

Abstract
The first examples of intramolecular Michael-type reactions of in situ-formed vinylnitroso compounds with carbon nucleophiles are reported. This methodology has been used to prepare a variety of ring systems including [3.2.1]-, [2.2.2]- and [2.2.1]-bridged carbobicyclic compounds, as well as a fused [5.5]-ring compound. Malonate anions have proven to be effective carbon nucleophiles in these conjugate addition reactions, and simple ester potassium enolates have also been successfully employed.
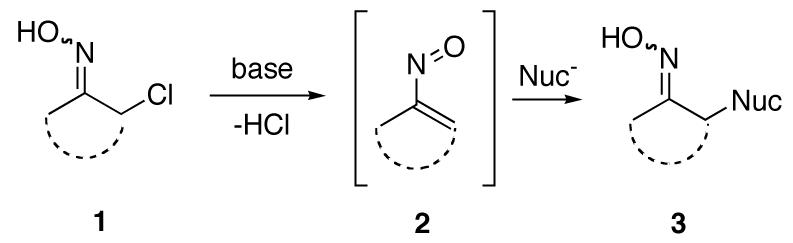
Although vinylnitroso compounds 2 have been known for many years, and are easily generated from cyclic and acyclic α-haloketoximes 1 (Scheme 1), these highly reactive, unstable species have found relatively little use in organic synthesis.1 The primary applications to date of vinylnitroso compounds have been as heterodienes in both inter- and intramolecular [4+2]-cycloadditions with olefins to produce 5,6-dihydro-1,2-oxazines.1,2 However, there are widely scattered examples in the literature of intermolecular conjugate additions of a variety of hetero and carbon nucleophiles to vinylnitroso compounds in a Michael-type process to produce adducts 3 in good yields. Among the hetero nucleophiles which have been used are amines, alcohols, azide, phosphines, and various thio compounds.1 Carbon nucleophiles which have been added to vinylnitroso compounds include inter alia electron rich arenes and heteroarenes,3 malonates,4 1,3-diketones, β-ketoesters, Grignard reagents,4 acetylides,4,5 and simple ketone enolates.6 Thus, it has been documented that these compounds can act as enolonium ion equivalents.7 In this communication we describe the first examples of intramolecular Michael-type conjugate additions of carbon nucleophiles to vinylnitroso compounds.8
Scheme 1.
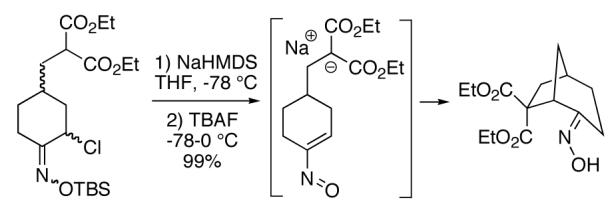
Our plan for the preparation of the requisite substrates to test the feasibility of this process was to rely on two key reactions developed in these laboratories, namely ring closing metathesis of vinyl chlorides9 and the regioselective conversion of vinyl chlorides to α-chloroketones with sodium hypochlorite.10,11 Thus, easily prepared chlorodiene 4 (see Supporting Information) was exposed to the second generation Grubbs ruthenium metathesis catalyst in hot toluene, leading to the cyclized vinyl chloride malonate 5 in high yield (Scheme 2). Subsequent treatment of this intermediate with 10% aqueous sodium hypochlorite in a 5:2 mixture of acetone/glacial acetic acid at 0 °C for 30 minutes afforded α-chloroketone 6 as a ∼1:1 mixture of diastereomers. It was found after some experimentation that the optimal way to generate the vinylnitroso species was from the corresponding Osilyloxime as developed by Denmark and coworkers.2 Therefore, α-chloroketone 6 was first transformed into oxime derivative 7 with commercially available O-TBS hydroxylamine. Compound 7 is a complex mixture of diastereomers, including oxime geometric isomers.
Scheme 2.
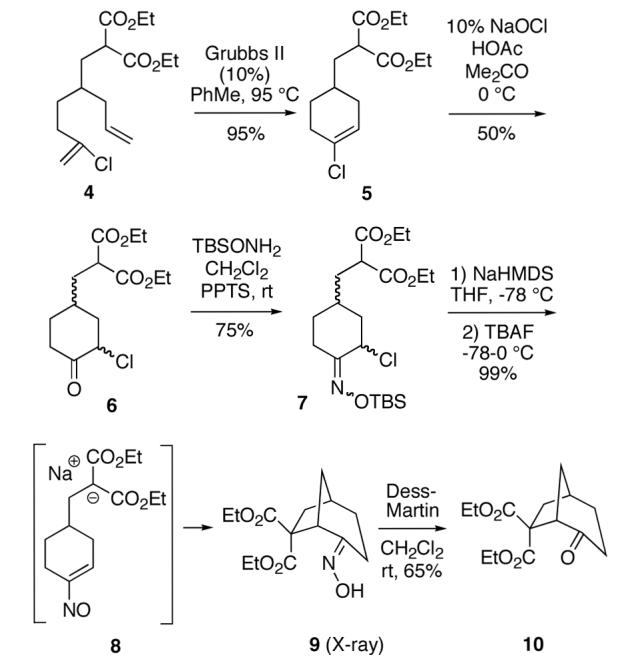
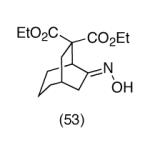
Since vinylnitroso compounds are so unstable, we decided it would be prudent to first form the tethered nucleophile prior to generating this reactive species. Thus, for the pivotal cyclization step it was best to initially deprotonate the malonate 7 with sodium hexamethyldisilazide in THF at low temperature, followed by addition of tetrabutylammonium fluoride,2 leading to formation of the desired [3.2.1]-bicyclic oxime diester 9 in nearly quantitative yield. Other bases such as sodium hydride or LDA gave poorer yields of cyclization product. Compound 9 is a single stereoisomer with the (E)-oxime configuration, as confirmed by X-ray analysis. We believe this cyclization occurs via the transient vinylnitroso intermediate 8. It should be noted that all attempts to directly cyclize chloro O-TBS oximes like 7, as well as the corresponding α-chloroketones 6, to the corresponding bridged systems by base treatment alone gave no reaction, thereby lending support to the intermediacy of a vinylnitroso compound in the cyclization event.
Although the oxime functionality in compounds like 9 can potentially be used in a variety of ways (eg Beckmann rearrangement, reduction, etc), one useful transformation is to produce the corresponding ketone. The conversion of 9 to 10 could be conveniently effected using Dess-Martin periodinane.12
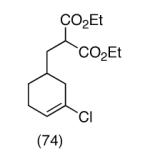
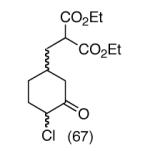
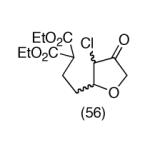
This strategy has been extended to prepare other types of ring systems as outlined in Table 1. We have previously reported that the vinyl chloride metathesis methodology is successful in forming 5-, 6- and 7-membered rings.9 Thus, the cyclopentenyl system shown in entry 1 was processed via the chemistry described in Scheme 2 to afford the corresponding bicyclo[2.2.1]-oxime. Similarly, the cyloheptenyl system (entry 2) could be used to form a bicyclo[3.2.2]-oxime ring system. A regioisomer of the vinylnitroso compound 8 was generated as shown in entry 3 to yield a [2.2.2]-bicyclic oxime. In addition we have found that fused ring systems can be produced in good yield by this strategy, as exemplified by the [5.5]-compound in entry 4. In all cases except for the cycloheptenyl-derived product, the oximes proved to be single geometric isomers, although the stereochemistry has not been definitively established.
Table 1.
| # | chlorodiene | metathesis product (yield, %) |
α-chloro- ketone (yield, %) |
chloro-O- TBS-oxime (yield, %) |
cyclization product (yield, %) |
|---|---|---|---|---|---|
| 1 |  |
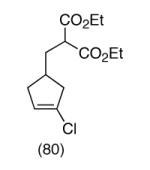 |
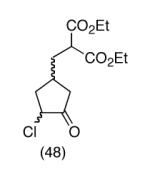 |
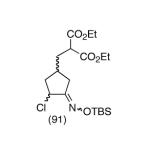 |
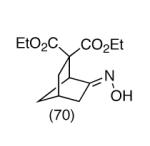 |
| 2 | 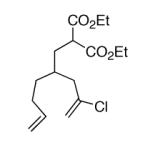 |
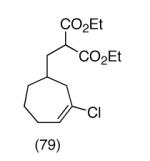 |
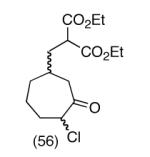 |
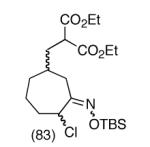 |
 |
| 3 | 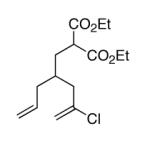 |
 |
 |
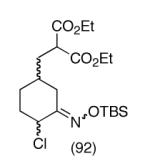 |
 |
| 4 | 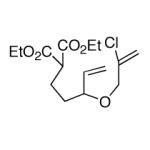 |
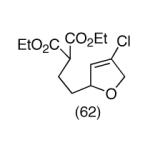 |
 |
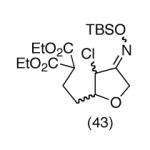 |
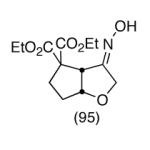 |
Reactions were conducted using small modifications of methodology described in Scheme 2. Detailed experimental procedures can be found in the Supporting Information.
Yields are unoptimized.
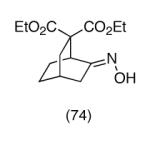
Finally, we have briefly explored the feasibility of employing carbon nucleophiles other than malonates in these cyclizations. Thus, monoester O-TBS oxime 11 was first enolized with potassium hexamethyldisilazide, followed by treatment with TBAF, to give a mixture of three [2.2.1]bicyclic oxime esters 12, 13 and 14 (∼8:7:10 ratio) in high total yield (Scheme 3). One oxime geometric isomer of the anti ester (12 or 13) can be isolated in pure form by chromatography, but 14 and the other anti isomer (12 or 13) were obtained as an inseparable mixture.
Scheme 3.
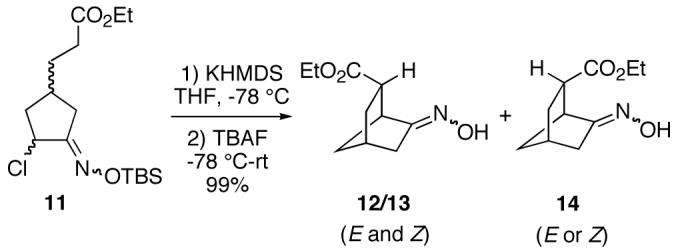
In conclusion, we have demonstrated that intramolecular conjugate additions of carbon nucleophiles to in situ-generated vinylnitroso compounds provides a novel approach to a wide array of highly functionalized bridged and fused ring systems. Work is currently underway on extending this methodology to formation of other types of ring systems. We are also actively investigating the use of a broader range of carbanions, as well as hetero-nucleophiles, in these cyclizations, and intend to apply the chemistry to synthesis of complex molecules.
Supplementary Material
Acknowledgment
We are grateful to the National Institutes of Health (CA-034303) for financial support of this research. We also thank Dr. Hemant Yennawar (Penn State Small Molecule X-Ray Crystallographic Facility) for the crystal structure determination of compound 9.
Footnotes
Supporting Information Available: Experimental procedures for the preparation of new compounds including copies of proton and carbon NMR spectra, as well as X-ray data for compound 9. This material is available free of charge on the Internet at http://pubs.acs.org.
References
- 1.For reviews of the chemistry of vinylnitroso compounds and lead references see: Gilchrist TL. Chem. Soc. Rev. 1983;11:53.Lyapkalo IM, Ioffe SL. Russ. Chem. Rev. 1998;67:467.
- 2(a).Denmark SE, Dappen MS. J. Org. Chem. 1984;49:798. [Google Scholar]; (b) Denmark SE, Dappen MS, Sternberg JA. J. Org. Chem. 1984;49:4741. [Google Scholar]; (c) Denmark SE, Dappen MS, Sear NL, Jacobs RT. J. Am. Chem. Soc. 1990;112:3466. [Google Scholar]
- 3.Plate R, Hermkens PHH, Smits JMM, Nivard RJF, Ottenheijm HCJ. J. Org. Chem. 1987;52:1047. [Google Scholar]
- 4.Ohno M, Torimitsu S, Naruse N, Okamoto M, Sakai I. Bull. Chem. Soc. Jpn. 1966;39:1129. [Google Scholar]
- 5.Trost BM, Barrett D. Tetrahedron. 1996;52:6903. [Google Scholar]
- 6.Oppolzer W, Battig K, Hudlicky T. Tetrahedron. 1981;37:4359. [Google Scholar]
- 7.For some examples of enolonium ion equivalents see: Fuchs PL. J. Org. Chem. 1976;41:2935.Wender PA, Erhardt JM, Letendre LJ. J. Am. Chem. Soc. 1981;103:2114. and references cited.
- 8.For a general review of intramolecular Michael reactions see: Little RD, Masjedizadeh MR, Wallquist O, McLoughlin JI. Org. React. 1995;47:315.
- 9(a).Chao W, Weinreb SM. Org. Lett. 2003;5:2505. doi: 10.1021/ol034775z. [DOI] [PubMed] [Google Scholar]; (b) Chao W, Meketa ML, Weinreb SM. Synthesis. 2004:2058. [Google Scholar]
- 10.VanBrunt MP, Ambenge RO, Weinreb SM. J. Org. Chem. 2003;68:3323. doi: 10.1021/jo020739m. [DOI] [PubMed] [Google Scholar]
- 11.Meketa ML, Mahajan YR, Weinreb SM. Tetrahedron Lett. 2005;46:4749. [Google Scholar]
- 12.Chaudhari SS, Akamanchi KG. Synthesis. 1999:760. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.