, PPE, and PPD, *p<0.05 compared to all other groups. For PPT, + with a solid bar underneath denotes main effect of lesion, p<0.05. Data are presented as mean % change from control (non-lesioned striata treated with vehicle) ± 1 SEM.
EXPERIMENT 5: THE ROLE OF IL-1β IN THE EXPRESSION OF L-DOPA-INDUCED DYSKINESIA
In order to test whether the reduction in ALO AIMs following CORT administration was mediated by IL-1β, rats received intrastriatal infusion of vehicle, 10-µg, or 100-µg of IL-1ra followed immediately by l-DOPA and then assessed for AIMs and rotations for 2 hours. As Figure 5 demonstrates, there was a dose-dependent reduction in ALO AIMs and rotations following intra-striatal injection of IL-1ra, however, with all doses included, this approached but did not achieve statistical significance, χ22=4.9,P>0.05 and [F(2,26)=2.89, P>0.05], respectively. Because our a priori prediction was that the high (100-µg) dose of IL-1ra would attenuate ALO AIMs, a planned comparison was also conducted between the high dose of IL-1ra and vehicle treated rats. Planned comparisons revealed that the suppressive effect of IL-1ra (100-µg) on ALO AIMs (χ12 =4.75,P< 0.05) and rotations [F(1,18)=6.10, P<0.05] was statistically significant.
Figure 5.
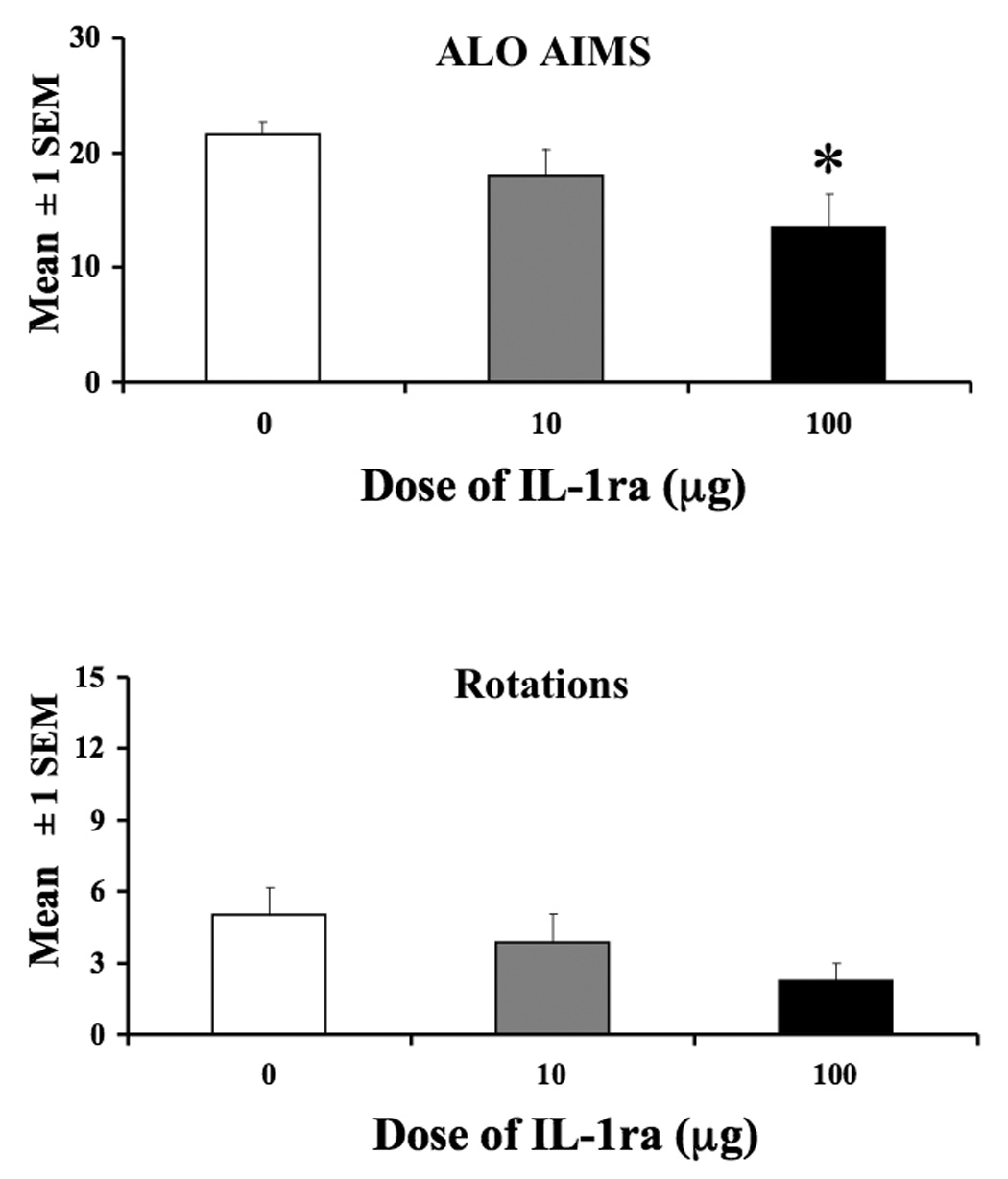
Role of IL-1β in the expression of l-DOPA-induced ALO AIMs and rotations. L-DOPA-primed rats were intrastriatally microinjected with IL-1ra (10-µg or 100-µg) or Vehicle (sterile saline) followed by an immediate systemic injection of l-DOPA (4 mg/kg + 15 mg/kg of benserazide) and assessed for AIMs and rotations for 2 hrs. Rats receiving 100-µg IL-1ra showed a reduction in AIMs and rotations compared to vehicle treated rats (* 100-µg vs. VEH, p<0.05). Data are presented as mean ± 1 SEM.
DISCUSSION
Convergent evidence supports the hypothesis that neuroinflammation contributes to the progressive loss of DA neurons in PD by direct recruitment of apoptotic pathways or through increased production of reactive oxygen species (Schulz et al., 1995; He et al., 2000; Anderson, 2001). While DA replacement therapy with l-DOPA provides unique symptomatic relief of PD-related movement disability, repeated administration leads to the development of LID (Jankovic, 2005). Traditional investigations of LID have focused primarily upon DA, GLUT and their signaling pathways. The results presented here suggest that corticosteroid signaling may moderate LID via inhibitory actions on inflammatory signaling pathways.
The purpose of the present series of studies was to examine whether exogenous CORT modulates the development and expression of LID in the hemi-parkinsonian rat. To do this, we first determined doses of exogenous CORT that mirror plasma CORT levels within the physiological range. This is important because the physiological and behavioral effects of CORT have been shown to be dependent upon dose and duration of corticosteroid exposure (Sapolsky et al., 1985a; Abraham et al., 2000; Nichols et al., 2005). We tested doses initially reported by Kalman and Spencer (2002) by analyzing plasma CORT levels at various time-points for 240 min. As depicted in Figure 1, each dose of exogenous CORT produced robust and statistically different levels of plasma CORT at nearly all time-points examined (240 min time-point is the exception). Importantly, CORT remained elevated for at least 2 hours, corresponding to the duration of behavioral monitoring employed in the current study.
As an initial investigation into the effects of CORT on LID, hemi-parkinsonian rats were primed with a dose of l-DOPA (4 mg/kg) that produces moderate AIMs (Lundblad et al., 2002; Winkler et al., 2002; Taylor et al., 2005; Putterman et al., 2007). These l-DOPA-primed rats were then tested multiple times to examine the effects of CORT on LID expression. As seen in the Vehicle-treated rats (Figure 2), l-DOPA led to a significant induction of ALO AIMs and contralateral rotations. Because plasma CORT levels peaked 30 min following CORT injections, rats were injected with CORT 30 min prior to l-DOPA treatment on test days. Exogenous CORT pretreatment produced a dose-dependent reduction (~50% with the highest dose) in the expression of ALO AIMs that did not significantly alter l-DOPA-induced rotations. To our knowledge, this is the first report of the anti-dyskinetic effects of CORT.
In order to extend these novel findings, we also examined whether chronic, exogenous CORT administration would attenuate the development of LID. To do this, separate, but equally disabled groups of l-DOPA-naïve rats were treated daily with Vehicle or CORT (1.25 or 3.75 mg/kg) over 9 days and examined periodically for the development of ALO AIMs and rotations. To assess the long-term influence of prolonged CORT on l-DOPA-induced behaviors, rats were then administered l-DOPA alone for 4 days and tested for AIMs and rotations on the 5th day. As demonstrated in Figure 3, adjunctive CORT treatment delayed the development of ALO AIMs without affecting rotations. This effect was more protracted with the higher dose of CORT in which ALO AIMs were consistently half of their vehicle counterparts, while in contrast, by day 8, rats receiving the low dose of CORT became increasingly dyskinetic. It is interesting to note that rats receiving the highest dose of CORT did not appear to reach Vehicle pretreatment levels, even on the final test day, 6 days after their last CORT injection. This observation suggests that higher doses of CORT may have enduring effects on LID even when rats are matched for similar levels of movement disability and DA depletion. Importantly, DA levels were similar across treatment groups (P>0.05; data not shown) suggesting that CORT did not modulate DA metabolism. Furthermore, CORT is a powerful inducer of locomotor activity (Wolkowitz, 1994) indicating that the reduction in ALO AIMs is likely not due to an overall decrease in motor activity. Taken together, the outcome of these behavioral experiments show for the first time that exogenous CORT administration attenuated both the expression and development of LID.
As a preliminary test of the mechanism(s) by which exogenous CORT reduced AIMs, striata from CORT-treated rats were divided for parallel determination of monoamines (verifying degree of lesion) and transcriptional changes using real-time RT-PCR. As described above (see methods for experiment 4), our transcriptional analysis focused on factors associated with dyskinesia (PPD, PPE, PPT), mRNA for DA receptors (D1R, D2R), and transcripts for key proinflammatory cytokines (IL-1, IL-6, TNF-α) to establish a potential mechanism(s) underlying the anti-dyskinetic properties of exogenous CORT because corticosteroid treatment has pronounced anti-inflammatory properties (Munck et al., 1984). While these inflammatory factors have been shown to be increased in animal models of PD and suppressed by exogenous administration of glucocorticoids (Arimoto & Bing, 2003; Garside et al., 2004; Kurkowska-Jastrzebska et al., 2004; Necela & Cidlowski, 2004;), their expression has not been investigated as a function of LID. As shown in Figure 4, IL-1β mRNA was significantly increased in the DA-depleted striatum following l-DOPA administration. Though a similar pattern of changes was observed in TNF-α and IL-6, these effects were far more variable across subjects and failed to achieve statistical significance. It should be noted, however, that the time of tissue collection (2 hr after l-DOPA administration) might explain the apparent ‘selective’ increase in IL-1β, and that evaluation of multiple time points could reveal a cascade of inflammatory cytokine expression involving multiple factors. The most effective anti-dyskinetic dose of CORT (3.75 mg/kg) completely reversed the increased IL-1β̣ mRNA observed on the lesion side while also blunting the LID-associated increases in striatal PPE and PPD mRNA in the DA-depleted striatum, indicating that CORT may influence the output of both the direct and indirect pathway. Indeed, a major strength of the current work is that IL-1β changed in parallel with PPE and PPD following l-DOPA administration and CORT pre-treatment providing a novel link between neuroinflammation and overactive striatal output (Cenci et al., 1998; Cenci, 2002; Tel et al., 2002).
It is important to note certain limitations of the current series of experiments. First, an alternative interpretation for CORT reducing ALO AIMs is that it interfered with DA transmission. This said, CORT did not alter the expression of D1/D2 receptors (Figure 4), nor did chronic treatment of CORT + l-DOPA alter DA levels in the striatum compared to rats that only received chronic l-DOPA, a preliminary indication that in our model CORT does not directly modify striatal DA processes. Second, changes in the reported increase in IL-1β mRNA cannot be directly ascribed to l-DOPA treatment as inflammation is a byproduct of the DA-depleting lesion (i.e., Whitton, 2007). Because the focus of this study was LID, all animals in the PCR experiment received l-DOPA treatment and vehicle-treated rats were not included. While these within-subjects (lesion vs. intact) comparisons can reduce variability, they may also minimize lesion effects. Studies are currently underway to further address these issues. In summary, these initial behavioral and cellular results supported that l-DOPA-induced IL-1β may play an important mechanistic role in the development and expression of LID.
In order to more directly test this hypothesis, IL-1ra was microinjected directly into the striata of l-DOPA-primed and treated rats. Consistent with our hypothesis, IL-1ra reduced the expression of ALO AIMs and rotations (see Figure 5), although this was only significant in rats treated with the high dose of IL-1ra (100 µg). Though the effects of IL-1ra were somewhat modest, this is not surprising given that l-DOPA administration activates the striatum very broadly (Mura et al., 2002; Svenningsson et al., 2002), while site-specific microinfusions are, by nature, limited to a discrete location within the striatum (see Figure 6 for injection placements). Moreover, changes in DA levels (table 2) are likely the result of a more posterior dissection necessitated by the location of striatal cannulation, although percent change from intact striatum remained approximately the same across studies (data not shown). Indeed, a considerable strength of the present work is that it provides novel evidence implicating endogenous expression of IL-1β in LID, while at the same time implicating the striatum as the site of action.
While the mechanism underlying CORT attenuation of LID is currently unknown, CORT may convey its anti-dyskinetic effects by acting on second messenger systems common to both IL-1β and LID. Indeed, it is generally accepted that l-DOPA administration leads to a transient, but marked, increase in extracellular striatal DA and GLUT that stimulates intrinsic D1 and D2 receptors ( Robelet et al., 2004; Cenci, 2007) and augments GLUT transmission via AMPA & NMDA receptors (Picconi et al., 2002; Santini et al., 2007). These concerted actions initiate LID through a cAMP/DARPP-32 and extracellular-regulated kinase (ERK)-dependent signaling pathway (Santini et al., 2007). It is at this point that the intersection may occur as ERK can activate both NF-kB and CREB, each of which can induce IL-1β (Jiang et al., 2004; Zhao & Brinton, 2004). In support of this, CORT has been shown to attenuate the activity of ERK, CREB, and NF-kB (Necela & Cidlowski, 2004; Yu et al., 2004; Li et al., 2005) lending to its possible anti-dyskinetic effects. Future mechanistic studies will be necessary to identify the mechanisms by which CORT suppresses IL-1β expression in the context of LID.
While it is not entirely clear to what extent l-DOPA directly enhances striatal IL-1, it appears to be transient. The idea that IL-1β augmentation is stimulus-dependent (l-DOPA) is consistent with previous reports showing that inflammatory transcripts are not elevated 18 hrs after the last l-DOPA injection (Konradi et al., 2004) nor are long-term changes in microglia observed (Lindgren et al., 2007). Regardless, microglial activation is not imperative as neurons within the striatum are capable of producing IL-1β in response to stress (Kwon et al., 2008), and multiple cell types in the CNS are known to express IL-1β (Vitkovic 2000). To what extent neuroinflammation and in particular IL-1β worsens LID is currently unknown, although it is well documented that IL-1β can activate ERK through a PKC-dependent pathway (Ginnan et al., 2006). Interestingly, IL-1β has been shown to exacerbate excitotoxicity (Stroemer & Rothwell, 1998; Allan, 2002) and promote seizure activity (Vezzani & Baram, 2007), an effect that might manifest as LID in DA-depleted rats. In this regard, LID might reflect a focal burst of excitation in the striatum that is akin to seizure activity. Indeed, changes in factors that promote excitotoxicity (i.e., Ca2+, energy metabolism) have been observed long after (18 hrs) l-DOPA administration (Konradi et al., 2004). This last observation might be the result of the ability of IL-1 to change the membrane potential of neurons (Ferri & Ferguson, 2003; Vezzani & Baram, 2007) thereby facilitating the activity of medium spiny neurons to produce LID behaviors.
There are two other considerations regarding CORT that should be pointed out. First, as the principal stress hormone, CORT plays an important role in regulating the stress response. However, CORT administration alone should not be equated with the stress response. More generally, stressor exposure results in array of endocrine and neurological changes that are dependent upon the nature, characteristics, and duration of the precipitating stimulus. Moreover, glucocorticoids with more profound anti-inflammatory properties such as dexamethasone, prednisone, and or hydrocortisone delivered in clinically established doses, which exceed the more physiologically relevant doses employed here, might have noticeably greater efficacy for tempering LID development and or expression.
While the precise mechanism by which IL-1β and CORT participate in the expression and attenuation of LID remains unclear, respectively, the current series of studies demonstrate for the first time that inflammatory factors may play a pivotal role in LID. Specifically, we have shown that CORT, a potent anti-inflammatory agent, attenuates the expression and development of LID using a well-validated rodent model (Lundblad et al., 2002; Eskow et al., 2007). Examination of transcriptional changes using real-time RT-PCR implicated a role for IL-1β, since mRNA for IL-1β was increased in the DA-depleted striatum of l-DOPA-treated rats, an effect that was completely abrogated by pretreatment with CORT. Finally, we showed that intrastriatal microinjection of IL-1ra reduced the expression of LID. Together, these findings suggest that striatal IL-1β may play a prominent role in the expression of LID. It should be noted, however, that increased IL-1β expression is often observed as just one inflammatory step in an overall cascade of neuroinflammatory signaling involving multiple cytokines, chemokines, and prostanoids (i.e., Bayon et al., 1998). In this regard, the present data open a wide range of questions regarding the role of inflammatory-related factors in the expression of LID. Our data, therefore, have significant implications for the development of new strategies in the treatment of LID.
Acknowledgements
This work was supported by grants from the American Parkinson Disease Association (C. Bishop), NIH NS059600 (C. Bishop), National Science foundation Grant No. 0549987 (T. Deak), and Center for Development and Behavioral Neuroscience at Binghamton University (C. Bishop & T. Deak). The authors would especially like to thank Sheri Zola for her excellent technical assistance during the running of these studies. We would also like to thank Kayla Wilt, Amy Steiniger, Anna Klioueva for their help with behavioral scoring and Joanne Brice for her help with tissue processing.
List of Abbreviations
- 6-OHDA
6-Hydroxydopamine
- AIMs
abnormal involuntary movements
- ALO
axial, forelimb, orolingual
- AMPA
α-amino-3-hydroxy-5-methylisoxazole-4- propionic acid receptor
- CAML
calcium modulating cyclophilin ligand
- cAMP
cyclic adenosine monophosphate
- CORT
corticosterone
- CREB
cyclic adenosine monophosphate response element-binding
- DA
dopamine
- DARPP-32
dopamine- and cyclic AMP-regulated phosphoprotein with molecular weight 32
- ERK
extracellular signal-regulated kinases
- GLUT
glutamate
- GR
glucocorticoid receptor
- IL-1
interleukin-1
- IL-1ra
interleukin-1 receptor antogonist
- IL-6
interleukin-6
- l-DOPA
L-3,4-dihydroxy-L-phenylalanine
- LID
l-DOPA-induced dyskinesia
- NF-kB
nuclear factor-kappa B
- NMDA
N-methyl-D-aspartate
- PD
Parkinson’s disease
- PPD
preprodynorphin
- PKC
protein kinase C
- PPE
preproenkephalin
- PPT
preprotachykinin
- RT-PCR
reverse-transcription polymerase chain reaction
- SNpc
substantia nigra pars compacta
- TH
tyrosine hydroxylase
- TNF-α
tumor necrosis factor alpha
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abraham I, Harkany T, Horvath K, Veenema A, Penke B, Nyakas C, et al. Chronic corticosterone administration dose-dependently modulates Abeta(1-42)-and NMDA-induced neurodegeneration in rat magnocellular nucleus basalis. Journal of Neuroendocrinology. 2000;12:486–494. doi: 10.1046/j.1365-2826.2000.00475.x. [DOI] [PubMed] [Google Scholar]
- Ahlskog J, Muenter M. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Movement Disorders. 2001;16:448–458. doi: 10.1002/mds.1090. [DOI] [PubMed] [Google Scholar]
- Allan S. Varied actions of proinflammatory cytokines on excitotoxic cell death in the rat central nervous system. Journal of Neuroscience Research. 2002;67:428–434. doi: 10.1002/jnr.10142. [DOI] [PubMed] [Google Scholar]
- Anderson J. Does neuronal loss in Parkinson's disease involve programmed cell death? BioEssays. 2001;23:640–646. doi: 10.1002/bies.1089. [DOI] [PubMed] [Google Scholar]
- Arimoto T, Bing G. Up-regulation of inducible nitric oxide synthase in the substantia nigra by lipopolysaccharide causes microglial activation and neurodegeneration. Neurobiology of Disease. 2003;12:35–45. doi: 10.1016/s0969-9961(02)00017-7. [DOI] [PubMed] [Google Scholar]
- Barnum CJ, Blandino P, Jr, Deak T. Adaptation in the corticosteron and hyperthermic responses to stress following repeated stressor exposure. Journal of Neuroendocrinology. 2007;19(8):632–642. doi: 10.1111/j.1365-2826.2007.01571.x. [DOI] [PubMed] [Google Scholar]
- Bayon Y, Alonso A, Hernandez M, Nieto M, Sanchez C. Mechanisms of cell signaling in immune-mediated inflammation. Cytokines, cellular & molecular therapy. 1998;4(4):275–286. [PubMed] [Google Scholar]
- Behl C, Lezoualc'h F, Trapp T, Widmann M, Skutella T, Holsboer F. Glucocorticoids enhance oxidative stress-induced cell death in hippocampal neurones in vitro. Endocrinology. 1997;138(101–106) doi: 10.1210/endo.138.1.4835. [DOI] [PubMed] [Google Scholar]
- Bibbiani F, Oh J, Kielaite A, Collins M, Smith C, Chase T. Combined blockade of AMPA and NMDA glutamate receptors reduces levodopa-induced motor complications in animals models of PD. Experimental Neurology. 2005;196:422–429. doi: 10.1016/j.expneurol.2005.08.017. [DOI] [PubMed] [Google Scholar]
- Bradford M. A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Analytical Biochemistry. 1976;72(1–2):248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Buck K, Ferger B. Intrastriatal inhibition of aromatic amino acid decarboxylase prevents l-DOPA-induced dyskinesia: a bilateral reverse in vivo microdialysis study in 6-hydroxydopamine lesioned rats. Neurobiology of Disease. 2007 doi: 10.1016/j.nbd.2007.08.010. [DOI] [PubMed] [Google Scholar]
- Cenci M. Transcription factors involved in the pathogenesis of l-DOPA-induced dyskinesia in a rat model of Parkinson's disease. Amino Acids. 2002;23:105–109. doi: 10.1007/s00726-001-0116-4. [DOI] [PubMed] [Google Scholar]
- Cenci M. Dopamine dysregulation of movement control in l-DOPA-induced dyskinesia. TRENDS in Neuroscience. 2007;30(5):236–243. doi: 10.1016/j.tins.2007.03.005. [DOI] [PubMed] [Google Scholar]
- Cenci M, Lee C, Bjorklund A. L-DOPA-induced dyskinesia in the rats is associated with striatal overexpression of prodynorphin- and glutamic acid decarboxylase mRNA. European Journal of Neuroscience. 1998;10(8):2694–2706. [PubMed] [Google Scholar]
- Chang J, Wachtel S, Kang U. Biochemical and anatomical characterization of forepaw adjusting steps in rats models of Parkinson's disease: studies on medial forebrain bundle and striatal lesions. Neuroscience. 1999;88(2):617–628. doi: 10.1016/s0306-4522(98)00217-6. [DOI] [PubMed] [Google Scholar]
- Cunningham C, Wilcockson D, Campion S, Lunnon K, Perry V. Central and systemic endotoxin challanges exacerbate the local inflammatory response and increases neuronal death during chronic neurodegeneration. Neurobiology of Disease. 2005;25(40):9275–9284. doi: 10.1523/JNEUROSCI.2614-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson's disease: Mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Deak T, Bordner K, McElderry N, Barnum C, Blandino P, Jr, Deak M, et al. Stress-induced increases in hypothalamic IL-1: a systematic analysis of multiplt stressor paradigms. Brain Research Bulletin. 2005;64:541–556. doi: 10.1016/j.brainresbull.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Du Y, Ma Z, Lin S, Dodel R, Gao F, Bales K, et al. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson's disease. PNAS. 2001;98(25):14669–14674. doi: 10.1073/pnas.251341998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupre K, Eskow K, Negron G, Bishop C. The differential effects of 5-HT1A receptor stimulationon dopamine receptor-mediated abnormal involuntary movements and rotations in the primed hemiparkinsonian rat. Brain Research. 2007;1158:135–143. doi: 10.1016/j.brainres.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Elenkov I, Chrousos G. stress hormones, proinflammatory and antiinflammatory cytokines, and autoimmunity. Annals of the New York Academy of Sciences. 2002;966:290–303. doi: 10.1111/j.1749-6632.2002.tb04229.x. [DOI] [PubMed] [Google Scholar]
- Eskow K, Gupta V, Alam S, Park J, Bishop C. The partial 5-HT1A agonist buspirone reduces the expression and development of l-DOPA-induced dyskinesia in rats and improves l-DOPA efficacy. Pharmacology, Biochemistry and Behavior. 2007;87:306–314. doi: 10.1016/j.pbb.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Ferrari C, Godoy M, Tarelli R, Chertoff M, Depino A, Pitossi F. Progressive neurodegeneration and motor disabilities induced by chronic expression of IL-1 beta in the substantia nigra. Neurobiology of disease. 2006;24:183–193. doi: 10.1016/j.nbd.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Ferri C, Ferguson A. Interleukin-1 beta depolarizes paraventricular nucleus parvocellular neurones. Journal of Neuroendocrinology. 2003;15:126–133. doi: 10.1046/j.1365-2826.2003.00870.x. [DOI] [PubMed] [Google Scholar]
- Gao H, Liu B, Zhang W, Hong J. Synergistic dopaminergic neurotoxicity of MPTP and inflammogen lipopolysaccharide: relevance to the etiology of Parkinson's disease. FASEB J. 2003;17:1957–1959. doi: 10.1096/fj.03-0203fje. [DOI] [PubMed] [Google Scholar]
- Garside H, Stevens A, Farrows S, Normand C, houle B, Berry A, et al. Glucorticoid ligans specify different interactions with NF-KB by allosteric effects on the glucocorticoid receptor DNA binding domain. Journal of Biological Chemistry. 2004;279(48):50050–50059. doi: 10.1074/jbc.M407309200. [DOI] [PubMed] [Google Scholar]
- Ginnan R, Guikema B, Singer H, Jourd'heuil D. PKC-delta mediates activation of ERK1/2 and induction of iNOS by IL-1 beta in vascular smooth muscle cells. American journal of Physiology. Cell physiology. 2006;290(6):C1583–C1591. doi: 10.1152/ajpcell.00390.2005. [DOI] [PubMed] [Google Scholar]
- Guyre P, Bodwell J, Munck A. Glucocorticoid actions on lymphoid tissue and the immune system: physiologic and therapeutic implications. Progress in clinical and biological research. 1984;142:181–194. [PubMed] [Google Scholar]
- He Y, lee T, Leong S. 6-Hydroxydopamine induced apoptosis of dopaminergic cells in the rat substantia nigra. Brain Research. 2000;858:163–166. doi: 10.1016/s0006-8993(99)02459-2. [DOI] [PubMed] [Google Scholar]
- Hirsch E, Hunot S, Damier P, Faucheux B. Glial cells and inflammation in Parkinson's disease: a role in neurodegeneration? Annals of Neurology. 1998;44:S115–S120. doi: 10.1002/ana.410440717. [DOI] [PubMed] [Google Scholar]
- Jiang B, Xu S, Hou X, Pimentel D, Brecher P, Cohen R. Temporal control of NF-kB activation by ERK differentially regulates interleukin-1beta-induced gene expression. Journal of Biological Chemistry. 2004;279(2):1323–1329. doi: 10.1074/jbc.M307521200. [DOI] [PubMed] [Google Scholar]
- Kalman B, Spencer R. Rapid corticosteroid-dependent regulation of mineralocorticoid receptor protein expression in rat brain. Endocrinology. 2002;143(11):4184–4195. doi: 10.1210/en.2002-220375. [DOI] [PubMed] [Google Scholar]
- Kilpatrick I, Jones M, Phillipson O. A semiautomated analysis method for catecholamines, indolamines, and some prominent metabolites in microdissected regions of the nervous system: an isocratic HPLC technique employing coulometric detection and minimal sample preparation. Journal of Neurochemistry. 1986;46(6):1865–1876. doi: 10.1111/j.1471-4159.1986.tb08506.x. [DOI] [PubMed] [Google Scholar]
- Konradi C, Westin J, Carta M, Eaton M, Kuter K, Dekundy A, et al. Transcriptome analysis in a rat model of L-DOPA-induced dyskinesia. Neurobiology of Disease. 2004;17:219–236. doi: 10.1016/j.nbd.2004.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurkowska-Jastrzebska I, Litwin T, Joniec I, Ciesielsak A, Przybylkowski A, Czlonkowski A, et al. Dexamethasone protects against dopaminergic neurons in a mouse model of Parkinson's disease. International Immunopharmacology. 2004;4(10–11):1307–1318. doi: 10.1016/j.intimp.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Kwon M, Seo Y, Lee J, Lee H, Jung J, Jang J, et al. The repeated immobilization stress increases IL-1beta immunoreactivities in only neuron, but not astrocyte or microglia in hippocampal CA1 region, striatum and paraventricular nucleus. Neuroscience Letters. 2008;430(3):258–263. doi: 10.1016/j.neulet.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Li X, Rana S, Kovacs E, Gamelli R, Chaudry I, Choudhry M. Corticosterone suppresses mesenteric lymph node T cells by inhibiting p38/ERK pateay and promotes bacterial translocation after alcohol and burn injury. Am J Physiol Regul Integr Comp Physio. 2005;289:R37–R44. doi: 10.1152/ajpregu.00782.2004. [DOI] [PubMed] [Google Scholar]
- Lindgren H, Rylander D, Ohlin K, Lundblad M, Cenci M. The 'motor complication syndrome' in rats with 6-OHDA lesions treated chronically with l-DOPA: relation to dose and route of administration. Behavioural Brain Research. 2007;177:150–159. doi: 10.1016/j.bbr.2006.09.019. [DOI] [PubMed] [Google Scholar]
- Livak K, Schmittgen T. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lundblad M, Andersson M, Winkler C, Kirik D, Wierup N, Cenci M. Pharmacological validation of behavioral measures of akinesia and dyskinesia in a rat model of Parkinson's disease. European Journal of Neuroscience. 2002;15(1):120–132. doi: 10.1046/j.0953-816x.2001.01843.x. [DOI] [PubMed] [Google Scholar]
- Mela FMM, Dekundy A, Danysz W, Morari M, Cenci M. Antagonsim of metabotropic glutamtate receptor type 5 attenuates l-DOPA-induced dyskinesia and its molecular and neurochemical corrleates in a rat model of Parkinson's disease. Journal of Neurochemistry. 2007;101(2):483–497. doi: 10.1111/j.1471-4159.2007.04456.x. [DOI] [PubMed] [Google Scholar]
- Metz G, Jadavji N, Smith L. Modulation of motor function by stress: a novel concept of the effects of stress and corticosterone on behavior. European Journal of Neuroscience. 2005;22(5):1190–1200. doi: 10.1111/j.1460-9568.2005.04285.x. [DOI] [PubMed] [Google Scholar]
- Mogi M, Harada M, Riederer P, Narabyashi H, Fujita J, Nagatsu T. Interleukin-1 beta growth factor and transforming growth factor-alpha are elevated in the brain from Parkinsonian patients. Neuroscience Letters. 1994;180:147–150. doi: 10.1016/0304-3940(94)90508-8. [DOI] [PubMed] [Google Scholar]
- Munck A, Guyre P, Holbrook N. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocrine Reviews. 1984;5(1):25–44. doi: 10.1210/edrv-5-1-25. [DOI] [PubMed] [Google Scholar]
- Mura A, Mintz M, Feldon J. Behavioral and anatomical effects of long-term l-dihydroxyphenylalanine (l-DOPA) administration in rats with unilateral lesions of the nigrostriatal system. Experimental Neurology. 2002;177(1):252–264. doi: 10.1006/exnr.2002.7976. [DOI] [PubMed] [Google Scholar]
- Necela B, Cidlowski J. Mechanisms of glucocorticoid receptor action in noninflammatory and inflammatory cells. Proceedings of the American Thoracic Society. 2004;1(3):239–246. doi: 10.1513/pats.200402-005MS. [DOI] [PubMed] [Google Scholar]
- Nichols N, Agolley D, Zieba M, Bye N. Glucorticoid regulation of glial responses during hippocampal neurodegeneration and regeneration. Brain Research Reviews. 2005;48:287–301. doi: 10.1016/j.brainresrev.2004.12.019. [DOI] [PubMed] [Google Scholar]
- Olsson M, Nikkhah G, Bentlage C, Bjorklund A. Forelimb akinesia in the rat Parkinson model: differential effects of dopamine agonists and nigral transplants as assessed by a new stepping test. The Journal of Neuroscience. 1995;15(5):3863–3875. doi: 10.1523/JNEUROSCI.15-05-03863.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson W. The rat brain in stereotaxic coordinates. 4th Ed. San Diego: Academic Press; 1998. [Google Scholar]
- Pfaffl M. A new mathmatical model for relative quantification in real-time RT-PCR. Nucleic Acids Research. 2001;29(9):e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picconi B, Pisani A, Barone I, Bonsi P, Centonze D, Bernardi G, et al. Pathological synaptic plasticity in the striatum: implications of Parkinson's disease. Neurotoxicoloy. 2005;26:779–783. doi: 10.1016/j.neuro.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Picconi B, Pisani A, Centonze D, Battaglia G, Storto M, Nicoletti F, et al. Striatal metabotropic glutamate receptor function following experimental parkinsonism and chronic levodopa treatment. Brain. 2002;125:2625–2646. doi: 10.1093/brain/awf269. [DOI] [PubMed] [Google Scholar]
- Putterman D, Munhall A, Kozell L, Belknap J, Johnson S. Evaluation of levodopa dose and magnitude of dopamine depletion as risk factors for levodopa-induced dyskinesia in a rat model of Parkinson's disease. The Journal of pharmacology and experimental therapeutics. 2007;323(1):27–284. doi: 10.1124/jpet.107.126219. [DOI] [PubMed] [Google Scholar]
- Robelet S, Melon C, Salin P, Kerkerian-Le Goff L. Chronic l-DOPA treatment increases extracellular glutamate levels and GLT1 expression in the basal ganglia in a rat model of Parkinson's disease. European Journal of Neuroscience. 2004;20:1255–1266. doi: 10.1111/j.1460-9568.2004.03591.x. [DOI] [PubMed] [Google Scholar]
- Santini E, Valjent E, Usiello A, Carta M, Borgkvist A, Girault J, et al. Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in l-DOPA-induced dyskinesia. Neurobiology of Disease. 2007;27(26):6995–7005. doi: 10.1523/JNEUROSCI.0852-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapolsky R. A Mechanism for glucocorticoid toxicity in the hippocampus: increased neuronal vulnerability to metabolic insults. The Journal of Neuroscience. 1985;5(5):1228–1232. doi: 10.1523/JNEUROSCI.05-05-01228.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapolsky R, Krey L, McEwen B. Prolonged glucocorticoid exposure reduces hippocampal neuron number: implications for aging. The Journal of Neuroscience. 1985a;5:1221–1226. doi: 10.1523/JNEUROSCI.05-05-01222.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz J, Matthews R, Mugit M, Browne S, Beal M. Inhibition of neuronal nitric oxide synthase by 7-nitroindazole protects against MPTP-induced neurotoxicity in mice. Journal of Neurochemistry. 1995;64(2):936–939. doi: 10.1046/j.1471-4159.1995.64020936.x. [DOI] [PubMed] [Google Scholar]
- Stocchi F, Nordera G, Marsden C. Strategies for treating patients with advanced Parkinson's disease with disastrous fluctuation and dyskinesias. Clinical Neuropharmacology. 1997;20:95–115. doi: 10.1097/00002826-199704000-00001. [DOI] [PubMed] [Google Scholar]
- Stroemer R, Rothwell N. Exacerbation of ischemic brain damage by localized striatal injection of IL-1 beta in the rat. Journal of Cerebral Blood Flow Metabolism. 1998;18:833–839. doi: 10.1097/00004647-199808000-00003. [DOI] [PubMed] [Google Scholar]
- Svenningsson P, Arts J, Gunne L, Andren P. Acute and repeated treatment with L-DOPA increase c-jun expression in the 6-hydroxydopamine-lesioned forebrain of rats and common marmosets. Brain Research. 2002;955(1–2):8–15. doi: 10.1016/s0006-8993(02)03289-4. [DOI] [PubMed] [Google Scholar]
- Taylor J, Bishop C, Walker P. Dopamine D1 and D2 receptor contributions to l-DOPA-induced dyskinesia in the dopamine-depleted rat. Pharmacology, biochemistry, and behavior. 2005;81(4):887–893. doi: 10.1016/j.pbb.2005.06.013. [DOI] [PubMed] [Google Scholar]
- Tel B, Zeng B, Cannizzaro C, Pearce R, Rose S, Jenner P. Alterations in striatal neuropeptide mRNA produced by repeated administration of l-DOPA, ropinirole or bromocriptine correlate with dyskinesia induction in MPTP-treated common marmosets. Neuroscience. 2002;115(4):1047–1058. doi: 10.1016/s0306-4522(02)00535-3. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Baram T. New roles for interleukin-1 Beta in the mechanisms of epilepsy. Epilepsy currents. 2007;7(2):45–50. doi: 10.1111/j.1535-7511.2007.00165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitkovic L, Bockaert J, Jacque C. "Inflammatory" cytokines: neuromodlators in normal brain? Journal of Neurochemistry. 2000;4(2):457–471. doi: 10.1046/j.1471-4159.2000.740457.x. [DOI] [PubMed] [Google Scholar]
- Viviani B, Bartesaghi S, Corsini E, Galli C, Marinovich M. Cytokines role in neurodegenerative events. Toxicology Letters. 2004;149:85–89. doi: 10.1016/j.toxlet.2003.12.022. [DOI] [PubMed] [Google Scholar]
- Whitton P. Inflammation as a causitive factor in the aetiology of Parkinson's disease. British Journal of Pharmacology. 2007;150(8):963–976. doi: 10.1038/sj.bjp.0707167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler C, Kirik D, Bjorklund A, Cenci M. L-DOPA-induced dyskinesia in the intrastriatal 6-hydroxydopamine model of Parkinson's disease: relation to motor and cellular parameters of nigrostriatal function. Neurobiology of Disease. 2002;10(2):165–186. doi: 10.1006/nbdi.2002.0499. [DOI] [PubMed] [Google Scholar]
- Wolkowitz O. Prospective controlled studies of the behavioral and biological effects of exogenous corticosteriods. Psychoneuroendocrinology. 1994;19(3):233–255. doi: 10.1016/0306-4530(94)90064-7. [DOI] [PubMed] [Google Scholar]
- Yu I, Lee S, Lee Y, Son H. Differential effects of corticosterone and dexamethasone on hippocampal neurogenesis in vitro. Biochemical and Biophysical Research Communications. 2004;317:484–490. doi: 10.1016/j.bbrc.2004.03.071. [DOI] [PubMed] [Google Scholar]
- Zhao L, Brinton R. Suppression of proinflammatory cytokines interleukin-1beta and tumor necrosis factor-alpha in astrocytes by a V1 vasopressin receptor agonist: a cAMP response element-binding protein-dependent mechanism. Journal of Neuroscience. 2004;24(9):2226–2235. doi: 10.1523/JNEUROSCI.4922-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]