

Abstract
Cell cycle markers have been shown to be upregulated and proposed to lead to apoptosis of postmitotic neurons in Alzheimer's disease (AD). Presenilin (PS) plays a critical role in AD pathogenesis, and loss-of-function studies in mice established a potent effect of PS in cell proliferation in peripheral tissues. Whether PS has a similar activity in the neuronal cell cycle has not been investigated. PS exhibits γ-secretase-dependent and -independent functions; the former requires aspartate 257 (D257) as part of the active site, and the latter involves the hydrophilic loop domain encoded by exon 10. We used two novel mouse models, one expressing the PS1 D257A mutation on a postnatal PS conditional knock-out background and the other deleting exon 10 of PS1, to dissect the γ-secretase-dependent and -independent activities of PS in the adult CNS. Whereas γ-secretase plays a dominant role in neuronal survival, our studies reveal potent neuronal cell cycle regulation mediated by the PS1 hydrophilic loop. Although neurons expressing cell cycle markers do not directly succumb to apoptosis, they are more vulnerable under stress conditions. Importantly, our data identify a novel pool of cytoplasmic p53 as a downstream mediator of this cellular vulnerability. These results support a model whereby the PS γ-secretase activity is essential in maintaining neuronal viability, and the PS1 loop domain modulates neuronal homeostasis through cell cycle and cytoplasmic p53 control.
Keywords: Alzheimer's disease, presenilin, γ-secretase, neurodegeneration, cell cycle events, mice
Introduction
Alzheimer's disease (AD) is the most common neurodegenerative disease in the aging population. AD is characterized by the deposition of β-amyloid peptides liberated from the amyloid precursor protein (APP) and neurofibrillary tangles composed of hyperphosphorylated tau. In addition to the pathological hallmarks, postmortem investigation reveals that molecules that are traditionally considered cell cycle proteins, such as Cyclins A, B, and D, can be detected at early stages of AD preceding neurodegeneration (Yang et al., 2003). This phenomenon, termed cell cycle events (CCEs), has also been replicated in AD mouse models with amyloid or tau pathologies (Andorfer et al., 2005; Yang et al., 2006). It has been postulated that unscheduled cell cycle reentry in postmitotic neurons is causal for neuronal cell death (for review, see Herrup et al., 2004). Mutations in presenilin genes (PSEN1 and PSEN2) lead to the majority of familial, early-onset cases of AD (FAD), establishing a critical role for presenilin (PS) in AD pathogenesis. Among many of the putative pathogenic activities reported, a recent paper documented that neurons harboring the PS1 M146V FAD mutation show Cyclin D1 upregulation, abnormal neuronal cell cycle, and enhanced neuronal apoptosis (Malik et al., 2008).
PS is required for γ-secretase cleavage of a multitude of substrates, in particular APP and Notch (De Strooper et al., 1998, 1999), and this activity requires the conserved aspartates at the 257 (D257) and 385 (D385) sites (Wolfe et al., 1999). In addition, PS has been shown to exhibit γ-secretase-independent activities through the large hydrophilic loop domain (Baki et al., 2001; Soriano et al., 2001; Kang et al., 2002), and these two events can be biochemically and genetically uncoupled (Xia et al., 2002). In light of the growing list of γ-secretase substrates and diverse pathways PS mediates (for review, see Sisodia and St George-Hyslop, 2002; Kopan and Ilagan, 2004; Vetrivel et al., 2006), the interpretation of pathogenic mechanisms by the FAD mutations is likely complex. However, increasing evidence supports the notion that the overall effects of these mutations are partial loss of both γ-secretase-dependent and -independent functions (for review, see De Strooper, 2007; Shen and Kelleher, 2007).
We reported that loss of PS in peripheral tissues leads to the activation of Cyclin D1, hyperproliferation, and skin tumorigenesis (Xia et al., 2001; Qyang et al., 2004). Inactivation of PS in postnatal neurons results in age-dependent neurodegeneration (Feng et al., 2004; Saura et al., 2004). Using a series of mouse models with either complete PS deficiency or specific disruption of the γ-secretase activity or the hydrophilic loop domain, and complemented with primary neuronal cultures and γ-secretase inhibitors, this study attempts to address the following questions. (1) Does PS regulate neuronal CCEs? (2) Are the neuronal CCEs mediated through γ-secretase-dependent or -independent mechanisms? (3) What is the functional consequence of PS-mediated neuronal CCEs?
Materials and Methods
Mouse models.
The references for the various PS alleles are as follows: exon 4 floxed (fl) allele (Feng et al., 2001); PS1 knock-out (Wong et al., 1997); PS2 knock-out (Donoviel et al., 1999); PS1 ΔE10 knock-in (Deng et al., 2006); and transgenic mice expressing wild-type or D257A mutant human PS1 (Qian et al., 1998; Xia et al., 2002). The CaMKIIα–Cre mice were also described previously (Tsien et al., 1996).
The breeding scheme for the generation of PS1 conditional knock-out on PS2 null background [PS conditional double-knock-out (PS cDKO)], PS cDKO expressing wild-type human PS1 (PS cDKO; hPS1), or the D257A mutant (PS cDKO; D257A) and their littermate controls are outlined below using hPS1 as an example. Note that PS2−/− mice were normal in all the assays applicable to this study and were used as controls for the PS cDKO animals.
 |
The breeding scheme for generating PS1 knock-out (PS1−/−), PS1 knock-out expressing wild-type or the D257A mutant human PS1 (PS1−/−; hPS1 or PS1−/−; D257A, respectively), and their littermate controls for embryonic neuronal culture studies are diagramed below using hPS1 as an example. Wild-type or heterozygous PS1 littermates were used as controls, and no differences between the two genotypes could be detected.
 |
Heterozygous interbreeding was set up to create homozygous PS1 ΔE10 and littermate wild-type controls for in vivo and primary culture studies. Table 1 is a summary of the mouse models used for in vivo and neuronal cultures studies.
Table 1.
Mouse models for in vivo and neuronal culture studies
| Mice | Genotype (color representation) | Objective |
|---|---|---|
| Control | PS1fl/fl; PS2−/− | Investigate γ-secretase-dependent effects in vivo |
| PS cDKO | Cre; PS1fl/fl; PS2−/− | |
| PS cDKO; hPS1 | Cre; PS1fl/fl; PS2−/−; hPS1 | |
| PS cDKO; D257A | Cre; PS1fl/fl; PS2−/−; hPS1D257A | |
| Control | PS1+/+ | Investigate γ-secretase-independent effects in vivo |
| PS1 ΔE10 | PS1ΔE10 /ΔE10 | |
| Control | PS1+/+ or PS1+/− | Study the mechanisms of γ-secretase-dependent and -independent effects in vitro |
| PS1−/− | PS1−/− | |
| PS1−/−; hPS1 | PS1−/−; hPS1 | |
| PS1−/−; D257A | PS1−/−; hPS1D257A | |
| PS1 ΔE10 | PS1ΔE10 /ΔE10 |
All animal experiments were performed in accordance with the Baylor College of Medicine Institutional Animal Care and Use Committee and with national regulations and policies.
Antibodies and reagents.
The following primary antibodies were used as follows: neuronal-specific nuclear protein (NeuN) (1:1000; Millipore Bioscience Research Reagents), Cyclin D1 (1:500; Abcam), bromodeoxyuridine (BrdU) (1:100; DakoCytomation), p53 (1:500; Abcam), Cyclin A (1:500; Abcam), cleaved caspase-3 (1:500; Abcam), and γ-tubulin (1:10,000; Sigma). The following secondary antibodies were used: cyanine 3 (1:500; Zymed), Oregon Green (1:500; Invitrogen), and HRP-conjugated anti-mouse and anti-rabbit (1:5000; Vector Laboratories). The γ-secretase inhibitor Compound E was a gift from Dr. Y. Li (Tarassishin et al., 2004). It was dissolved in DMSO and used at a final concentration of 50 nm. Quercetin (Sigma), a potent Cyclin D1 inhibitor, was dissolved in DMSO and used at a final concentration of 10 μm (Malik et al., 2008).
Three-dimensional magnetic resonance imaging.
Mice were imaged using a 9.4 T Bruker Avance Biospec spectrometer with a 21 cm horizontal bore and a 35 mm volume resonator coil (Bruker Biospin) and maintained on 1–2% isoflurane during the imaging session. The body temperature of the mice was monitored with a rectal temperature probe and maintained at 37°C using an air heater (Small Animal Instruments). Additionally, the respiration rate of the animals was monitored with a pressure transducer placed underneath the animals during imaging (Small Animal Instruments). Three-dimensional (3D) rapid acquisition with relaxation enhancement (RARE) image datasets of the brain were obtained for each mouse with the following: RARE factor of 8; repetition time, 2000 ms; echo time, 45.2 ms; field of view, 30 × 30 × 30 mm; a matrix size of 128 × 128 × 128; and two signal averages. The acquisition time was 2 h 16 min 32 s for each mouse. All pixels attributable to the cortex in the 3D volume were labeled in each slice, and volume analysis was performed using the AMIRA software (Mercury Computer Systems) (Redwine et al., 2003). Cerebellar volume was also quantified as an internal control, which remained constant among all genotypes and across all ages.
BrdU injection and brain immunohistochemistry.
BrdU (Sigma) was dissolved in 0.9% saline. Each animal was injected intraperitoneally at a dose of 200 mg/kg daily for 4 consecutive days (adapted from Wojtowicz and Kee, 2006). Mice underwent trans-cardiac perfusion with PBS containing heparin and fixed with 4% paraformaldehyde (PFA) at 4°C overnight. After embedding the brains in paraffin wax, sections were cut at 10 μm thickness. The sections were deparaffinized in xylene and then rehydrated with 100, 95, and 70% ethanol and water. If sections were to be stained with BrdU, treatment with 2N HCl for 20 min before staining was used to retrieve the BrdU antigen. Protein antigen retrieval was performed using 0.1 m sodium citrate, pH 7.4, for 10 min at a constant boil, and sections were cooled at room temperature for 30 min thereafter, followed by blocking with 5% goat serum for 1 h. Primary antibodies were applied at 4°C overnight in 2.5% goat serum. Secondary antibodies were applied for 1 h at 37°C in 2.5% goat serum. The sections were mounted using ProlongGold with 4′,6′-diamidino-2-phenylindole (Invitrogen). Pictures were taken with a Carl Zeiss LSM510 laser scanning confocal microscope equipped with an Axiocam MRC digital camera, and the images were analyzed using NIH Image J software. Quantification of BrdU-, Cyclin D1-, and Cyclin A-positive neurons was performed by counting the number of marker positive neurons per equal area in 15 sections per animal and three animals per genotype.
Western blot analysis.
Forebrain samples or cultured cells were homogenized using 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) lysis buffer (1% CHAPS, 50 mm Tris, pH 7.6, 150 mm NaCl, and 2% sodium azide) containing complete protease inhibitor mixture (Roche Diagnostics). After three sets of 10 pulses of sonication, the homogenates were spun at 14,000 rpm for 15 min. Protein concentrations were determined using Bio-Rad Dc Protein Assay. Ten micrograms of protein were loaded on a 10% SDS-PAGE gel run at 100 V for 2 h at room temperature and transferred onto a nitrocellulose membrane (Bio-Rad) for 1 h. Membranes were blocked 1 h using 5% nonfat dry milk in TBS containing 0.1% Tween 20 (TBST) (Sigma). Cyclin D1 primary antibody incubation was done in 5% milk in TBST; p53 primary antibody was added to 5% BSA in TBST and incubated overnight at 4°C with constant agitation. After three washes with PBS with 0.1% Tween 20, secondary antibody application was performed at room temperature for 1 h using 5% milk in TBST, followed by three additional washes with PBST (PBS with 0.1% Tween20). Bands were visualized using Immobilon Western ECL system (Millipore).
Primary embryonic neuronal cultures and treatment.
Timed mating was set up, and the day when a vaginal plug was observed was considered to be embryonic day 0.5 (E0.5). E15.5 embryos were collected from pregnant females, and primary neuronal cultures were prepared from combined neocortex and hippocampi of the embryonic brains. The brain tissues were dissected under a microscope, and the cells were mechanically dissociated, resuspended in Neurobasal medium (Invitrogen) with DNase, spun down at 1000 × g, resuspended in Neurobasal medium containing 2 mm glutamine and penicillin/streptomycin, and plated on poly-d-lysine-coated 60 mm dishes (for Western blotting) or 24-well plates (for all other experiments). Specifically, if cultures were to be used for hydrogen peroxide (H2O2) treatment followed by lactate dehydrogenase (LDH) measurement, the cells were plated at a density of 5 × 105 cells per well. If the cells were to be used for immunohistochemistry followed by confocal microscopy, the cells were plated at a density of 3 × 104 cells per well. Twenty-four hours after plating, the medium was replaced with Neurobasal medium containing 1% B27 supplement (Invitrogen) to promote neuronal survival and discourage non-neuronal cell growth. Subsequently, the medium was changed every other day. For neurons treated with DMSO or Compound E (dissolved in DMSO), the treatment was begun concurrently with the initial plating of the cells.
For LDH measurements, the H2O2 treatment was performed in neurons cultured 10 d in vitro (DIV) for 24 h at concentrations ranging from 0 to 100 μm in 700 μl of Neurobasal medium containing 1% B27, DMSO, Quercetin, or Compound E when applicable. After the treatment, samples, each plated in 50 μl triplicates, were removed and placed in a 96-well plate. Fifty microliters of substrate mix (Cytotox 96; Promega) were added to each well, followed by a 30 min incubation at room temperature protected from light. The reaction was aborted with 50 μl stop solution per well, and absorption was measured at 490 nm using a Biotek 96-well plate reader and analyzed using KC junior software. Basal LDH release was obtained from wells with no H2O2 treatment for the 24 h incubation period for background correction. Total LDH (100%) was obtained by treating wells with lysis solution for 1 h before LDH measurement. All values are given as percentage of total LDH release.
For time course experiments, the neurons were subjected to 1 mm H2O2 treatment for specified times. To inhibit new protein synthesis, cycloheximide at 2 mg/ml (Sigma) was added to the Neurobasal medium and applied concurrently with H2O2. For immunostaining, medium was aspirated off, plates were washed with TBS twice, and 4% PFA was added for 20 min in TBST overnight. Subsequently, the plates were washed twice with TBST (0.1% Triton X-100) and incubated for 20 min to permeabilize the cells. Nonspecific interactions were blocked with 10% goat serum in TBST. The primary antibody incubation was performed in 5% TBST at 4°C overnight, followed by three TBST washes and incubation with secondary antibody in 5% TBST for 1 h at room temperature. Images were acquired using a Carl Zeiss LSM 520 laser confocal microscope and analyzed using NIH Image J software. Cyclin D1-positive neurons were counted in each field using NIH Image J nuclei counter. Total neuronal counts were obtained per section, and percentage Cyclin D1-positive neurons were calculated. Percentage nuclear p53 was obtained using NIH ImageJ to attain nuclear and cytoplasmic staining intensity in a set area (50 × 50 pixels).
Statistical analysis.
All statistical analysis was performed using the Student's t test. Statistical significance is indicated in the figures: *p < 0.05, **p < 0.01, ***p < 0.001. Data are presented as average ± SEM.
Results
γ-Secretase is the major but not the sole contributor to neuronal viability
Previous studies have established that conditional inactivation of PS1 in postnatal forebrain neurons (PS1 cKO) by CaMKIIα–Cre transgene expression resulted in only subtle phenotypes (Feng et al., 2001; Yu et al., 2001). However, deleting PS2 on conditional PS1 null background (herein referred to as PS cDKO) led to profound neuronal cell loss (Feng et al., 2004; Saura et al., 2004). Magnetic resonance imaging (MRI) of live animals was used to quantify the degree of neurodegeneration by measuring the cortical volumes of control and PS cDKO mice at 2, 6, and 12 months of age (Fig. 1A). Consistent with previous studies (Feng et al., 2004; Saura et al., 2004), when measured at 2 months of age, no difference in cortical volume was visible (Fig. 1A, quantified in B). However, at 6 months, the cortical volume of the PS cDKO animals was decreased by 15%, and, in 12-month-old animals, this decrease was a striking 40% (Fig. 1A,B). Mice at 6 months were chosen for additional analysis because it represents an early stage of neurodegenerative processes.
Figure 1.
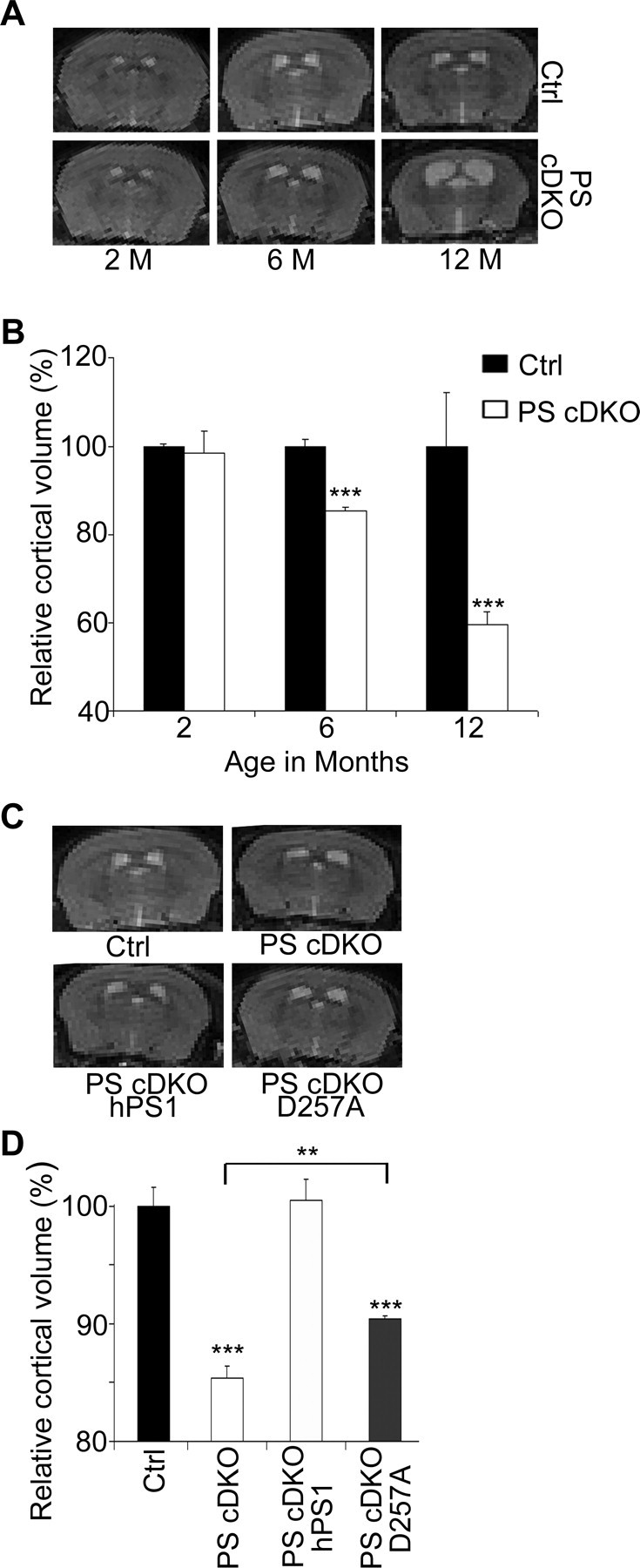
3D MRI analysis of cortical volumes of PS animals. A, Representative 2D images of littermate PS2 null control (Ctrl) and PS cDKO animals at 2, 6, and 12 months of age. B, Quantification of relative 3D cortical volumes showing age-dependent reduction in PS cDKO mice. Control at 2 months is set at 100%. C, Representative 2D images of 6-month-old control (Ctrl), PS cDKO, PS cDKO; hPS1, and PS cDKO; D257A animals. D, Quantification of 3D cortical volumes relative to the control. PS cDKO animals showed ∼15% reduction when compared with the controls, which was completely rescued by introduction of a wild-type PS1 transgene (PS cDKO; hPS1). Expressing a γ-secretase-deficient PS1 D257A transgene on PS cDKO background (PS cDKO; D257A) was not sufficient to completely rescue the neurodegenerative phenotype (Ctrl vs PS cDKO; D257A, p < 0.001) but resulted in significant increase in the cortical volume (PS cDKO vs PS cDKO; D257A, p < 0.01). All statistical analysis was performed using Student's t test. **p < 0.01, ***p < 0.001. n = 3 per genotype.
To determine the contribution of γ-secretase to neuronal survival, we expressed either the human wild-type PS1 transgene (hPS1, line 17-3) or the γ-secretase-deficient transgene PS1 D257A (D257A, line 7) with similar levels of expression onto PS cDKO background (PS cDKO; hPS1 or PS cDKO; D257A), respectively. The PS1 D257A mutant has been shown to be defective in γ-secretase processing but preserve γ-secretase-independent activities (Xia et al., 2002). MRI analysis revealed that expression of the wild-type human PS1 resulted in complete rescue because the cortical volumes were indistinguishable from that of the control animals (Fig. 1C,D, compare Ctrl with PS cDKO; hPS1). Interestingly, although reduced cortical volume was readily detectable, a partial rescue of the PS cDKO phenotype was evident in animals expressing the PS1 D257A transgene (Fig. 1D, compare PS cDKO with PS cDKO; D257A). We conclude, therefore, that although the lack of γ-secretase activity is the major contributor to neuronal cell loss in PS cDKO animals, other PS1 domains may also play a functional role in preserving neuronal viability.
Neuronal cell cycle activation is γ-secretase independent and is not sufficient to cause neurodegeneration
Neuronal CCEs have been observed in various neurodegenerative conditions, including AD, and have been proposed to trigger neuronal cell loss. Our previous studies identified a potent role for PS in promoting cell proliferation in peripheral tissues (Xia, 2001; Qyang et al., 2004), and this activity may involve the PS1 hydrophilic loop domain and the β-catenin/Cyclin D1 pathway (Soriano et al., 2001; Kang et al., 2002). To examine whether PS plays a similar role in the neuronal cell cycle and whether CCEs contribute to neuronal cell death, we performed Cyclin D1 immunostaining in PS cDKO mice and PS cDKO expressing wild-type or D257A mutant PS1 (Fig. 2A). As expected, control brains were devoid of Cyclin D1 expression. Consistent with our finding that PS is a negative regulator of Cyclin D1 in peripheral tissues, we found that both Cyclin D1 (Fig. 2A) and Cyclin A (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material) were aberrantly expressed in the forebrain of PS cDKO animals. Interestingly, expression of either wild-type human PS1 or the D257A mutant completely rescued the aberrant expression of Cyclin D1 (Fig. 2A) and Cyclin A (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material), demonstrating a γ-secretase-independent effect. To directly test whether the neuronal CCE is mediated by the PS1 loop, we performed similar cell cycle marker immunostaining in a strain of knock-in mice in which the exon 10 of PS1 encoding the hydrophilic loop domain is deleted (PS1 ΔE10) (Deng et al., 2006). Indeed, abundant Cyclin D1 (Fig. 2B) and Cyclin A (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material) expressing neurons could be detected in homozygous PS1 ΔE10 mice. The immunostaining data were validated by Western blot analysis of forebrain protein lysates (Fig. 2C). The fact that neuronal CCE can be rescued by the γ-secretase defective PS1 D257A despite persistent neurodegeneration and that prominent CCE is present in γ-secretase competent PS1 ΔE10 animals without overt neuronal loss prompted us to conclude that neuronal CCE is γ-secretase independent and that aberrant expression of cell cycle proteins is not a direct correlate of neuronal cell death in vivo.
Figure 2.
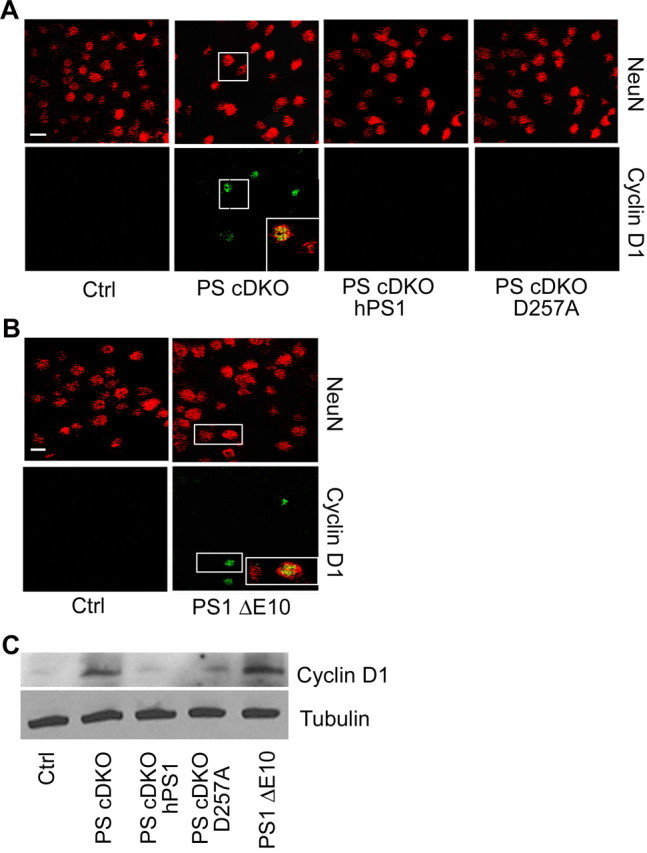
Cyclin D1 activation in PS mutant animals. A, Representative images of NeuN and Cyclin D1 immunostaining of 6-month-old control (Ctrl), PS cDKO, PS cDKO; hPS1, and PS cDKO; D257A animals. Inset shows representative NeuN/Cyclin D1 double-positive cells in PS cDKO mice. B, Representative images of NeuN and Cyclin D1 immunostaining of 6-month-old control (Ctrl) and PS1 ΔE10 brains with NeuN/Cyclin D1 double-positive cells shown in inset. C, Western blot analysis of Cyclin D1 protein levels in forebrain samples of PS mutants. γ-Tubulin was used as a loading control.
To further evaluate the functional consequences of neuronal CCE, we examined BrdU incorporation in PS cDKO and PS1 ΔE10 animals, which either do or do not possess neurodegenerative phenotype, respectively. Daily BrdU injections for 4 d followed by immunostaining 1, 7, or 14 d later revealed that, interestingly, BrdU-positive neurons could only be detected in PS cDKO animals (Fig. 3A) but not in PS1 ΔE10 animals (data not shown), suggesting that neuronal CCEs are not a direct cause for DNA replication. However, ∼80% of BrdU-positive cells also expressed Cyclin D1 (Fig. 3C), supporting the view that CCE serves as an important trigger for BrdU incorporation. Quantification of BrdU-positive PS cDKO neurons at 1, 7, and 14 d after injection revealed that most of the neurons remained BrdU positive after 7 d, but the number of BrdU-positive neurons was reduced by ∼50% at day 14 (Fig. 3B). Double labeling of BrdU with active caspase-3 showed a time-dependent increase of BrdU and active caspase-3 double-positive cells (Fig. 3D, quantified in E). Interestingly, the active caspase-3 was predominately localized to the nucleus (Fig. 3D), indicating that the cell death may be primarily mediated by activation of the nuclear targets of caspase-3. These results combined suggest that additional insults, absent in PS1 ΔE10 but present in PS cDKO, are required for the CCE neurons to begin DNA replication and that BrdU-incorporating neurons remain viable for a certain period of time but then eventually succumb to apoptosis.
Figure 3.

BrdU, Cyclin D1, and active caspase-3 immunostaining of PS cDKO animals. A, Representative images of BrdU-positive neurons in PS cDKO animals 24 h (PS cDKO, day 1), 7 d (PS cDKO, day 7), and 14 d (PS cDKO, day 14) after the last BrdU injection. Mature neurons are marked by NeuN. BrdU incorporation in mature neurons, marked as NeuN/BrdU double positive, is highlighted in insets. B, Quantification of percentage of BrdU-positive neurons at 1, 7, and 14 d after injection. Fifteen sections from each animal and n = 3 animals per group were analyzed. C, Representative images of Cyclin D1 and BrdU colabeling in PS cDKO mice 1 and 7 d after the last BrdU injection. D, Representative images of BrdU and active caspase-3 costaining. E, Quantification of BrdU/active caspase-3 double-positive cells at 1, 7, and 14 d after injection. Data were analyzed using Student's t test. *p < 0.05, **p < 0.01. Scale bar, 10 μm.
Cell cycle events promote neuronal cell death under stress conditions
The above in vivo studies suggest that loss of γ-secretase is a major contributor to the neurodegenerative phenotype seen in PS cDKO animals. However, considerable rescue by expression of the γ-secretase defective PS1 D257A mutant indicates a role for γ-secretase-independent domains. Although activation of neuronal CCE is an obvious candidate for the γ-secretase-independent activity, our in vivo results clearly show that CCEs alone are not sufficient to cause neurodegeneration. To seek additional understanding of the in vivo phenotypes and to explore the functional consequences of neuronal CCEs, we used primary neuronal cultures. The early embryonic lethality of PS1 and PS2 double-null embryos precludes the derivation of PS-deficient neuronal cultures (Donoviel et al., 1999; Herreman et al., 1999). However, PS1 is known to be the major presenilin during development (Shen et al., 1997; Wong et al., 1997), and neurons derived from PS1 null animals have been used extensively to study PS function. We therefore chose to culture neurons isolated from E15.5 of the following PS1 genotypes (summarized in Table 1): PS1 knock-out (PS1−/−), PS1 knock-out expressing wild-type or D257A mutant of human PS1 (PS1−/−; hPS1 or PS1−/−; D257A, respectively), as well as homozygous PS1 ΔE10. Examination of neuronal morphology and measurement of neuronal viability using the lactate dehydrogenase (LDH) release assay failed to detect overt differences in any of the genotypes when compared with littermate controls (data not shown).
We first assessed cell cycle marker expression in the above neurons cultured 5 DIV. Strikingly, immunostaining showed that, although control neurons lacked Cyclin D1, prominent Cyclin D1 expression could be detected in the nucleus and cytoplasm in ∼85% of PS1−/− and PS1 ΔE10 neurons (Fig. 4A, quantified in B). Similar to that of the mouse brain, aberrant Cyclin D1 activation was rescued by expressing either the wild-type PS1 or the D257A mutant PS1 (Fig. 4A). Western blot analysis of Cyclin D1 protein levels confirmed the immunostaining data (Fig. 4C). Similar results were also obtained when Cyclin A was analyzed (supplemental Fig. S1B, available at www.jneurosci.org as supplemental material). The γ-secretase-independent regulation of neuronal CCE was further confirmed by treating the control neurons with Compound E, a potent γ-secretase inhibitor (Tarassishin et al., 2004), showing that inhibition of γ-secretase had no effect on Cyclin D1 expression (supplemental Fig. S2, available at www.jneurosci.org as supplemental material).
Figure 4.

Cyclin D1 expression in neuronal cultures derived from PS1 mutants. A, Representative images of NeuN and Cyclin D1 immunostaining of primary neuronal cultures isolated from E15.5 of various PS mutants. Ctrl, PS1+/+ or PS1+/− (combined as no differences could be detected); PS1−/−, PS1 knock-out; PS1−/− hPS1, PS1 knock-out expressing wild-type human PS1; PS1−/− D257A, PS1 knock-out expressing D257A mutant of human PS1; PS1 ΔE10, PS1 knock-in mice with deletion of exon 10. Neuronal nuclei were marked with an anti-NeuN antibody. NeuN/Cyclin D1 double-positive cells could be found in PS1−/− and PS1 ΔE10 neurons but not in PS1−/−; hPS1 or PS1−/−; D257A neurons. Scale bar, 10 μm. B, Quantification of percentage of neurons positive for Cyclin D1. Approximately 1000 cells were counted from five independent experiments. ***p < 0.001, Student's t test. C, Representative Western blotting of Cyclin D1 levels in various PS cultures. γ-Tubulin was used as a loading control.
Although the basal survival of PS1 null and PS1 ΔE10 neurons was not affected compared with their littermate controls, we reasoned that lack of γ-secretase or abnormal CCE may render them susceptible under stress conditions. We therefore investigated the effects of these functional domains on neuronal survival by treating the cultured neurons with 25–100 μm H2O2 and measuring the degree of cell death using the LDH release assay (Fig. 5A). Although control neurons were resistant to 25 μm H2O2, PS1−/− neurons showed significant cell death (Fig. 5A). The increased LDH release could be rescued by expressing wild-type human PS1 but not the PS1 D257A mutant (supplemental Fig. S3A, available at www.jneurosci.org as supplemental material). Although profound CCE can be observed in PS1 ΔE10 neurons, they were resistant to 25 μm H2O2, similar to control (Fig. 5A). These results support the view that, at low concentrations of H2O2, γ-secretase is the major determinant of neuronal viability. This assessment was further confirmed by treating the neurons with the γ-secretase inhibitor Compound E in combination with 25 μm H2O2, which resulted in significant LDH release in both control neurons and PS1 ΔE10 neurons (supplemental Fig. S3B, available at www.jneurosci.org as supplemental material). However, at higher concentrations of H2O2 (75 and 100 μm) (Fig. 5A), the effect of the hydrophilic loop became evident because PS1 ΔE10 neurons had significantly higher LDH release compared with the controls. To directly test that neuronal CCEs were responsible for the increased cell death, we treated PS1 ΔE10 neurons with Quercetin, a potent cell cycle inhibitor (Park et al., 2005; Malik et al., 2008). Although Quercetin treatment only partially reduced cell death in PS1 null neurons, it restored the viability of PS1 ΔE10 neurons to near wild-type levels (Fig. 5B). These results indicate that aberrant neuronal CCEs are indeed responsible for the increased vulnerability in PS1 ΔE10 neurons and also partially contribute to the cell death in the absence of PS1.
Figure 5.

Increased H2O2-induced cell death in PS1 null and PS1 ΔE10 neurons and effects of the cell cycle inhibitor Quercetin. A, E15.5 primary neurons from control (Ctrl), PS1−/−,] and PS1 ΔE10 mice were cultured for 10 DIV and treated with 25, 50, 75, and 100 μm H2O2 for 24 h. Cell death was evaluated by measuring the release of LDH into the culture medium with the value measured from cell lysates set at 100%. B, Percentage of LDH release in the absence (nontreated or NT, filled bars) or presence of 10 μm Quercetin (Qct, open bars). Data represent an average ± SEM of six independent experiments. Statistical analysis was performed using Student's t test. *p < 0.05, **p < 0.01, ***p < 0.001.
The cell culture studies indicate that, as found in vivo, loss of γ-secretase is the major factor rendering the neurons susceptible to cytotoxic stress. However, under high stress (75 and 100 μm H2O2) conditions, both γ-secretase and the hydrophilic loop are required for neuronal integrity. Our data suggest that rescue of neuronal CCE by the PS1 D257A mutant may be responsible for the partial rescue of the neurodegenerative phenotype in PS cDKO; D257A animals compared with PS cDKO mice.
p53 is a downstream effecter of the PS loop domain
The tumor suppressor p53 is a stress-induced protein with a well established role in cell cycle checkpoint control. It is also the most important determinant of neuronal viability (for review, see Miller et al., 2000). Because PS has been shown to modulate p53 expression (Alves da Costa et al., 2002, 2003, 2006), we investigated whether basal p53 expression is altered as a function of PS1 genotypes. Surprisingly, we found that both PS1−/− and PS1 ΔE10 neurons showed prominent expression of p53 protein under nonstimulated conditions (Fig. 6A). The aberrant p53 expression in PS1−/− neurons was rescued by expressing either the wild-type or the D257A mutant PS1 (supplemental Fig. S4A, available at www.jneurosci.org as supplemental material), implicating a γ-secretase-independent effect. This assessment was further reinforced by the inability to induce p53 by γ-secretase inhibitor treatment (supplemental Fig. S4B, available at www.jneurosci.org as supplemental material). Interestingly, p53 was found to localize to the cytoplasm instead of the nucleus (Fig. 6A). Elevated p53 protein in PS1 null and PS1 ΔE10 neurons was also seen by Western blot analysis (Fig. 6B). Coimmunostaining using anti-Cyclin D1 and anti-p53 antibodies showed that the two proteins were colocalized in both PS1−/− and PS1 ΔE10 neurons (Fig. 6C). However, although Cyclin D1 was expressed in both nucleus and cytoplasm, p53 was predominantly cytoplasmic. The γ-secretase independence and the coexistence of neuronal CCEs and p53 suggest that the two events are linked. Indeed, aberrant basal expression of p53 was blocked by treating the cells with Quercetin (Fig. 6C), demonstrating that p53 is downstream of neuronal CCEs. This γ-secretase-independent coactivation of CCEs and p53 was validated in vivo by double immunostaining of mouse brain tissues using anti-Cyclin D1 and p53 antibodies, which revealed that the two proteins can be detected in the same set of cells in PS cDKO and PS1 ΔE10 brains, whereas both proteins were absent in PS cDKO mice rescued with wild-type PS1 or PS1 D257A (supplemental Fig. S5, available at www.jneurosci.org as supplemental material).
Figure 6.
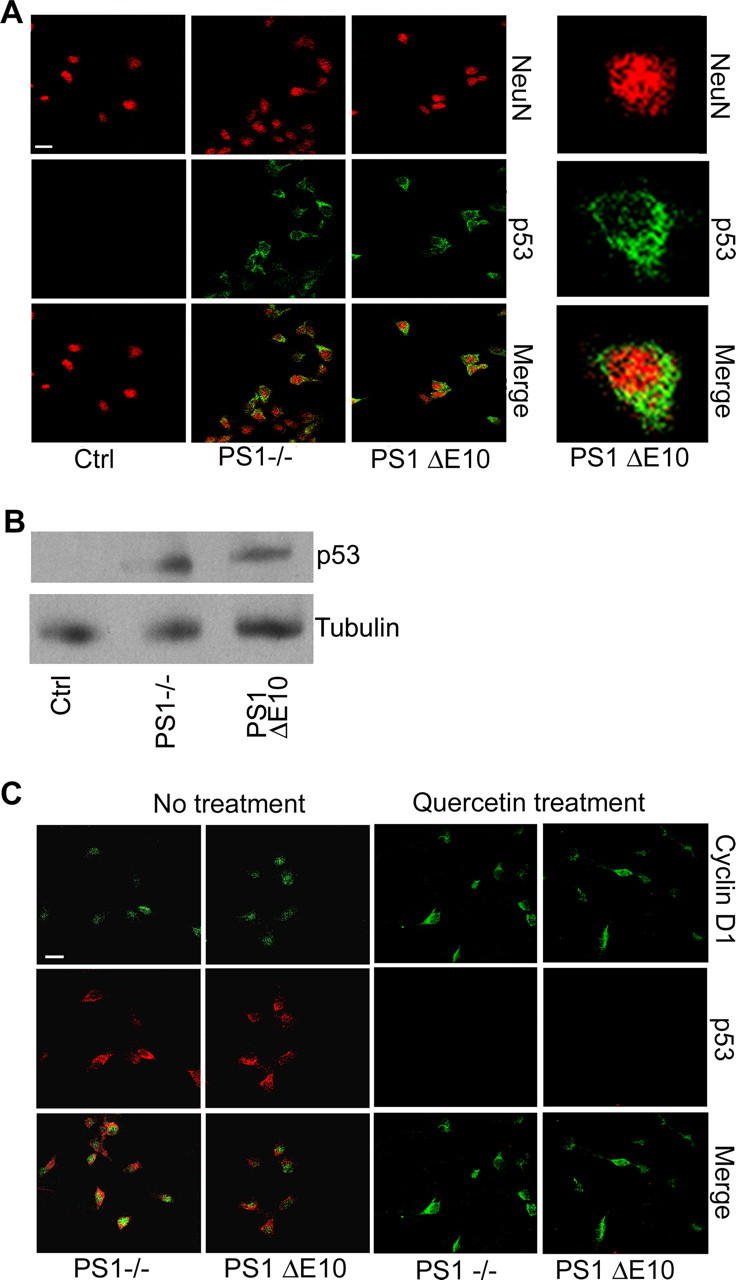
Cytoplasmic p53 accumulation and colocalization with Cyclin D1 in PS1 null and PS1 ΔE10 mutants. A, Primary neurons were stained for NeuN and p53 after 5 d in culture. PS1−/− and PS1 ΔE10 neurons expressed high levels of cytoplasmic p53. Right column is an enlarged view showing cytoplasmic p53 localization in PS1 ΔE10 neurons. NeuN staining marks the nucleus of mature neurons. B, Representative Western blotting of p53 levels in control, PS1 null, and PS1 ΔE10 cultures. γ-Tubulin was used as a loading control. C, Costaining for p53 and Cyclin D1 reveal that they are expressed in the same set of neurons in PS1−/− and PS1 ΔE10 cultures under basal conditions. Aberrant p53 expression was blocked by the cell cycle inhibitor Quercetin. Scale bar, 10 μm.
Because cell death induction by p53 likely requires nuclear localization, we wondered whether the cytoplasmic p53 present in PS1 null and PS1 ΔE10 neurons was capable of translocating to the nucleus under stress conditions. Because p53 is known to be induced by stress, we treated the neurons with cycloheximide, a potent protein synthesis inhibitor, after the H2O2 induction to prevent new p53 synthesis and to allow specific follow up of the cytoplasmic pool of p53. Inhibition of new p53 synthesis was demonstrated by the absence of p53 in control neurons after 60 min of H2O2 treatment (data not shown). Although p53 was exclusively cytoplasmically localized before H2O2 treatment (Fig. 7, 0 min), distinct nuclear staining of p53 could be detected in PS1 null and PS1 ΔE10 neurons as early as 15 min after treatment and lasted through the treatment period of 60 min (Fig. 7). We thus conclude that the cytoplasmic pool of p53 in the absence of PS1 or its hydrophilic loop, and not new protein synthesis, was responsible for its nuclear accumulation under stress conditions. Because nuclear p53 can induce proapoptotic genes and initiate the apoptotic cascade, our results suggest that the cytoplasmic p53, although inactive under resting conditions, contributes to stress-induced cell death in PS1−/− and PS1 ΔE10 neurons by translocating to the nucleus.
Figure 7.

Cytoplasmic p53 in PS1−/− and PS1 ΔE10 neurons translocates to nucleus during oxidative stress. Representative images of NeuN and p53 immunostaining in the presence of cycloheximide, showing that previously synthesized cytoplasmic p53 in PS1−/− and PS1 ΔE10 neurons translocates to the nucleus at 15, 30, and 60 min after treatment with H2O2. Scale bar, 10 μm.
Discussion
Presenilin plays a pivotal role in AD pathogenesis because mutations in PSEN1 and PSEN2 lead to early onset of FAD. The large number (>150 in PSEN1 alone) and widespread distribution of the FAD mutations are indicative of a partial loss of function by the disease-causing mutations, and this interpretation is supported by abundant experimental evidence. Originally discovered as part of the γ-secretase complex essential for APP and Notch processing, PS has been shown to be responsible for the intramembraneous cleavage of an increasing list of type I transmembrane proteins (Kopan and Ilagan, 2004; Wang et al., 2006). In addition to this important γ-secretase activity, which requires the aspartates at the 257 and 385 sites of PS1 (Wolfe et al., 1999), PS has been reported to possess γ-secretase-independent functions, including the regulation of β-catenin/Cyclin D1 (Soriano et al., 2001; Kang et al., 2002), intracellular protein turnover (Esselens et al., 2004; Wilson et al., 2004; Repetto et al., 2007), and endoplasmic reticulum calcium (Tu et al., 2006). In both γ-secretase-dependent and -independent activities, FAD-linked presenilin mutations have been shown to result in partial loss of function (for review, see De Strooper, 2007; Shen and Kelleher, 2007). Accordingly, studying the mechanisms underlying the physiological functions of PS will provide pathogenic insights.
Besides the pathological hallmarks of β-amyloid plaques and neurofibrillary tangles, aberrant activation of cell cycle markers can be found in postmitotic neurons in AD, and these CCEs precede neuronal cell death (Yang et al., 2003). Although cell cycle reentry in postmitotic neurons has been postulated to lead to neurodegeneration in AD, this cause–effect relationship cannot be established in humans. We reported previously that loss of PS1 or reduction of total PS dosage results in Cyclin D1 activation and hyperproliferation in peripheral tissues (Xia et al., 2001; Qyang et al., 2004). Mice with postnatal inactivation of PS develop neurodegeneration (Feng et al., 2004; Saura et al., 2004). These data combined prompted us to hypothesize that PS may have a similar role in modulating neuronal cell cycle and that neuronal CCEs may contribute to the neurodegenerative phenotype seen in PS conditional knock-out mice.
Immunohistochemical staining showed that, indeed, significant activation of cell cycle markers, including Cyclin D1 and Cyclin A, can be detected accompanying neurodegeneration in PS cDKO animals. However, CCEs were also evident in PS1 ΔE10 brains. The absence of neuronal cell loss in these animals argues against a causal role of CCE in neuronal cell death. This assessment is further substantiated by genetic rescue demonstrating that expression of PS1 D257A on PS cDKO background can completely suppress CCE despite significant persistence of neurodegeneration. As such, neuronal viability is primarily determined by PS γ-secretase activity, whereas neuronal CCEs, activated by the loss of the PS1 hydrophilic loop domain, are not obligatory for neurodegeneration. Nevertheless, partial rescue of the cortical volume by PS1 D257A indicates that γ-secretase-independent activity, which has been shown to be preserved in the D257A mutant (Xia et al., 2002), also plays a functional role in neuronal survival.
Previous studies have established a role of the PS1 loop domain in regulating β-catenin stability and downstream Cyclin D1 transcription (Soriano et al., 2001; Kang et al., 2002, 2005). Therefore, activation of Cyclin D1 in PS loss of function and PS1 ΔE10 brains is likely mediated through deregulated β-catenin signaling. Consistent with a partial loss-of-function mechanism, a recent report showed that neurons derived from PS1 M146V FAD knock-in mice exhibit impaired β-catenin/Cyclin D1 signaling pathway and abnormal neuronal cell cycle profiles (Malik et al., 2008). Because expression of Cyclins can be either the trigger of or in response to the cell cycle and are often coactivated, it is possible that Cyclin A is ectopically elevated in response to Cyclin D1 and impaired cell cycle control. It is interesting to note that, although CCE are prominent features in both PS cDKO and PS1 ΔE10 neurons, they show different BrdU profiles. Although a subset of PS cDKO neurons are BrdU positive, the same BrdU injection protocol failed to label any neurons in PS1 ΔE10 brains. These results indicate that additional insults are required for the CCE-positive neurons to undergo active DNA synthesis. Even neurons incorporating BrdU do not succumb to immediate death as revealed by the retention of most of the labeled neurons 7 d after injection. However, approximately half of the BrdU-positive neurons are lost 14 d after injection, and the remaining ones exhibit abnormal morphology and ∼80% of the cells are positive for active caspase-3, suggesting that the BrdU-incorporating neurons are destined to die over a period of time. Although caspase-3 is known to be a cytoplasmic protein, nuclear localization of cleaved and active caspase-3 has been reported in multiple cell death systems, in particular in a rat model of Parkinson's disease (Jeon et al., 1999). The nuclear staining of active caspase-3 in PS cDKO mice suggests that PS deficiency-induced neurodegeneration likely involves activation of nuclear targets of caspase-3. Our results combined support a model whereby CCEs represent an early stage of neuronal impairment. A series of downstream insults is necessary to trigger the CCE-positive neurons to reenter the cell cycle, leading to eventual neuronal cell death. This model is compatible with the fact that CCEs precede neuronal cell loss in human AD (Yang et al., 2003) and that cell cycle markers are activated in the absence of neurodegeneration in APP transgenic mice (Yang et al., 2006).
Consistent with this “multiple insult” hypothesis, our genetic and pharmacological studies using primary neuronal cultures reveal that loss of PS1 does not affect basal neuronal survival. It rather renders the neurons more susceptible to oxidative stress, and multiple PS-mediated pathways are involved in response to stress levels. In particular, at low H2O2 concentration (25 μm), neuronal death is conferred by the loss of γ-secretase. However, both γ-secretase-dependent and -independent activities are necessary to support neuronal survival under high stress conditions (75 and 100 μm H2O2), as demonstrated by the significantly increased LDH release in PS1 ΔE10 neurons. The mechanisms mediating the neuronal viability by the two PS domains are distinct. Although defective PS substrate processing and signaling should be considered as the cause for the neuronal vulnerability in the absence of γ-secretase, neuronal CCE and cytoplasmic p53 accumulation likely trigger γ-secretase-independent neuronal cell death.
p53 immunoreactivity can be detected in degenerating neurons of AD, and its activation has been shown to precede neuronal cell death (Culmsee et al., 2003). PS and other components of the γ-secretase complex have been reported to modulate the expression of p53, and this pathway has been proposed to mediate PS-dependent cell death (Alves da Costa et al., 2002, 2003, 2006; Dunys et al., 2007). Motivated by these findings, we examined p53 levels in our various PS alleles. Surprisingly, we did not see a significant regulation of p53 by γ-secretase. Instead, we uncovered a novel cytoplasmic pool of p53 that is subject to PS1 loop regulation. This pool is inactive under resting conditions. However, it is capable of translocating to the nucleus under stress conditions and mediates neuronal apoptosis. The fact that aberrant p53 expression can be blocked by the cell cycle inhibitor Quercetin places p53 downstream of neuronal CCEs. This γ-secretase-independent regulation of p53 is different from work published by the Checler group (Alves da Costa et al., 2002, 2003, 2006; Checler et al., 2007; Dunys et al., 2007), which documented a requirement for the γ-secretase complex and involvement of APP-mediated transcriptional regulation. Although the reason for this discrepancy is not currently understood, it is important to point out that p53 is a stress-induced protein and is subject to multiple levels of regulation through multiple pathways. Because p53 plays a critical role in neuronal survival, the fact that the PS1 null neurons are more vulnerable compared with that of PS1 ΔE10 despite similar accumulation of the cytoplasmic p53 supports the idea that additional mechanisms, which may involve APP-mediated p53 transcription as reported by the Checler group, contribute to the higher H2O2-induced cell death in PS1-deficient neurons.
Our data combined support a model in which distinct genetic and biochemical pathways govern γ-secretase-dependent and -independent functions of PS, and these two pathways differentially contribute to neuronal vulnerability. Although loss of γ-secretase activity and its associated substrate processing and signaling play a major role in the neurodegenerative process, absence of the hydrophilic loop domain leads to neuronal CCE and cytoplasmic p53 accumulation, likely sensitizing the neurons to exogenous and potentially endogenous insults and leading to stress-induced cell death. This model may have direct implications for AD and other neurodegenerative diseases in general. Postmitotic neurons are exposed to various environmental and intrinsic insults throughout their lifetime. Although each individual insult renders the neurons vulnerable, we propose that the accumulation of multiple damages over a threshold level is required for the neurons to degenerate. It is conceivable that PS deficiency, PS FAD mutations, genetic risk factors such as ApoE4, or aging provides intrinsic insults or increases the sensitivity to environmental insults. Each factor contributes to neuronal vulnerability, the combination of which leads to neurodegeneration and AD pathogenesis.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants AG20670, AG21141, NS40039 (H.Z.), and AG29977 (R.G.P.). V.K.-M. is a Baylor Research Advocates for Student Scientists Scholar and trainee of NIH Training Grant T32 GM08307. We thank Drs. S. Sisodia (University of Chicago, Chicago, IL) and S. Tonegawa (Massachusetts Institute of Technology, Cambridge, MA) for the PS1 floxed mice and the CaMKIIα–Cre transgenic line, respectively, and Dr. Y. Li (Memorial Sloan Kettering Cancer Center, New York, NY) for Compound E. We are grateful to N. Aithmitti for expert technical support and members of the Zheng laboratory for constructive discussions. We acknowledge R. Atkinson and the Mental Retardation and Developmental Disabilities Research Center at Baylor College of Medicine for confocal imaging support.
References
- Alves da Costa C, Paitel E, Mattson MP, Amson R, Telerman A, Ancolio K, Checler F, Mattson MP. Wild-type and mutated presenilins 2 trigger p53-dependent apoptosis and down-regulate presenilin 1 expression in HEK293 human cells and in murine neurons. Proc Natl Acad Sci USA. 2002;99:4043–4048. doi: 10.1073/pnas.062059899. Erratum (2002) 99:9081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves da Costa C, Mattson MP, Ancolio K, Checler F. The C-terminal fragment of presenilin 2 triggers p53-mediated staurosporine-induced apoptosis, a function independent of the presenilinase-derived N-terminal counterpart. J Biol Chem. 2003;278:12064–12069. doi: 10.1074/jbc.M212379200. [DOI] [PubMed] [Google Scholar]
- Alves da Costa C, Sunyach C, Pardossi-Piquard R, Sévalle J, Vincent B, Boyer N, Kawarai T, Girardot N, St George-Hyslop P, Checler F. Presenilin-dependent gamma-secretase-mediated control of p53-associated cell death in Alzheimer's disease. J Neurosci. 2006;26:6377–6385. doi: 10.1523/JNEUROSCI.0651-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci. 2005;25:5446–5454. doi: 10.1523/JNEUROSCI.4637-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baki L, Marambaud P, Efthimiopoulos S, Georgakopoulos A, Wen P, Cui W, Shioi J, Koo E, Ozawa M, Friedrich VL, Jr, Robakis NK. Presenilin-1 binds cytoplasmic epithelial cadherin, inhibits cadherin/p120 association, and regulates stability and function of the cadherin/catenin adhesion complex. Proc Natl Acad Sci U S A. 2001;98:2381–2386. doi: 10.1073/pnas.041603398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Checler F, Sunyach C, Pardossi-Piquard R, Sévalle J, Vincent B, Kawarai T, Girardot N, St George-Hyslop P, da Costa CA. The gamma/epsilon-secretase-derived APP intracellular domain fragments regulate p53. Curr Alzheimer Res. 2007;4:423–426. doi: 10.2174/156720507781788945. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Siewe J, Junker V, Retiounskaia M, Schwarz S, Camandola S, El-Metainy S, Behnke H, Mattson MP, Krieglstein J. Reciprocal inhibition of p53 and nuclear factor-κB transcriptional activities determines cell survival or death in neurons. J Neurosci. 2003;23:8586–8595. doi: 10.1523/JNEUROSCI.23-24-08586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Tarassishin L, Kallhoff V, Peethumnongsin E, Wu L, Li YM, Zheng H. Deletion of presenilin 1 hydrophilic loop sequence leads to impaired γ-secretase activity and exacerbated amyloid pathology. J Neurosci. 2006;26:3845–3854. doi: 10.1523/JNEUROSCI.5384-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B. Loss-of-function presenilin mutations in Alzheimer disease. Talking point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007;8:141–146. doi: 10.1038/sj.embor.7400897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- Donoviel DB, Hadjantonakis AK, Ikeda M, Zheng H, Hyslop PS, Bernstein A. Mice lacking both presenilin genes exhibitearlyembryonic patterning defects. Genes Dev. 1999;13:2801–2810. doi: 10.1101/gad.13.21.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunys J, Kawarai T, Sevalle J, Dolcini V, George-Hyslop PS, Da Costa CA, Checler F. p53-Dependent Aph-1 and Pen-2 anti-apoptotic phenotype requires the integrity of the gamma-secretase complex but is independent of its activity. J Biol Chem. 2007;282:10516–10525. doi: 10.1074/jbc.M611572200. [DOI] [PubMed] [Google Scholar]
- Esselens C, Oorschot V, Baert V, Raemaekers T, Spittaels K, Serneels L, Zheng H, Saftig P, De Strooper B, Klumperman J, Annaert W. Presenilin 1 mediates the turnover of telencephalin in hippocampal neurons via an autophagic degradative pathway. J Cell Biol. 2004;166:1041–1054. doi: 10.1083/jcb.200406060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng R, Rampon C, Tang YP, Shrom D, Jin J, Kyin M, Sopher B, Miller MW, Ware CB, Martin GM, Kim SH, Langdon RB, Sisodia SS, Tsien JZ. Deficient neurogenesis in forebrain-specific presenilin-1 knockout mice is associated with reduced clearance of hippocampal memory traces. Neuron. 2001;32:911–926. doi: 10.1016/s0896-6273(01)00523-2. [DOI] [PubMed] [Google Scholar]
- Feng R, Wang H, Wang J, Shrom D, Zeng X, Tsien JZ. Forebrain degeneration and ventricle enlargement caused by double knockout of Alzheimer's presenilin-1 and presenilin-2. Proc Natl Acad Sci U S A. 2004;101:8162–8167. doi: 10.1073/pnas.0402733101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herreman A, Hartmann D, Annaert W, Saftig P, Craessaerts K, Serneels L, Umans L, Schrijvers V, Checler F, Vanderstichele H, Baekelandt V, Dressel R, Cupers P, Huylebroeck D, Zwijsen A, Van Leuven F, De Strooper B. Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc Natl Acad Sci U S A. 1999;96:11872–11877. doi: 10.1073/pnas.96.21.11872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrup K, Neve R, Ackerman SL, Copani A. Divide and die: cell cycle events as triggers of nerve cell death. J Neurosci. 2004;24:9232–9239. doi: 10.1523/JNEUROSCI.3347-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon BS, Kholodilov NG, Oo TF, Kim SY, Tomaselli KJ, Srinivasan A, Stefanis L, Burke RE. Activation of caspase-3 in developmental models of programmed cell death in neurons of the substantia nigra. J Neurochem. 1999;73:322–333. doi: 10.1046/j.1471-4159.1999.0730322.x. [DOI] [PubMed] [Google Scholar]
- Kang DE, Soriano S, Xia X, Eberhart CG, De Strooper B, Zheng H, Koo EH. Presenilin couples the paired phosphorylation of beta-catenin independent of axin: implications for beta-catenin activation in tumorigenesis. Cell. 2002;110:751–762. doi: 10.1016/s0092-8674(02)00970-4. [DOI] [PubMed] [Google Scholar]
- Kang DE, Yoon IS, Repetto E, Busse T, Yermian N, Ie L, Koo EH. Presenilins mediate phosphatidylinositol 3-kinase/AKT and ERK activation via select signaling receptors. Selectivity of PS2 in platelet-derived growth factor signaling. J Biol Chem. 2005;280:31537–31547. doi: 10.1074/jbc.M500833200. [DOI] [PubMed] [Google Scholar]
- Kopan R, Ilagan MX. Gamma-secretase: proteasome of the membrane? Nat Rev Mol Cell Biol. 2004;5:499–504. doi: 10.1038/nrm1406. [DOI] [PubMed] [Google Scholar]
- Malik B, Currais A, Andres A, Towlson C, Pitsi D, Nunes A, Niblock M, Cooper J, Hortobágyi T, Soriano S. Loss of neuronal cell cycle control as a mechanism of neurodegeneration in the presenilin-1 Alzheimer's disease brain. Cell Cycle. 2008;7:637–646. doi: 10.4161/cc.7.5.5427. [DOI] [PubMed] [Google Scholar]
- Miller FD, Pozniak CD, Walsh GS. Neuronal life and death: an essential role for the p53 family. Cell Death Differ. 2000;7:880–888. doi: 10.1038/sj.cdd.4400736. [DOI] [PubMed] [Google Scholar]
- Park CH, Chang JY, Hahm ER, Park S, Kim HK, Yang CH. Quercetin, a potent inhibitor against beta-catenin/Tcf signaling in SW480 colon cancer cells. Biochem Biophys Res Commun. 2005;328:227–234. doi: 10.1016/j.bbrc.2004.12.151. [DOI] [PubMed] [Google Scholar]
- Qian S, Jiang P, Guan XM, Singh G, Trumbauer ME, Yu H, Chen HY, Van de Ploeg LH, Zheng H. Mutant human presenilin 1 protects presenilin 1 null mouse against embryonic lethality and elevates Abeta1–42/43 expression. Neuron. 1998;20:611–617. doi: 10.1016/s0896-6273(00)80999-x. [DOI] [PubMed] [Google Scholar]
- Qyang Y, Chambers SM, Wang P, Xia X, Chen X, Goodell MA, Zheng H. Myeloproliferative disease in mice with reduced presenilin gene dosage: effect of gamma-secretase blockage. Biochemistry. 2004;43:5352–5359. doi: 10.1021/bi049826u. [DOI] [PubMed] [Google Scholar]
- Redwine JM, Kosofsky B, Jacobs RE, Games D, Reilly JF, Morrison JH, Young WG, Bloom FE. Dentate gyrus volume is reduced before onset of plaque formation in PDAPP mice: a magnetic resonance microscopy and stereologic analysis. Proc Natl Acad Sci U S A. 2003;100:1381–1386. doi: 10.1073/pnas.242746599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repetto E, Yoon IS, Zheng H, Kang DE. Presenilin 1 regulates epidermal growth factor receptor turnover and signaling in the endosomal-lysosomal pathway. J Biol Chem. 2007;282:31504–31516. doi: 10.1074/jbc.M704273200. [DOI] [PubMed] [Google Scholar]
- Saura CA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, Chattarji S, Kelleher RJ, 3rd, Kandel ER, Duff K, Kirkwood A, Shen J. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004;42:23–36. doi: 10.1016/s0896-6273(04)00182-5. [DOI] [PubMed] [Google Scholar]
- Shen J, Kelleher RJ., 3rd The presenilin hypothesis of Alzheimer's disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci U S A. 2007;104:403–409. doi: 10.1073/pnas.0608332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell. 1997;89:629–639. doi: 10.1016/s0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- Sisodia SS, St George-Hyslop PH. gamma-Secretase, Notch, Abeta and Alzheimer's disease: where do the presenilins fit in? Nat Rev Neurosci. 2002;3:281–290. doi: 10.1038/nrn785. [DOI] [PubMed] [Google Scholar]
- Soriano S, Kang DE, Fu M, Pestell R, Chevallier N, Zheng H, Koo EH. Presenilin 1 negatively regulates beta-catenin/T cell factor/lymphoid enhancer factor-1 signaling independently of beta-amyloid precursor protein and Notch processing. J Cell Biol. 2001;152:785–794. doi: 10.1083/jcb.152.4.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarassishin L, Yin YI, Bassit B, Li YM. Processing of Notch and amyloid precursor protein by gamma-secretase is spatially distinct. Proc Natl Acad Sci U S A. 2004;101:17050–17055. doi: 10.1073/pnas.0408007101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996;87:1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease-linked mutations. Cell. 2006;126:981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetrivel KS, Zhang YW, Xu H, Thinakaran G. Pathological and physiological functions of presenilins. Mol Neurodegener. 2006;1:4. doi: 10.1186/1750-1326-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Tang P, Wang P, Boissy RE, Zheng H. Regulation of tyrosinase trafficking and processing by presenilins: partial loss of function by familial Alzheimer's disease mutation. Proc Natl Acad Sci U S A. 2006;103:353–358. doi: 10.1073/pnas.0509822102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CA, Murphy DD, Giasson BI, Zhang B, Trojanowski JQ, Lee VM. Degradative organelles containing mislocalized α- and β-synuclein proliferate in presenilin-1 null neurons. J Cell Biol. 2004;165:335–346. doi: 10.1083/jcb.200403061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtowicz JM, Kee N. BrdU assay for neurogenesis in rodents. Nat Protoc. 2006;1:1399–1405. doi: 10.1038/nprot.2006.224. [DOI] [PubMed] [Google Scholar]
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- Wong PC, Zheng H, Chen H, Becher MW, Sirinathsinghji DJ, Trumbauer ME, Chen HY, Price DL, Van der Ploeg LH, Sisodia SS. Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature. 1997;387:288–292. doi: 10.1038/387288a0. [DOI] [PubMed] [Google Scholar]
- Xia X, Qian S, Soriano S, Wu Y, Fletcher AM, Wang XJ, Koo EH, Wu X, Zheng H. Loss of presenilin 1 is associated with enhanced beta-catenin signaling and skin tumorigenesis. Proc Natl Acad Sci U S A. 2001;98:10863–10868. doi: 10.1073/pnas.191284198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X, Wang P, Sun X, Soriano S, Shum WK, Yamaguchi H, Trumbauer ME, Takashima A, Koo EH, Zheng H. The aspartate-257 of presenilin 1 is indispensable for mouse development and production of beta-amyloid peptides through beta-catenin-independent mechanisms. Proc Natl Acad Sci U S A. 2002;99:8760–8765. doi: 10.1073/pnas.132045399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Mufson EJ, Herrup K. Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer's disease. J Neurosci. 2003;23:2557–2563. doi: 10.1523/JNEUROSCI.23-07-02557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Varvel NH, Lamb BT, Herrup K. Ectopic cell cycle events link human Alzheimer's disease and amyloid precursor protein transgenic mouse models. J Neurosci. 2006;26:775–784. doi: 10.1523/JNEUROSCI.3707-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Saura CA, Choi SY, Sun LD, Yang X, Handler M, Kawarabayashi T, Younkin L, Fedeles B, Wilson MA, Younkin S, Kandel ER, Kirkwood A, Shen J. APP processing and synaptic plasticity in presenilin-1 conditional knockout mice. Neuron. 2001;31:713–726. doi: 10.1016/s0896-6273(01)00417-2. [DOI] [PubMed] [Google Scholar]