

Abstract
The latency period for lung tumor progression offers a window of opportunity for therapeutic intervention. Herein, we studied the effect of oral silibinin (742 mg/kg body wt, 5 days/wk for 10 wks) on the growth and progression of established lung adenocarcinomas in A/J mice. Silibinin strongly decreased both tumor number and tumor size, an antitumor effect that correlates with reduced antiangiogenic activity. Silibinin reduced microvessel size (50%, p<0.01) with no change in the number of tumor microvessels and reduced (by 30%, p<0.05) formation of nestin-positive new microvessels in tumors. Analysis of several proteins involved in new blood vessel formation showed that silibinin decreased tumor expression of IL-13 (47%) and TNF-α (47%), and increased TIMP-1 (2-fold) and TIMP-2 (7-fold) expression, without significant changes in VEGF levels. HIF-1α expression and nuclear localization were also decreased by silibinin treatment. Cytokines secreted by tumor cells and tumor associated macrophages (TAMs) regulate angiogenesis by activating NF-κB and STAT. Silibinin decreased phosphorylation of p65NF-κB (ser276) (38%, p<0.01) and STAT3 (ser727) (16%, p<0.01) in tumor cells and decreased the lung macrophage population. Angiopoietin-2 (Ang-2) and Ang-receptor tyrosine kinase (Tie-2) expression were increased by silibinin. Therapeutic efficacy of silibinin in lung tumor growth inhibition and regression by antiangiogenic mechanisms appears to be mediated by decreased TAMs and cytokines, inhibition of HIF-1α, NF-κB and STAT3 activation, and up-regulation of the angiogenic inhibitors, Ang-2- and Tie-2.
Keywords: Silibinin, lung tumor, angiogenesis, macrophages, cytokines, NF-κB, STAT3
Introduction
Lung cancer is the leading cause of cancer death in both men and women in the United States, with an estimated 213,380 new lung cancer cases and 160,390 associated deaths in 2007 (1). The 5-year survival rate of 14% has shown little improvement over the last 30 years, even with the development of molecularly targeted therapies such as epidermal growth factor receptor (EGFR) inhibitors. Tobacco exposure has been implicated in 90% of lung carcinomas; compared to never smokers, smokers have a 20-fold greater risk of developing lung cancer (2). Because smoking is the major risk factor for developing lung cancer and most smokers have small pulmonary nodules, strategies for inducing nodule regression or preventing their further growth should decrease the number of patients diagnosed with advanced malignant disease.
Efforts are being made towards identifying dietary supplements to prevent and treat lung cancer. One such agent, silibinin, inhibits the growth of various cancer cell lines and primary tumors in several chemically-induced rodent models, including mouse lung (3-7). Silibinin is a flavonolignan, a major component in the silymarin complex of flavonolignans and polyphenols present in milk thistle (Silybum marianum) seeds. Silymarin has been extensively used in patients with liver disease for decades (8). Silibinin has shown strong anticancer efficacy against SHP-77 and A549 lung cancer cells, where it inhibits cell growth and induces cell cycle arrest (9). It also inhibits the invasion of lung cancer cells via down-regulating PI3K-Akt and MAPK signaling pathways and decreased production of urokinase-plasminogen activator and matrix metalloproteinase-2 (10,11). Silibinin inhibits in vivo growth of A549 xenografts in nude mice, reduces systemic toxicity of doxorubicin, and reduces doxorubicin-induced chemo-resistance by inhibiting NF-κB signaling (12).
Angiogenesis, the formation of new blood vessels, is required early on in tumor development. It is essential for tumor expansion, progression, and metastasis (13). Angiogenesis is a complex process that involves degradation of the basement membrane and invasion of the stroma by endothelial cells, which then proliferate, migrate and become organized into capillary structures (14). Tumor associated macrophages (TAMs) constitute an important interface between tumor cells and the immune system, and influence neoplastic growth and progression in several ways (15). Macrophage infiltration affects angiogenesis by influencing the production of angiogenic cytokines and growth factors including interleukins, TNF-α and IFN-γ (16). Therefore, modulation of TAMs may be critical in nascent vessel formation in tumors, and could be targeted to inhibit tumor angiogenesis.
Recently, we showed that dietary silibinin markedly reduced the growth and progression of urethane-induced primary lung tumors in A/J mice. In a previous study, silibinin was fed to mice 2 weeks after carcinogen administration to model a chemopreventive treatment regimen (7). We now examine the chemotherapeutic effect of silibinin on established advanced lung tumors in this animal model. Tumors at this stage are adenocarcinomas with malignant characteristics such as invasiveness, nuclear dysmorphology, and cellular heterogeneity. Our findings are novel and significant because this chemopreventive agent that is known to inhibit cell proliferation and induce apoptosis did not effect these biological activities in a therapeutic setting. Instead, silibinin targeted vessel size and nascent tumor microvessel formation via its effects on TAMs, known modulators of angiogenesis.
Materials and Methods
Chemicals
Urethane (ethyl carbamate) and silibinin were purchased from Sigma (St. Louis, MO). Purity (>98%) of silibinin was confirmed by high-performance liquid chromatography, as previously described (17). Mice were fed AIN-76A rodent diet pellets (Dyets Inc, Bethlehem, PA).
Urethane-induced lung tumorigenesis experimental design
A/J male mice (4-6 wks of age) were purchased from the Jackson Laboratory (Bar Harbor, ME) and housed under standard laboratory conditions in the Center of Laboratory Animal Care at the University of Colorado Denver. Animal care was performed in accordance with institutional guidelines, and all animal treatments performed under an institutionally approved animal protocol. Mice (n=25, 8 wks of age) were given a single i.p. injection of 1 mg/g body wt of urethane in saline, as previously described (7). Urethane-induced lung tumors harbor a mutation in codon 61 of the Kras proto-oncogene (18). Thirty two wks after urethane injection, 5 mice were sacrificed and their lung tumors dissected, counted, and diameters measured with digital calipers, and pooled tumors weighed to determine total tumor burden/mouse. The remaining 20 mice were randomly divided into 2 groups (n=10 mice per group) and gavaged with normal saline (control group) or silibinin 742 mg/kg body wt 5 days/wk for 10 wks. This dose of silibinin was extrapolated from the dietary consumption of A/J mice exposed to 1% silibinin (w/w) and fed AIN-76A diet in our previous study (7). At 43 wks, mice were sacrificed by i.p. pentobarbital injection, lung tumors harvested, and the parameters mentioned above were recorded from 5 mice in each group. Lungs from the remaining 5 mice from each group were perfused, formalin fixed, and paraffin embedded for histological and immunohistochemical (IHC) analyses. Plasma samples from all mice were collected by cardiac puncture into heparinized tubes, and stored at -80°C.
Immunohistochemistry (IHC)
Fixed lung samples were sectioned (5 μm), deparaffinized, rehydrated, and subjected to heat-induced antigen retrieval in citrate buffer (pH 6.0) for 30 min at 90°C. Nonspecific binding sites were blocked with serum-free blocking reagent (DAKO), and antibodies against proliferating cell nuclear antigen (PCNA, 1:400, mouse monoclonal, Dako, Carpinteria, CA), CD31 (platelet-derived endothelial cell adhesion molecule, 1:200, goat polyclonal, Santa Cruz Biotechnology, Santa Cruz, CA), nestin (1:200, mouse monoclonal, Abcam, Cambridge, MA), pSTAT3 (ser727) (1:200, rabbit polyclonal, Abcam), inducible nitric oxide synthase (iNOS; 1:200, BD-Transduction Laboratory, San Diego, CA), cyclooxygenase-2 (COX-2; 1:2000, goat polyclonal, Santa Cruz Biotechnology), p65NF-κB (ser276) (1:200 rabbit polyclonal, Cell Signalling, Beverly, MA), HIF-1α (1:200, mouse monoclonal, Novus, Littleton, CO) and the macrophage specific marker, F4/80 (1:100, Caltag laboratories, Burlingame, CA) were applied overnight at 4°C followed by appropriate secondary antibodies. Proteins were visualized using 3, 3′-diaminobenzidine as previously described (7). Sections were counterstained with hematoxylin. Apoptotic cells were identified by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining using Dead End Colorometric TUNEL system (Promega Corp).
The PCNA, TUNEL, p65NF-κB (ser276) and pSTAT (ser727) -positive cells (brown) were quantified by counting the numbers of stained cells compared to the total number of cells in five randomly selected tumor fields, at 400X magnification. HIF-1α nuclear staining was visualized at 1000X using an oil immersion lens. Blood vessels (CD31-positive) and newly formed vessels (nestin-positive) were quantified as the mean number of positive vessels per 400X field in five randomly selected tumor fields. CD-31 positive blood vessel diameter of circle-shaped vessel cross sections was measured using Spot Advanced software on an Olympus BX-41 micrscope equipped with an Olympus U-TVX-1 camera. Intensity of COX-2 and iNOS immunoreactivities were scored as 0 (no staining), 1+ (weak staining), 2+ (moderate staining), 3+ (strong staining) or 4+ (very strong staining) as recently published (7). Since most epithelial cells express these proteins basally, intensity of staining described increases in protein expression more clearly than did ratios of expressing cells to non-expressing cells.
Immunoblot analysis
Lung tumor lysates from 3-4 individual mice from each group, were prepared and analyzed by immunoblotting, as previously described (7). Primary antibodies against PCNA, cyclin D1, VEGF, TIMP-1 and Tie-2 (Santa Cruz Biotechnology); p65NF-κB (ser276), total p65NF-κB (ser536), pSTAT3 (ser727), total STAT3 and phospho-Tie-2 receptor (Tyr992) (Cell Signalling, Beverly, MA); HIF-1α (Novus Biologicals, Inc); nestin, angiopoietin 2 and TNFα (Abcam, Cambridge, MA); TIMP-2 (Chemicon, Temecula, CA), and IL-13 (Sigma-Aldrich) and secondary antibodies against anti-rabbit IgG (Cell Signalling) or anti-mouse IgG (GE Healthcare, Piscataway, NJ) were used. Equal protein loading was determined by stripping and reprobing membranes with anti-β actin primary antibody (Sigma Aldrich). Protein bands were visualized by enhanced chemiluminescence detection.
Mouse angiogenesis antibody array
Two randomly selected lung tumor lysates from each group were applied to a Mouse Angiogenesis Antibody Array (RayBiotech Inc, Norcross, GA) to analyze the expression of angiogenesis-related molecules according to the manufacturer's protocol. Expression of each protein was represented in duplicate on the membrane. Duplicate dots identifying each protein were scanned using Adobe Photoshop software and quantified by ScionImage Program. The mean intensity of these dots (arbitrary units) was determined for intergroup comparisons.
ELISA assay for IL-13
Plasma samples from control and silibinin-fed group were applied to a mouse IL-13 ELISA kit, following vendor's protocol (RayBiotec Inc, Norcross, GA) to quantify secreted IL-13. The assay is based on quantitative sandwich enzyme immunoassay using an antibody specific for mouse IL-13 coated onto a microplate for solid phase ELISA. Briefly, 100 μl plasma samples (collected from 4-5 individual mice/group) were assayed, and absorbance of the developing color was determined by microplate reader at 450 nm wavelength. IL-13 concentration was extrapolated from a recombinant mouse IL-13 standard curve.
Statistical and microscopic analyses
Statistical analyses were carried out by using Sigma Stat software version 2.03 (Jandel Scientific, San Rafael, CA). All statistical tests were two-sided, and p<0.05 was considered statistically significant. The differences between controls (urethane treated group) and silibinin-fed groups were analyzed by unpaired two-tailed Student's t-test and one-way ANOVA followed by a Bonferroni t-test for pair-wise multiple comparisons. Densitometric analysis of immunoblots was performed using the Scion Image program (NIH, Bethesda, MD). IHC staining was visualized with a Zeiss Axioscope 2 microscope (Carl Zeiss, Inc, Jena, Germany), and photographs were captured with a Carl Zeiss AxioCam MrC5 camera at 400X or 1000X magnification.
Results
Silibinin inhibits urethane-induced lung tumor progression in A/J mice
A/J mice given a single 1 mg/g body wt i.p. injection of urethane develop microadenomas (<0.3 mm) within 3 wks, macroscopic adenomas (tumors <1.5 mm with uniform nuclei that aren't invasive) within 16 wks, and adenocarcinomas (invasive tumors with nuclear dysmorphology and cellular heterogeneity) within 30 wks (19, 20). Metastasis is rare in this model as the mice die from oxygen deprivation before tumors progress to that stage. To determine whether silibinin could act as a chemotherapeutic agent in this model, we examined the effect of oral silibinin (742 mg/kg body wt, 5 days/wk for 10 wks, a dose determined to be equivalent to the 1% wt/wt dietary consumption of silibinin) treatment on mice with 32-wk established lung tumors; also, a group of mice were sacrificed 32 wks after urethane injection as a reference control (Fig. 1A). Mice receiving silibinin had 33% (p<0.05) fewer tumors than age-matched 43-wk controls; this control group developed slightly more tumors than a 32-wk control group (Fig. 1B). From 32 - 43-wks, a high tumor growth rate (p<0.001) was observed. Silibinin decreased tumor burden (pooled tumor wt/mouse) by 37% (p<0.05) compared to mice receiving vehicle (Fig. 1C). When tumors were categorized according to size, smaller tumors (<1.5 mm) progressed to a larger size (≥1.5 mm) from 32 wks to 43 wks (p<0.05-0.001) (Fig. 1D). Silibinin treatment significantly decreased the number of larger tumors (>2.5 mm) by 37% (p<0.01), and caused a 50% (p<0.01) decrease in the number of tumors between of 1.5 and-2.5 mm diameter compared to the 43-wk control group (Fig. 1D). The decreased number of tumors in the silibinin group indicates regression, whereas the decreased number of larger tumors in the silibinin group indicates its growth inhibitory effects. For molecular analysis, we used samples from the 43-wk control and silibinin-treated groups.
Figure 1.

Silibinin inhibits urethane-induced lung tumorigenesis in A/J mice. A, Eight wk old male A/J mice (n=25) were given a single i.p. injection of 1 mg urethane/g body weight. Thirty-two wks later, 5 mice were sacrificed and the remaining 20 mice randomly divided into 2 groups (n=10 mice per group), and gavaged with saline (control group) or silibinin 742 mg/kg body weight for 5 days/wk for 10 wks and sacrificed. B, lung tumors dissected from all groups and lung tumor multiplicity/mouse determined. C, tumors were pooled together for each mouse and weighed to determine burden. D, tumor diameters were measured with digital calipers under a dissecting microscope, and tumors were grouped by diameter: <1.5 mm, 1.5-2.5 mm and >2.5 mm. Columns, mean; error bars, SEM for each group. $, p<0.05; #, p<0.01; *, p<0.001 versus 43 wks for tumor size; SB, silibinin
Histopathological characteristics of lung tissue and tumors
Histopathological examination of lung tissue and tumors in A/J mice at 32 and 43 wks after treatment with a single urethane injection showed well-vascularized adenocarcinomas that exhibited invasiveness, nuclear dysmorphology, decreased cytoplasm/nuclear ratio, and undifferentiated cellular orgainization (data not shown). In the 43 wk groups, most tumors were large (>1.5-2.5 mm in size), and the surrounding alveolar spaces contained numerous large foamy macrophages. Airways and alveolar spaces immediately adjacent to the tumors were compressed. Bronchus-associated lymphatic tissue (BALT) was present adjacent to large tumors. When sections from untreated tumor bearing mice were compared to those from mice given silibinin for 10 wks prior to harvest, few differences besides the reduction in tumor size were evident, but large alveoli with characteristic ‘emphysematous’ hooked alveolar walls appeared in the silibinin-treated samples. Since silibinin decreased tumor number, these large open spaces may indicate areas of tumor regression. To determine whether tumor regression left fibrotic deposits, as described in other tumor models, pentachrome stains were performed on lung sections from control and silibinin treated mice. However, no differences in staining for collagen I, mucin, ground substance, muscle, or elastic fibers were observed between the two groups (data not shown).
Effect of silibinin on lung tumor cell proliferation and apoptosis
Since we observed tumor growth inhibition by silibinin, tumors from both control and silibinin-treated groups were analyzed for PCNA and TUNEL IHC staining. No significant difference in PCNA immunoreactivity (Supplementary Fig. 1A) was observed in the silibinin-fed group (39% ± 1.97) as compared to control (43%±2.34). To confirm our IHC results, tumor lysates from control and silibinin groups were analyzed by western immunoblotting for PCNA and cyclin D1. Little change in PCNA and cyclin D1 expression was observed in silibinin-fed group of tumors as compared to control tumors (Supplementary Fig. 1B). TUNEL staining, performed to assess the apoptotic effect of silibinin in tumors, showed similar numbers of TUNEL-positive cells in both silibinin and control groups (Supplementary Fig. 1C). Additionally, we did not observe any considerable effect of silibinin on ERK1/2 and Akt phosphorylation in lung tumors (data not shown). These results suggest that silibinin does not considerably affect cell proliferation and apoptosis in established lung tumors although a trend toward lower proliferative and higher apoptotic rates was observed. Since small differences in tumor proliferative rates could amount to big differences in tumor size over 10 wks, the significance of these slight changes cannot be accurately assessed.
Silibinin inhibits angiogenesis in urethane-induced lung tumors
Since significant antiproliferative and apoptotic effects of silibinin on lung tumors were not detected, we assessed the effect of silibinin on tumor angiogenesis using CD-31 staining, a marker for endothelial cells (both established and nascent). Image analysis followed by quantitative counting of CD-31 positive vessels did not reveal significant differences in tumor microvessel density between control and silibinin-fed groups (data not shown). However, vessel cross-sectional area in the silibinin-fed decreased by 50% (p<0.01; Fig. 2A). Nestin, a stem cell marker, is expressed on newly formed microvessels in advanced tumors but is lost upon endothelial cell maturation (21). Tumors in mice gavaged with silibinin showed fewer nestin-positive microvessels (30% decrease, p<0.05) as compared to control tumors (Fig. 2B). Western immunoblotting for nestin in tumor lysates confirmed that less nestin was expressed in mice treated with silibinin (Fig. 2C). Overall, silibinin had little significant effect on tumor cell proliferation and apoptosis but may suppress tumor angiogenesis by inhibiting increased microvessel size and the formation of new microvessels.
Figure 2.
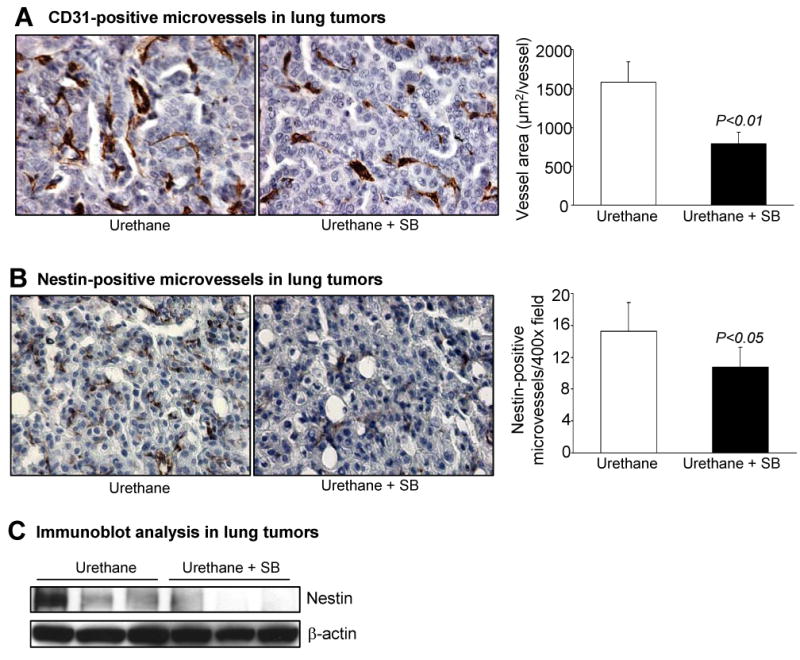
Silibinin inhibits angiogenesis in urethane-induced lung tumors. Tumor-bearing lungs harvested from A/J mice after 43 wks of urethane (n = 5 mice/group) injection were analyzed by IHC staining for CD31 and nestin, as described in Materials and Methods. A, IHC of CD31-positive (brown) endothelial cells from urethane (control) and urethane + silibinin treated groups (left). Microvessel diameter was quantified in 5 randomly selected fields in tumors from each of 5 different mice in both groups. Columns, mean; error bars, SEM for each group (right). B, nestin-positive (brown), newly formed endothelial cells from urethane (control) and urethane + silibinin treated groups of lung tumors (left). Microvessel numbers were quantified by measuring the nestin-positive cells in 5 randomly selected fields at 400X magnification in tumors from each of 5 different mice in each group (right). Columns, mean; error bars, SEM for each group (right). C, lung tumor lysates from three individual randomly chosen mice from each group were analyzed for nestin protein levels by immunoblotting. Membranes were stripped and reprobed with β-actin as loading control. SB, silibinin. Magnification 400X.
Silibinin modulates angiogenesis-related cytokines in tumors and decreases tumor-associated macrophages
Mechanisms of inhibition of angiogenesis by silibinin in lung tumors were explored by performing protein array analysis for biomolecules that regulate angiogenesis in tumor lysates. Silibinin treatment decreased levels of several interleukins and cytokines including IL-1α (34%), -6 (44%), -9 (29%), -13 (47%) and -16 (44%), as well as IFN-γ (16%) and TNF-α (47%) in tumors compared to controls (Fig. 3A). Expression levels of TIMP-1 and TIMP-2 were increased 2- and 7-fold, respectively, by silibinin (Fig. 3A). Results for the most strongly effected proteins, IL-13, TNF-α, TIMP-1 and TIMP-2, were confirmed by western blot analysis, which showed changes similar to those observed in the antibody arrays (Fig. 3B; densitometry confirmed decreases of 14% and 31% in IL-13 and TNF-α, respectively and increases of 1.5 and 3-fold in TIMP-1 and TIMP-2, respectively.
Figure 3.

Silibinin modulates angiogenesis-related cytokines in lung tumors and decreases tumor-associated macrophages. A, lung tumor lysates from control and silibinin-treated mice at 43 wks post urethane injection were analyzed for angiogenesis related protein expression with an angiogenesis antibody array kit, as described in Material and Methods. Reactive protein spots were visualized by enhanced chemiluminescence detection. Densitometric analysis of angiogenesis related protein dot intensity was adjusted with positive control, and the data shown as percent or fold change by silibinin treatment. B, three lung tumor samples from each group were analyzed for IL-13, TNFα, TIMP-1, TIMP-2 and VEGF protein levels by immunoblotting. Membranes were stripped and reprobed with β-actin as a loading control. C, Elisa for mouse IL-13 determined plasma IL-13 levels from control and silibinin treated groups using recombinant mouse IL-13 as a standard. Columns, mean of four samples; error bars, SEM for each group. D, the presence of TAMs were assayed by F4/80 IHC (brown staining) of tumor-bearing lungs harvested from control and silibinin-treated A/J mice after 43 wks (n = 5 mice/group). Macrophage numbers were quantified by counting brown staining cells in the pulmonary stroma in 5 randomly selected fields at 400X magnification from each of 5 different samples in both groups. Columns, mean; error bars, SEM for each group (right). SB, silibinin; IL, interleukin; IFN, interferon; TNF, tumor necrosis factor; TIMP, tissue inhibitor of metalloproteinase; VEGF, vascular endothelial growth factor.
IL-13 plays an important role in regulating angiogenesis, and circulating IL-13 could functionally affect tumor angiogenesis. To test this, we measured levels of IL-13 in mouse plasma by ELISA, and observed that the silibinin treated group had less circulating IL-13 than controls (317.8 ± 43.4 pg/ml in control group versus 137.5 ± 10.6 pg/ml in silibinin group; 57% decrease; p<0.01; Fig. 3C). Silibinin treatment did not influence VEGF or Fas ligand expression either in the protein array or western blot analysis (Fig. 3B). Since macrophages critically modulate cytokine secretion (22), and several cytokines decreased upon silibinin treatment, we used F4/80 staining of macrophages to examine whether silibinin affected the number of macrophages in tumor bearing lungs. In sections from control mice, macrophages were more populous around tumors compared to sections from the silibinin-treated group. Quantification of these macrophages showed a 38% (p<0.05) decrease in TAM number following silibinin feeding (Fig. 3D). Overall, these results suggest that silibinin targets TAMs to suppress the angiogenic tumor microenvironment.
Effect of silibinin on HIF-1α, iNOS and COX-2 expression in lung tumors
HIF-1α, iNOS and COX-2 can promote tumor angiogenesis (23, 24). To examine whether the antiangiogenic effects of silibinin were mediated at least in part by HIF-1α, iNOS and COX-2, we performed IHC analysis of these proteins in lung tumors. Lung tumors from mice in the control group displayed more cells containing HIF-1α positive nuclei (8% ± 1.5 in the control group compared to 2% ± 0.5 (p<0.01) in the silibinin-treated group; Fig. 4A). The effect of silibinin on HIF-1α expression was also analyzed by immunoblotting tumor lysates, which exhibited less 25% HIF-1α protein in the silibinin-treated group of tumors (Fig. 4A). Our previous studies showed that urethane-induced mouse lung tumors usually express high levels of iNOS and COX-2 enzymes (25,26), but no effects of silibinin on iNOS and COX-2 immunoreactivity by either IHC or immunoblot analysis were noted in these samples (data not shown). These results suggest a selective effect of silibinin on HIF-1α rather than on iNOS or COX-2 in these advanced tumors.
Figure 4.

Silibinin inhibits activation of HIF-1α, p65NF-κB and STAT3 in urethane-induced lung tumor cell nuclei. Tumor-bearing lungs harvested from A/J mice after 43 wks of urethane (n = 5 mice/group) and analyzed by using IHC for HIF-1α (A), p65NF-κB (ser276) (B) and pSTAT3 (ser727) (C), as detailed in Materials and Methods (left). HIF-1α, p65NF-κB (ser276) and pSTAT3 (ser727) immunoreactivities in tumors were quantified by counting (brown) positive cells in 5 randomly selected fields at 400X magnification from each of 5 different samples in both groups. The percent positive cells was determined as the number of positive-stained cells X 100/total number of cells counted. Columns, mean; error bars, SEM for each group (right). Final magnification 1000X for HIF-1α and 400X for phospho-p65NF-κB and phospho-STAT3. (A-C, right), three lung tumor samples were randomly taken from each group and analyzed for HIF-1α, p65NF-κB (ser276), p65NF-κB (ser536), p65NF-κB, pSTAT3 (ser727) and STAT3 protein levels by immunoblotting total cell lysates. Bands were visualized by enhanced chemiluminescence detection. Membranes were stripped and reprobed with β-actin as loading control. SB, silibinin; NF-κB, nuclear factor kappa B; STAT, signal transducer and activator of transcription factor.
Silibinin inhibits the activation of p65NF-κB in urethane-induced lung tumors
In addition to HIF-1α, the NF-κB transcription factor also regulates expression of many genes that control angiogenesis (27,28). High levels of NF-κB expression have been reported in non-small cell lung cancer cell lines compared to normal bronchial epithelial cell lines (29). In order to examine the status of NF-κB activity in urethane-induced lung tumors, IHC staining for phospho-p65NF-κB (ser276) was performed. Thirty-eight percent less nuclear immunoreactivity was observed in tumors from silibinin-treated mice (Fig. 4B). When phospho-p65NF-κB (ser276 and ser536) levels were examined by western blot in tumors lysates, the silibinin group had lower expression of both p65(ser276, 15%) and p65(ser536, 40%) than controls, with no effect on total p65 in tumors (Fig. 4B). These results indicate silibinin inhibits the activity of the angiogenic cytokine-NF-κB loop in urethane-induced lung tumors.
Silibinin inhibits the activation of STAT3 in urethane-induced lung tumors
Since we observed a down-regulation of cytokine expression by silibinin treatment in urethane-induced tumors, inhibited STAT signaling was anticipated. STAT3 was originally discovered as a mediator of cytokine signaling pathways that play an active role in oncogenesis, inflammation and angiogenesis (30). Accordingly, we analyzed STAT3 activity in lung tumors by assessing phospho-STAT3 (ser727) by IHC. Fifty eight percent of the tumor cells had nuclear staining of phospho-STAT3 in the silibinin-treated group as compared to 70% (p<0.01) in control group (Fig. 4C). Western immunoblot analysis of phospho-STAT3 (ser727) and total STAT3 in tumor lysates showed that, consistent with the IHC data, control tumor samples had more phospho-STAT3 (ser727) than the silibinin-treated group with no change in total STAT3 protein levels (Fig. 4C). These results revealed inhibition by silibinin of STAT3 (ser727) phosphorylation reduced its nuclear translocation and, thus, its transcriptional activity in lung tumors.
Silibinin enhances Angiopoietin-2 and Tie-2 levels in urethane-induced lung tumors
Angiopoietin-2 (Ang-2) is the ligand for the Tie-2 (tyrosine kinase) receptor in endothelial as well as immune cells. Ang-2 can act as both an antagonist and agonist after binding to the Tie-2 receptor (31). Transgenic overexpression of Ang-2 impairs angiogenesis as well as vasculogenesis (31). In the present study, lung tumors from silibinin-treated mice expressed higher levels of Tie-2 (1.5 fold increase, p<0.05) protein, compared to control tumors (Fig. 5). We do not observe any considerable change in phospho-Tie-2 (tyr992) levels, adjusted with total Tie-2 level, by silibinin treatment (Fig. 5). Consistent with immunoblot data, we also observed increased Tie-2 protein in lung tumors lysate in silibinin-treated mice using a Kinexus assay, a global protein kinase antibody-based assay system (data not shown). Concurrently, we observed enhanced Ang-2 expression (1.6 fold increase, p<0.01) in lung tumors from silibinin-treated mice as compared to the controls (Fig. 5). These results suggest that silibinin increased Ang-2 and Tie-2 levels as well as enhancing Ang-2 activated Tie-2 receptor signaling, which may in part be responsible for destabilizing and regressing tumor vasculature.
Figure 5.

Silibinin enhances angiopoietin-2 and Tie-2 levels in urethane-induced lung tumors. Three lung tumor samples were randomly taken from each group and analyzed for pTie-2(Tyr992), Tie-2 and angiopoietin-2 protein levels by immunoblotting. Bands were visualized by enhanced chemiluminescence detection. Membranes were stripped and reprobed with β-actin as a loading control. Densitometric analysis of band intensity for each protein was adjusted with β-actin. Columns, mean intensity of three bands; error bars, SEM for each group. SB, silibinin; Ang-2, angiopoietin-2.
Discussion
The focus of the present study was to assess the chemotherapeutic potential and related mechanisms of oral silibinin on urethane-induced advanced lung tumors in A/J mice, a model of human adenocarcinoma. Previously we showed that silibinin was a potent chemopreventive agent in preventing tumor formation and adenoma to adenocarcinoma prevention in this model (7). Our novel findings are that silibinin reduced tumor number, tumor burden, and progression of adenocarcinomas without adverse effects on mice. Tumor angiogenesis, as opposed to tumor cell proliferation or survival was targeted by silibinin. Silibinin did not exert any significant effect on tumor microvessel density, but rather inhibited increases in vessel size and inhibited new microvessel growth. This antiangiogenic effect of silibinin was accompanied by a decrease in the number of TAMs as well as reduced levels of cytokines known to promote inflammation and angiogenesis (Fig. 6). Enzymes important for inhibiting metalloproteinases such as TIMP-1 and TIMP-2 were up-regulated by silibinin treatment of mice bearing advanced lung tumors. Activation of the transcription factors, HIF-1α, NF-κB and STAT3, was also inhibited by silibinin. Finally, a higher content of Ang-2 and Tie-2 proteins were observed; sustained levels of these proteins impair microvessel development (31).
Figure 6.

Potential anti-angiogenic mechanism of silibinin in advanced lung tumor cells in A/J mice. Silibinin inhibits the production and secretion of cytokines and interleukins via inhibition of tumor associated macrophages. Further, silibinin inhibits the activation of transcription factors (NF-κB/STAT/HIF-1α) but induces the expression of Ang-2/Tie2 to inhibit the angiogenesis in urethane induced-advanced lung tumors in A/J mice. The green color scheme follows the events involved in driving angiogenesis, and all red color schemes show the effect of silibinin.
In this study, A/J mice were given urethane, and after 33 wks, treated with oral silibinin for 10 wks. Lung tumors at this stage were mainly adenocarcinomas i.e., invasive tumors with nuclear dysmorphology and cellular heterogeneity. Silibinin feeding for 10 wks reduced lung adenocarcinoma multiplicity as well as tumor burden, and this was accompanied by significant antiangiogenic effects. In a recently completed chemoprevention study in this urethane-induced lung tumorigenesis model we observed that in early lung lesions, silibinin inhibits tumor growth and progression by strongly inhibiting tumor microvessel density and tumor cell proliferation (7). However, in the present therapeutic experimental design, in which tumors were adenocarcinomas with established tumor vasculature prior to silibinin treatment, silibinin suppressed the growth of existing tumor vasculature as well as microvessel size. Slight but not significant differences were seen in tumor cell proliferation and apoptosis. We also did not observe any considerable effect of silibinin on ERK1/2 and Akt phosphorylation. It appears that at later stage of lung tumor progression, silibinin has no effect on mitogenic and survival pathways. Increased cellular heterogeneity and further mutations in key growth and cell death regulatory pathway components may render cells in these advanced tumors resistant to the effects of silibinin seen in our previous chemoprevention study. These observations suggest that silibinin can target tumors at different stages of progression through its antiangiogenic effects.
Another novel finding is that silibinin inhibited infiltration of macrophages into tumor bearing lungs; macrophages were sparse around the tumors from silibinin-treated mice as compared to those in untreated controls. This observation suggested that silibinin may target TAMs as part of its mechanism in blocking angiogenesis. Numerous studies showed macrophage involvement in promoting tumor angiogenesis (15). Other agents can also influence these macrophages to down-modulate angiogenesis, such as linomide and thiol containing compounds (32, 33). Activated macrophages create a proangiogenic microenvironment by secreting high levels of cytokines, but tumor cells at advanced stages of neoplasia also express high levels of angiogenic cytokines possibly in response to TAMs (16, 34). Silibinin treatment decreased tumor expression of many inflammatory and angiogenic cytokines, including IL-1α, IL-6, IL-9, IL-13, IL-16, IFN-γ and TNF-α which may represent a combined effect of silibinin on macrophages as well as tumor cells. Metalloproteinase activity is required for paving a path for growing microvessels, which can be regulated by TIMPs (35). Silibinin treatment increased expression of inhibitors of metalloproteinases TIMP-1 and TIMP-2, providing support for another mechanistic explanation of the inhibition of tumor microvessel growth and size by silibinin.
Angiogenesis is a highly orchestrated process involving sprouting of new capillary-like structures from the existing vasculature that mature into new blood vessels (36). It can be triggered and modified by many factors, including cytokines, growth factors and their receptors, chemokines, and extracellular matrix macromolecules (36). These factors regulate many transcription factors, including HIF-1α, NF-κB and STAT (23, 24). Hypoxia within tumors stimulates production of HIF-1α, among other pro-angiogenic molecules. HIF-1α is low in normal cells, but reaches high intracellular concentrations in many cancers, and these concentrations strongly correlate with poor prognosis and resistance to therapy (37). Silibinin significantly decreased the number of HIF-1α-positive nuclei, indicating inhibition of its transcriptional activity. Therefore, it is likely that down-regulation of HIF-1α, in part plays a role in the antiangiogenic effect of silibinin on advanced lung tumors.
Recently, activated macrophages have been linked to NF-κB activation in urethane-induced lung carcinogenesis (34). To our knowledge, our finding of inhibition of STAT3 activation is the first report showing STAT3 signaling in urethane-induced lung tumors in mice. Pulmonary macrophages can produce various cytokines, such as TNFα, interleukins, and IFN-γ (as observed in the present study), which in turn activate NF-κB and STAT3 signaling in tumors. As anticipated, we observed activation of both NF-κB and STAT3 in tumors, as indicated by increased levels of their phosphorylated forms, namely, p65 NF-κB (ser276 and ser536) and pSTAT3 (ser727). Silibinin treatment inhibited macrophage infiltration in tumor bearing lungs and inhibited activation of both NF-κB and STAT3 in tumors, which correlates with the diminished size of microvessels as well as the decrease in nestin-positive newly formed microvessels.
We were surprised to note that even after inhibition of HIF-1α, NF-κB and STAT3 signaling by silibinin in tumors, we observed no effect on expression of VEGF, iNOS and COX-2, potential targets of these transcription factors that usually play important roles in tumor angiogenesis. This is relevant because in our previous chemoprevention study, silibinin decreased expression of all three of these proteins (VEGF, iNOS and COX-2) as well as angiogenesis in developing lung tumors (7). This suggests that lung tumor progression in this animal model recruits different angiogenic mediators in early stages of tumor development (microadenoma to adenoma) than late adenocarcinoma stages. Further, we observed that silibinin enhances expression of Ang-2 and Tie-2 receptor tyrosine kinase factors which regulate vessel stabilization and angiogenesis (31). Ang-1, the ligand for the Tie-2 receptor, promotes angiogenesis and recruits pericytes to stabilize vessels (38). However, Ang-2 is a conditional antagonist and agonist for Tie-2 receptor, whose systemic over expression leads to tumor vessel regression without concomitant inhibition of VEGF (31). Similar results were observed with silibinin treatment; we saw no change in VEGF levels but observed more Ang-2 and Tie-2 levels. This could be a potential antiangiogenic mechanism of silibinin on established lung adenocarcinoma.
In summary, oral silibinin showed antitumor effects in urethane-induced and established lung adenocarcinomas most likely by decreasing microvessel size and inhibiting newly formed microvessel growth in tumors. The decrease in TAM infiltration into lungs as well as lower levels of angiogenic cytokines, and greater TIMP-1 and TIMP-2 concentrations, along with the inhibition of HIF-1α, NF-κB and STAT3 activation, could account for the antiangiogenic effects of silibinin. Additionally, elevating levels of Ang-2 and Tie-2 without changing VEGF amounts could have led to microvessel regression in tumors by silibinin. Overall, our findings here, together with our earlier studies (7), suggest that silibinin is a promising agent for intervention in human lung cancer oncogenesis.
Supplementary Material
Acknowledgments
Grant support: This work was supported by RO1 grant CA 113876 and CA 33497 from the National Cancer Institute
Abbreviations
- IL
interleukin
- TNFα
tumor necrosis factor α
- IFNγ
interferon γ
- TIMP
tissue inhibitor of metalloproteinase
- VEGF
vascular endothelial growth factor
- HIF-1α
hypoxia inducing factor 1α
- NF-κB
nuclear factor kappa B
- STAT
signal transducer and activator of transcription
- Ang-2
angiopoietin-2
- TAM
tumor associated macrophage
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics 2007. CA Cancer J Clin. 2007;57:43–6. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Karp DD, Tsao AS, Kim ES. Nonsmall-cell lung cancer: chemoprevention studies. Semin Thorac Cardiovasc Surg. 2003;15:405–20. doi: 10.1053/s1043-0679(03)00100-x. [DOI] [PubMed] [Google Scholar]
- 3.Tyagi A, Raina K, Singh RP, et al. Chemopreventive effects of silymarin and silibinin on N-butyl-N-(4-hydroxybutyl) nitrosamine induced urinary bladder carcinogenesis in male ICR mice. Mol Cancer Ther. 2007;6:3248–55. doi: 10.1158/1535-7163.MCT-07-2006. [DOI] [PubMed] [Google Scholar]
- 4.Raina K, Blouin MJ, Singh RP, et al. Dietary feeding of silibinin inhibits prostate tumor growth and progression in transgenic adenocarcinoma of the mouse prostate model. Cancer Res. 2007;67:11083–91. doi: 10.1158/0008-5472.CAN-07-2222. [DOI] [PubMed] [Google Scholar]
- 5.Roy S, Kaur M, Agarwal C, Tecklenburg M, Sclafani RA, Agarwal R. p21 and p27 induction by silibinin is essential for its cell cycle arrest effect in prostate carcinoma cells. Mol Cancer Ther. 2007;6:2696–707. doi: 10.1158/1535-7163.MCT-07-0104. [DOI] [PubMed] [Google Scholar]
- 6.Gu M, Singh RP, Dhanalakshmi S, Agarwal C, Agarwal R. Silibinin inhibits inflammatory and angiogenic attributes in photocarcinogenesis in SKH-1 hairless mice. Cancer Res. 2007;67:3483–91. doi: 10.1158/0008-5472.CAN-06-3955. [DOI] [PubMed] [Google Scholar]
- 7.Singh RP, Deep G, Chittezhath M, et al. Effect of silibinin on the growth and progression of primary lung tumors in mice. J Natl Cancer Inst. 2006;98:846–55. doi: 10.1093/jnci/djj231. [DOI] [PubMed] [Google Scholar]
- 8.Singh RP, Agarwal R. Prostate cancer chemoprevention by silibinin: bench to bedside. Mol Carcinog. 2006;45:436–42. doi: 10.1002/mc.20223. [DOI] [PubMed] [Google Scholar]
- 9.Sharma G, Singh RP, Chan DC, Agarwal R. Silibinin induces growth inhibition and apoptotic cell death in human lung carcinoma cells. Anticancer Res. 2003;23:2649–55. [PubMed] [Google Scholar]
- 10.Chu SC, Chiou HL, Chen PN, Yang SF, Hsieh YS. Silibinin inhibits the invasion of human lung cancer cells via decreased productions of urokinase-plasminogen activator and matrix metalloproteinase-2. Mol Carcinog. 2004;40:143–9. doi: 10.1002/mc.20018. [DOI] [PubMed] [Google Scholar]
- 11.Chen PN, Hsieh YS, Chiou HL, Chu SC. Silibinin inhibits cell invasion through inactivation of both PI3K-Akt and MAPK signaling pathways. Chem Biol Interact. 2005;156:141–50. doi: 10.1016/j.cbi.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 12.Singh RP, Mallikarjuna GU, Sharma G, et al. Oral silibinin inhibits lung tumor growth in athymic nude mice and forms a novel chemocombination with doxorubicin targeting nuclear factor kappaB-mediated inducible chemoresistance. Clin Cancer Res. 2004;10:8641–7. doi: 10.1158/1078-0432.CCR-04-1435. [DOI] [PubMed] [Google Scholar]
- 13.Folkman J. Angiogenesis. Annu Rev Med. 2006;57:1–18. doi: 10.1146/annurev.med.57.121304.131306. [DOI] [PubMed] [Google Scholar]
- 14.Handsley MM, Edwards DR. Metalloproteinases and their inhibitors in tumor angiogenesis. Int J Cancer. 2005;115:849–60. doi: 10.1002/ijc.20945. [DOI] [PubMed] [Google Scholar]
- 15.Redente EF, Orlicky DJ, Bouchard RJ, Malkinson AM. Tumor signaling to the bone marrow changes the phenotype of monocytes and pulmonary macrophages during urethane-induced primary lung tumorigenesis in A/J mice. Am J Pathol. 2007;170:693–708. doi: 10.2353/ajpath.2007.060566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kimura YN, Watari K, Fotovati A, et al. Inflammatory stimuli from macrophages and cancer cells synergistically promote tumor growth and angiogenesis. Cancer Sci. 2007;98:2009–18. doi: 10.1111/j.1349-7006.2007.00633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao J, Agarwal R. Tissue distribution of silibinin, the major active constituent of silymarin, in mice and its association with enhancement of phase II enzymes: implications in cancer chemoprevention. Carcinogenesis. 1999;20:2101–8. doi: 10.1093/carcin/20.11.2101. [DOI] [PubMed] [Google Scholar]
- 18.You M, Candrian U, Maronpot RR, Stoner GD, Anderson MW. Activation of the Ki-ras protooncogene in spontaneously occurring and chemically induced lung tumors of the strain A mouse. Proc Natl Acad Sci U S A. 1989;86:3070–4. doi: 10.1073/pnas.86.9.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malkinson AM. Primary lung tumors in mice as an aid for understanding, preventing, and treating human adenocarcinoma of the lung. Lung Cancer. 2001;32:265–79. doi: 10.1016/s0169-5002(00)00232-4. [DOI] [PubMed] [Google Scholar]
- 20.Malkinson AM. Primary lung tumors in mice: an experimentally manipulable model of human adenocarcinoma. Cancer Res. 1992;52:2670s–6s. [PubMed] [Google Scholar]
- 21.Kleeberger W, Bova GS, Nielsen ME, et al. Roles for the stem cell associated intermediate filament nestin in prostate cancer migration and metastasis. Cancer Res. 2007;67:9199–206. doi: 10.1158/0008-5472.CAN-07-0806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sinha P, Clements VK, Ostrand-Rosenberg S. Interleukin-13-regulated M2 macrophages in combination with myeloid suppressor cells block immune surveillance against metastasis. Cancer Res. 2005;65:11743–51. doi: 10.1158/0008-5472.CAN-05-0045. [DOI] [PubMed] [Google Scholar]
- 23.Singh RP, Agarwal R. Inducible nitric oxide synthase-vascular endothelial growth factor axis: a potential target to inhibit tumor angiogenesis by dietary agents. Curr Cancer Drug Targets. 2007;7:475–83. doi: 10.2174/156800907781386632. [DOI] [PubMed] [Google Scholar]
- 24.Singh RP, Agarwal R. Tumor angiogenesis: a potential target in cancer control by phytochemicals. Curr Cancer Drug Targets. 2003;3:205–17. doi: 10.2174/1568009033481985. [DOI] [PubMed] [Google Scholar]
- 25.Kisley LR, Barrett BS, Dwyer-Nield LD, Bauer AK, Thompson DC, Malkinson AM. Genetic ablation of inducible nitric oxide synthase decreases mouse lung tumorigenesis. Cancer Res. 2002;62:6850–6. [PubMed] [Google Scholar]
- 26.Bauer AK, Dwyer-Nield LD, Malkinson AM. High cyclooxygenase 1 (COX-1) and cyclooxygenase 2 (COX-2) contents in mouse lung tumors. Carcinogenesis. 2000;21:543–50. doi: 10.1093/carcin/21.4.543. [DOI] [PubMed] [Google Scholar]
- 27.Lee K, Roth RA, LaPres JJ. Hypoxia, drug therapy and toxicity. Pharmacol Ther. 2007;113:229–46. doi: 10.1016/j.pharmthera.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 28.Van Waes C. Nuclear factor-kappaB in development, prevention, and therapy of cancer. Clin Cancer Res. 2007;13:1076–82. doi: 10.1158/1078-0432.CCR-06-2221. [DOI] [PubMed] [Google Scholar]
- 29.Baby J, Pickering BF, Vashisht Gopal YN, Van Dyke MW. Constitutive and inducible nuclear factor-kappaB in immortalized normal human bronchial epithelial and non-small cell lung cancer cell lines. Cancer Lett. 2007;255:85–94. doi: 10.1016/j.canlet.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 30.Jiang H, Harris MB, Rothman P. IL-4/IL-13 signaling beyond JAK/STAT. J Allergy Clin Immunol. 2000;105:1063–70. doi: 10.1067/mai.2000.107604. [DOI] [PubMed] [Google Scholar]
- 31.Cao Y, Sonveaux P, Liu S, et al. Systemic overexpression of angiopoietin-2 promotes tumor microvessel regression and inhibits angiogenesis and tumor growth. Cancer Res. 2007;67:3835–44. doi: 10.1158/0008-5472.CAN-06-4056. [DOI] [PubMed] [Google Scholar]
- 32.Vukanovic J, Isaacs JT. Linomide inhibits angiogenesis, growth, metastasis, and macrophage infiltration within rat prostatic cancers. Cancer Res. 1995;55:1499–504. [PubMed] [Google Scholar]
- 33.Koch AE, Burrows JC, Polverini PJ, Cho M, Leibovich SJ. Thiol-containing compounds inhibit the production of monocyte/macrophage-derived angiogenic activity. Agents Actions. 1991;34:350–7. doi: 10.1007/BF01988728. [DOI] [PubMed] [Google Scholar]
- 34.Stathopoulos GT, Sherrill TP, Cheng DS, et al. Epithelial NF-kappaB activation promotes urethane-induced lung carcinogenesis. Proc Natl Acad Sci U S A. 2007;104:18514–9. doi: 10.1073/pnas.0705316104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramer R, Eichele K, Hinz B. Upregulation of tissue inhibitor of matrix metalloproteinases-1 confers the anti-invasive action of cisplatin on human cancer cells. Oncogene. 2007;26:5822–7. doi: 10.1038/sj.onc.1210358. [DOI] [PubMed] [Google Scholar]
- 36.Plank MJ, Sleeman BD. Tumor-induced angiogenesis: A review. J Theoretical Med. 2003;5:137–53. [Google Scholar]
- 37.Li XF, Carlin S, Urano M, Russell J, Ling CC, O'Donoghue JA. Visualization of hypoxia in microscopic tumors by immunofluorescent microscopy. Cancer Res. 2007;67:7646–53. doi: 10.1158/0008-5472.CAN-06-4353. [DOI] [PubMed] [Google Scholar]
- 38.Hawighorst T, Skobe M, Streit M, et al. Activation of the tie2 receptor by angiopoietin enhances tumor vessel maturation and impairs squamous cell carcinoma growth. Am J Pathol. 2002;160:1381–92. doi: 10.1016/S0002-9440(10)62565-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.