

Abstract
During the past decade, the immune and endocrine systems have been discovered to interact in controlling physiologic processes as diverse as cell growth and differentiation, metabolism, and even human and animal behavior. The interaction between these two major physiological systems is a bi-directional process. While it has been well documented that hormones, including prolactin (PRL), growth hormone (GH), insulin-like growth factor-I (IGF-I), and thyroid-stimulating hormone (TSH), regulate a variety of immune events, a great deal of data have accumulated supporting the notion that cytokines from the innate immune system also affect the neuroendocrine system. Communication between these two systems coordinates processes that are necessary to maintain homeostasis. Proinflammatory cytokines often act as negative regulatory signals that temper the action of hormones and growth factors. This system of ‘checks and balances’ is an active, ongoing process, even in healthy individuals. Dysregulation of this process has been implicated as a potential pathogenic factor in the development of co-morbid conditions associated with several chronic inflammatory diseases, including type 2 diabetes, cardiovascular disease, cerebrovascular disease, inflammatory bowel disease, rheumatoid arthritis, major depression and even normal aging. Over the past decade, research in our laboratory has focused on the ability of the major proinflammatory cytokines, tumor necrosis factor (TNF)α and interleukin (IL)-1β, to induce a state of IGF resistance. This review will highlight these and other new findings by explaining how proinflammatory cytokines induce resistance to the major growth factor, insulin-like growth factor-I (IGF-I). We also highlight that IGF-I can induce resistance or reduce sensitivity to brain TNFα and discuss how TNFα, IL-1β and IGF-I interact to regulate several aspects of behavior and cognition.
Keywords: IGF-I, TNF-α, IL-1β, innate immune system, resistance, receptor crosstalk, sickness behavior
1. Introduction
The immune and endocrine systems have historically been studied independently of one another, with immunologists attending immunology meetings and endocrinologists talking only to other endocrinologists. Stunning advances have been made in both of these disciplines during the past 50 years. These discoveries have revealed that health and wellness is maintained only when these two great systems interact with one another in an appropriate manner. Changes in one system, such as activation of the innate immune system, cause reciprocal changes in the neuroendocrine system. Accumulating data now provide overwhelming evidence that these two ‘sensory’ systems continuously interact to regulate diverse physiological processes ranging from cell growth and differentiation to brain-controlled symptoms of sickness that occur following infection. Indeed, the four cardinal characteristics of inflammation – heat, pain, redness and swelling – have long been known to be regulated by anti-inflammatory steroids. Cytokine and hormone secretion is a dynamic process that results in an ever-changing cellular microenvironment. Cells are equipped with both hormone and pathogen-associated molecular pattern (PAMP) receptors that interpret this intricate, interactive network of signals in order to respond in a physiologically appropriate fashion. Within a single cell, this integration is made possible by a process called intracellular crosstalk in which the cellular signaling machinery is shared among distinct ligand-activated pathways. Intracellular enzymes and substrates are often affected by entirely unrelated ligands, leading to signal amplification. Alternatively, activation of one pathway may interfere with signal transduction of a second, or subsequent, pathway. This type of receptor crosstalk may result in cellular resistance to a given ligand. Collectively, this means that cellular responses may vary tremendously depending on many factors, such as the relative ligand concentration, whether the ligand originates from the immune or endocrine system, the number of immune receptors on the cell and signaling kinetics following activation of each receptor.
Both the immune and endocrine systems constantly survey the body and its environment to coordinate adaptive mechanisms that ensure homeostasis is maintained. This ability to adapt is necessary for optimal health and well being because it allows the body to fend off pathogenic insults, divert metabolic resources during times of nutritional insufficiency and cope with physical and psychological stressors. The immune and endocrine systems regulate adaptive processes in a bi-directional manner, and efficient regulation is accomplished by the secretion and action of cytokines [1, 2] and hormones [2, 3]. Moreover, homeostasis requires both positive interactions, such as potentiation of IL-4 induced upregulation of the type II IgE receptor by IGF-I in human B-cells [4] and negative interactions, such as IGF-I dependent down-regulation of interferon-γ receptor 2 chain on human T lymphocytes [5].
It has been well documented that immune processes are modulated by the endocrine system, as our group has demonstrated for GH, IGF-I and PRL [6]. In fact, glucocorticoid hormones have potent anti-inflammatory properties on cells of the innate immune system. More recently, the concept of proinflammatory cytokine-induced hormone resistance has been thrust into the limelight because of the implication of this interaction in human disorders ranging from type 2 diabetes to clinical depression. The most studied example is TNF-α-induced insulin resistance, which was first described in the early 1990’s. However, it is now known that proinflammatory cytokines also induce a state of resistance in other important hormone systems, including glucocorticoids [7, 8], GH [9] and IGF-I [10]. Proinflammatory cytokines suppress proliferation and induce apoptosis in a variety of cancer cells [11, 12]. In skeletal muscle, proinflammatory cytokines induce wasting [13]. In the brain, they act to induce behavioral and motivational changes associated with sickness [14] and depression [15]. A common thread in all of these examples is the induction of IGF resistance by proinflammatory cytokines.
Hormone resistance induced by proinflammatory cytokines may be part of the body’s global negative regulatory mechanism to dampen hormone action. Another possibility is that hormone resistance has evolved as a means to regulate the body’s utilization of limited resources. This adaptive process may have helped to manage and sustain the three most rudimentary processes: (1) growth and energy storage, (2) reproduction and (3) survival, including recovery from injury and infection. One action of proinflammatory cytokines is to restrict growth and energy storage, diverting resources for survival of the organism. Redistribution of energy resources may have partially been achieved via inhibition of the anabolic effects of IGF-I by proinflammatory cytokines. But, in modern, industrialized societies where medications are abundant and food is plentiful, proinflammatory cytokine-induced hormone resistance may have become maladaptive. Many chronic human diseases, and even aging, are associated with an elevation of proinflammatory cytokine levels. Chronically elevated proinflammatory cytokines may act to stifle the beneficial actions that hormones and growth factors have on health and well being. This review will discuss the underlying mechanisms of proinflammatory cytokine-induced hormone resistance with an emphasis on the interaction between two major proinflammatory cytokines, TNF-α and IL-1β, and an important growth factor, IGF-I.
2. The IGF System and Mechanisms of Control: Expression, Sensitivity and Resistance
The IGFs display pleiotropic properties, including the ability to promote cellular proliferation, differentiation and metabolism/hypertrophy (e.g., nutrient transport, energy storage, gene transcription and protein synthesis) [16]. These actions are common to both IGF-I and IGF-II and are all mediated by either the type 1 IGF receptor (IGF-1R) or the insulin receptor (InsR), which have similar structure but differing ligand preferences (Table 1). Both receptors are ligand-activated tyrosine kinases and are so closely related that hybrid receptors naturally occur with full ligand-activated signal transduction capability. From a practical perspective, this means that IGF-I, IGF-II and insulin have the same effects on cells with differing sensitivities that are proportionate to their relative affinities for the receptors and dependent on the abundance of the type of receptor that is present on the target cell [16]. IGF/insulin receptor cross-reactivity is widely accepted and exploited for the formulation of in vitro cell growth supplements, which generally contain very high concentrations of the less-expensive insulin in lieu of IGF-I. With the ubiquitous presence of receptors and ligands, it is not surprising that the IGF system, in combination with growth hormone, accounts for 83% of postnatal body growth [17]. In fact, a recent study identified an allelic variant of IGF-I as a major determinant of body size in dogs. These workers found a single nucleotide polymorphism in the IGF-I gene that is common to all small breeds of dogs while absent in giant breeds [18].
Table 1.
Members of the IGF System
| Ligands | F.W. | Structure | Primary Role | Location : source |
|---|---|---|---|---|
| IGF-I and IGF-II (70a.a. and 67a.a.) |
7.6 | Monomeric active forms | Drive metabolism, proliferation, survival, differentiation | All body fluids : all tissues |
| Insulin | 5.8 | Monomeric active form | Drives nutrient metabolism for storage | Circulation / pancreas |
| Receptors | Relative Affinities | Location | ||
| IGF-1R | 304 | Heterotetramer 2 α / 2 β | IGF-I > IGF-II >> Insulin 1 : 2 : 200 | One or both present on most, if not all, cells of the body |
| InsR (a/b isoforms) | 307 | Heterotetramer 2 α / 2 β | Insulin ≥ IGF-II > IGF-I 1 : 2 : 30 | |
| Natural Antagonists | Relative Affinities | Location : source | ||
| IGFBPs (1–6) | 25–30 | Monomers active | IGF-II ≥ IGF-I ≠ insulin | Body fluids, cell surfaces : all tissues |
| IGF-2R | 270 | Dimer | IGF-II >>>> IGF-I ≠ insulin | |
| sIGF-2R | 250 | Dimer | IGF-II >>>> IGF-I ≠ insulin | |
 | ||||
IGF activity is controlled by a variety of mechanisms. Usually 5,000–20,000 hormone receptors are present on cells. Combined concentrations of the two IGF ligands in circulation are generally above 25 nM (~ 200 ng/ml) for mammals, which is much higher than that of insulin. Humans generally have higher circulating concentrations of IGFs. For example, 25–34-year-old adult humans have ~250 ng/ml (~33 nM) IGF-I in blood, which declines > 30% over the next 2 decades [166]. IGF-II in the serum of young (28-yr-old) adult humans is ~1200 ng/ml (~160 nM)[167]. These levels are much greater than the Kd (<1 nM) of the IGF-1R. Without additional modes of regulation, cells would be continuously activated by the IGFs. In contrast, insulin levels are below the Kd of its receptor but insulin concentrations rise toward the receptor Kd after a meal (example; insulin is < 0.1 nM = 0.58 ng/ml during fasting but rise to levels near the receptor Kd with feeding [168]). Thus insulin activity is primarily controlled extracellularly by changes in ligand concentration, however, control of IGF-1R activation is maintained by other specific extracellular proteins; 1) A signal deficient decoy receptor, the IGF-2R, which binds and prevents IGF-II from activating the IGF-1R. and 2) Six high-affinity, and several low affinity, IGFBPs that bind both ligands. Together, these binding proteins serve as direct extracellular negative regulators that maintain circulating ‘free’ IGF-I levels at or below 1 nM. At this concentration, small changes in free IGF result in proportionate changes in IGF-1R association and intracellular signaling. Each component within this system is independently regulated at the transcriptional level, which is particularly relevant to IGF-I and the IGFBPs whose extracellular levels are finely adjusted by a variety of inputs. An additional level of control exists for both the IGFs and insulin within the cell, where signaling from two or more factors can interact in either agonistic or antagonistic modes.
Sizes are approximate and based on amino acid composition of the mature protein (Swiss-Prot; ExPASy). Human and mouse IGF-I have 70 amino acids in the mature peptide; with only 4 non-conserved residues. IGF-I and IGF-II have high (>90 %) sequence conservation between the ligands of various species, human IGF-I and IGF-II are used for most studies since they react with the receptors of other species
It should be pointed out that, until rather recently, it was believed that supra-physiologic levels of IGFs did not have any damaging biological consequences. However, based on data initially generated in C. elegans and eventually expanded to mammals, it was discovered that circulating IGF-I concentrations are inversely related to life span [19]. IGF also increases cell proliferation, and increased IGF levels have been positively correlated with tumor progression [20]. FOXO, a member of the forkhead family of transcription factors, appears to be an important molecular convergence point for IGF-I signaling. FOXO inhibits cellular proliferation and is associated with increased life span, and inactivation of this transcription factor by IGF promotes cell proliferation. FOXO-deficient mice experience hematopoietic abnormalities characterized by increased cell cycling and apoptosis [21]. Further, new studies in C. elegans indicate that the extended life-span associated with calorie restriction is mediated by pha-4, which is an orthologue of the human FOXA family of transcription factors [22]. The paradox that IGF-I is critical for growth and development and yet inversely related to life span is further perpetuated by recent studies demonstrating beneficial effects of IGF-I in the treatment of behavioral characteristics of depression [23, 24]. In these studies, central administration of IGF-I [23] or IGF-I binding protein inhibitors [24] displayed anti-depressant-like properties. Another example of the complexity of IGF-I physiology is demonstrated in the Ames dwarf mice. These mice have a profound reduction in circulating IGF-I levels that is associated with extended life span and maintenance of cognitive function until late in life. Unexpectedly, IGF-I expression in the brain is actually elevated in these mice, indicating a potentially important beneficial role for IGF-I expression in the central nervous system [25]. It is possible that the elevated levels of IGF-I that are correlated with shortened life span are actually a byproduct of compensation for an environment that is characterized by IGF-I resistance, much like elevated insulin concentrations that often occur in type 2 diabetic patients who display insulin resistance. At a minimum, these disparate relationships exemplify the need to learn more about IGF-I physiology and the importance of physiological mechanisms that precisely control IGF-I biological activity.
IGF activity is modulated by three distinct modes. (1) Control of ligand expression: Secretion of IGF-I and thus serum levels are sensitive to nutritional and endocrine control. Numerous studies have made it very clear that IGF-I is present in high concentrations in body fluids (≥ 100 ng/ml in serum) and that its concentrations are positively related to growth rate [16]. However, the local concentration of IGF-I, rather than circulating IGF-I (which is mostly derived from the liver), is the primary causative factor for growth. IGF-I administration is now a frequent tool used to enhance growth rate of children with short stature, much as insulin is used to control type 1 and 2 diabetes [26], whereas antisense RNA administration is a promising cancer therapy by targeted diminution of IGF-I expression [27].
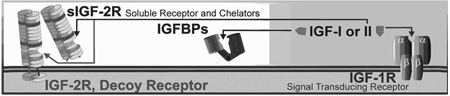
2) Extracellular control of IGF sensitivity: Unique to IGF-I and IGF-II, but absent for insulin, is a second mode of regulating IGF activity. A group of high affinity IGF binding proteins (IGFBPs) are present in extracellular fluids, and they bind both IGF-I and IGF-II (Table 1). Additionally, a decoy receptor, the type 2 IGF receptor (IGF-2R), is present on cell surfaces and in the circulation that chelates only IGF-II (Table 1, inset B). Chelation of IGF by either the IGFBPs or the IGF-2R prevents their association with the IGF-1R. These chelators are saturable, which means that their inhibitory ability can be overcome by additional ligand. This would be a pure case of chelator-induced hormone insensitivity; wherein maximal responsiveness of the cell is unaltered, but half-maximal ligand concentration is increased (see Fig. 6). The IGFBPs are responsible for maintaining a large circulating pool of IGF-I and IGF-II with less than 5% ‘free/active’ hormone [28]. Thus, approximately 20-fold greater total hormone levels are needed to cause the same biological action than would be required in the absence of the IGFBPs. Soluble IGF-1Rs, IGFBPs and competitive IGF-1R antagonists that induce IGF insensitivity are potential pharmaceutical treatments to depress IGF activity with particular relevance as a treatment for IGF-dependent tumors [28]. The main limitations of an extracellular pharmacological approach to diminish IGF activity are the lack of specificity and reversibility. Such an approach is problematical because treatments that depress IGF sensitivity block all of the pleiotropic actions of IGFs and the actions of IGFs can be overcome by additional ligand production by most tissues in the body.
Fig. 6. Interaction between the immune and IGF systems.

There is an important negative feedback between the immune and IGF systems. Proinflammatory cytokines decrease IGF sensitivity by enhancing IGFBP production and induce IGF resistance by decreasing signaling through the IRS → Akt pathway (left chart inset). In contrast, there is considerable evidence that IGFs increase cell survival by blocking caspase induction. IGFs depress proinflammatory cytokine signaling by increasing IL-10 secretion and expanding evidence suggests that IGFs directly depress proinflammatory cytokine signaling via JNK and NF-κB pathways (right chart inset). An appropriate balance between the immune and IGF systems is necessary for normal growth, development and quality of life.
3) IGF resistance is an important phenomenon that the body uses to regulate specific actions of IGFs. Unlike decreased sensitivity, hormone resistance is characterized by a diminished maximal response to the ligand (see Fig. 6), which may or may not be accompanied by a change in sensitivity. With IGF’s ubiquitous presence, differential expression of the receptor is a natural means for targeting IGF activity. For instance, hepatocytes and adipocytes have very few IGF-1Rs (IGF resistant), but abundant InsRs, and these cells are highly responsive to insulin. In contrast, skeletal muscle myoblasts have few InsRs (insulin resistance) but express abundant IGF-1Rs, and myoblasts differentiate into mature into myofibers with both receptors, albeit with a greater number of InsRs [29]. In the case of muscle development, responsiveness changes from predominantly IGF to predominantly insulin in a differentiation-specific manner. Clearly, receptor type and receptor number determine normal cellular responsiveness to members of the IGF family, whereas ligand concentration determines extent of the response.
Independent of receptor expression, in vitro model systems show that high extracellular levels of glucose [30] or alcohol [31, 32] induce IGF resistance in muscle, the latter probably contributing to alcoholic myopathy. Rodent models extend this phenomenon to show that skeletal muscle of type 2 diabetic mice with insulin resistance are also IGF resistant [33, 34], attesting to the common intracellular signaling pathways shared by the IGFs and insulin. Similarly, obese Zucker rats have both insulin and IGF resistant muscle [35]. Obese humans elicit signs of IGF resistance that are independent of type 2 diabetic insulin resistance [35], and IGF resistance is also found in human patients with Turner’s syndrome [36]. Turner’s syndrome is a condition found only in women when one of the two X chromosomes is missing, leading to deficiency in height and ovarian development. IGF resistance may also result from antibodies directed toward the IGF-1R [37, 38]. Although not fully understood, it is clear that IGF resistance plays a role in growth retardation and metabolic disorders and that resistance can be caused by a variety of factors: receptor distribution, diabetes, obesity, nutrient imbalances, IGF-1R mutation, IGF-1R antibodies or post-receptor signaling deficits. Several lines of investigation are aimed at manipulating IGF resistance for cancer therapy, including development of antibodies against the IGF-1R that prevent ligand binding [28], the use of antisense RNA for targeted diminution of IGF-1R expression [27] and the development of pharmaceutical agents that specifically target intracellular signaling pathways initiated by the activated IGF-1R [20, 28].
Temporary IGF resistance can result from intracellular receptor crosstalk as a means for physiological regulation of IGF activity. Proinflammatory cytokine-induced IGF resistance is the main theme of this review. Receptor crosstalk provides an exquisite means for the cell to integrate and regulate specific signaling pathways. The two major pathways that are activated by the IGF-1R are the MAPK (specifically, ERK-1/2) and PI3-K pathways (Figure 1). Each requires the assembly of different receptor-associated scaffolds. Although a generalization, many cell types require IGF-induced ERK-1/2 activation for cellular proliferation because of its ability to drive the cyclin loop [39]. The ERK-1/2 pathway has different intermediates that play the same role in various cell types, but overall they are fairly linear [40]. In contrast, the PI3-K pathway quickly diverges, with Akt as the major branching point. Various downstream components of the PI3-K pathway are associated with enhancing specific cellular events, including metabolism, transcription, hypertrophy and differentiation. Although seemingly distinct, the pathways can interact. For example, activation of PI3-K followed by Akt activation can enhance cell proliferation by activating the cyclin loop. Similarly, Akt activation may be needed for cell proliferation due to its survival-promoting property: i.e., cells must survive to divide. Alternatively, cells require enhanced transcription for differentiation. This interaction links the pathways that are involved in the transition from transcription to differentiation. Similarly, all cells increase in size prior to cell division, thereby linking hypertrophy to proliferation. IGF resistance could result from interference with any of the various steps downstream of IGF-1R activation, and the biological outcome would depend on where the interaction occurs. It is therefore possible for pharmaceutical-induced IGF resistance to be used to target specific aspects of IGF activity. As detailed in the sections below, proinflammatory cytokine-induced IGF resistance is a highly developed natural process that is being exploited therapeutically. Cancer cell proliferation can be depressed by proinflammatory cytokines; an effect that is mediated by receptor crosstalk that leads to intracellularly-mediated IGF resistance. Skeletal muscle differentiation and hypertrophy can be controlled by proinflammatory cytokine-induced IGF resistance, and proinflammatory cytokine-induced cognitive and behavioral effects are inhibited by exogenous IGF-I, suggesting involvement of IGF-induced proinflammatory cytokine resistance in the brain. To understand the interactions between the IGF and inflammatory systems, it is necessary to begin to define the intracellular mechanisms of action of proinflammatory cytokines.
Fig. 1. Intracellular signaling pathways for IGF.

Upon binding IGF, the IGF-1R recruits scaffold proteins that direct the intracellular signaling down two major IGF-responsive pathways. Acting via Grb2 and SOS recruitment, IGF enhances cell proliferation by activation of the loop of cyclins and their cyclin-dependent kinases (left). Alternatively, recruitment of PI3-K via IRS proteins activates Akt (formally known as protein kinase B), which is a master switch involved in survival, hypertrophy, differentiation and transcriptional activation (right). These two pathways are not independent, as illustrated by the ability of IGF to enhance cell proliferation via Akt - TSC - p27 - CDK or Akt – forkhead - cyclin. Presumably, all these branches could be activated in most types of cells, but the requirement of any given pathway for a respective physiological response depends on what primary path is limiting. For instance, IGF-dependent Erk activation is limiting for myoblast proliferation [89, 165], but IGF-dependent PI3-K activation is the restrictive critical pathway for breast tumor cell proliferation [85].
3. Proinflammatory Cytokine Signaling
TNFα and IL-1β have been shown to induce IGF resistance (Table 2) by diminishing downstream signaling in myoblasts [41–43] and breast tumor epithelial cells [10]. Closer examination of the signaling pathways for both cytokines reveals that they share several signaling components but also possess distinct differences. For example, TNFα receptor associating factor (TRAF) serves as an adaptor protein for both TNF-R1 and IL-1R1 [44, 45]. Consistent with this function, TRAF regulates cell survival, differentiation and proliferation by the activation of downstream effectors, including NF-κB, and induction of the MAPKs, the p38 stress-activated protein kinase (SAPK) and c-Jun N terminal kinase (JNK; Fig. 2) [46–48]. Recent results show that the common downstream kinase JNK plays an important role in mediating TNFα- and IL-1β-induced hormone resistance [49] and consequently acts as potential therapeutic target for several inflammatory disorders. Signal transduction pathways for TNF-R1 and IL-1R1 also share other signaling components, such as induction of ceramide [50]. Ceramide is produced following acid sphingomyelinase (A-SMase) or neutral (N-SMase) activation (Fig. 2, right) and serves as a second messenger for both receptors, [51, 52], although the mechanism by which TNFα activates N-SMase is different than that for IL-1. These results are supported by findings which show that ceramide, its analogs and exogenous cell permeable SMases mimic the biological activities of both cytokines [51, 53, 54]. Consistent with these data, results from our laboratory show that C2-ceramide and exogenous N-SMase mimic cytokine inhibition of IGF-I-induced protein synthesis and myogenic factor expression in myoblasts [43].
Table 2.
Proinflammatory Cytokines Implicated in IGF-Resistance
| Ligands | (Synonyms) | F.W.1 | Structure | |
|---|---|---|---|---|
| TNFα | (Cachectin) | 17.3 | Trimer active form | |
| IL-1β | 17.4 | Monomeric active form | ||
| Receptors | Notes | |||
| TNF-R1 | (p55/p60, CD120a antigen) |
h/m 48/48 | Homodimer (ligand recruitment of 3 dimers) |
Murine receptor binds murine and human TNFα |
| TNF-R2 | (h/m p75/p80, CD120b antigen) |
h/m 46/48 | Homodimer (ligand recruitment of 3 dimers) |
Murine receptor binds only murine TNFα |
| IL-1R-1 | (p80, CD121a antigen) |
65 | Heterodimer with IL-1RAcP | Member of the toll-like receptor family |
| IL-1RacP | 63 | Heterodimer with IL-1R-1/2 | ||
| Natural Antagonists | Notes | |||
| sTNF-R2 | 25 | Sequesters TNFα to temper activity | ||
| (Etanercept2) | 150 | Designed antagonist | ||
| IL-1ra | (IRAP) | 17 | Competes with IL-1β to temper receptor activation | |
| IL-1R-2 | (IL-1RII, CD121b antigen) |
44 | As a receptor, forms a heterodimer with IL-1RacP, may sequester IL-1RacP to reduce IL-1β activity | |
 | ||||
Controlling cytokine activity is critical for wellness and regulating inflammatory responses. Most cytokine receptors are present at 100–1,000 receptors per cell, or at about 10% the level of hormone receptors. TNF-R1 and TNF-R2 have Kds of 0.02 and 0.4 nM, respectively. Concentrations of circulating ligand, such as TNFα, are generally less than 0.0005nM(0.009ng/ml). Similarly the IL-1R-1 and IL-1R-2 bind with affinities all less than 3 nM. Similar to TNFα, serum levels in healthy individuals of IL-1β are below the Kd of the receptors (example; IL-1β is 0.0008 ng/ml (0.00005 nM) in serum of adult humans [167]). These facts indicate that receptor activation parallels changes in ligand availability. During inflammation, circulating levels of the cytokines increase to concentrations adequate to activate cellular events. Although proinflammatory cytokine concentrations are below receptor Kds, additional controls exist, similar to the IGF system. Proinflammatory cytokines have both decoy receptors and soluble negative regulators that control the amount of active ‘free’ cytokine in the extracellular space.
Human - h, mouse - m. Size of monomeric forms based on amino acid composition of the mature protein (Swiss-Prot; ExPASy).
Etanercept consists of two extracellular binding domains of TNF-R2 linked to the Fc portion of human IgG1 (weight approximate).
Fig. 2. Intracellular signaling pathways for two proinflammatory cytokines directly implicated in IGF/insulin resistance and sickness behaviors.

TNFα and IL-1β bind to three well-characterized receptors (TNF-R1 and TNFR2 bind TNFα and IL-1β-R1 binds IL-1β). All three receptors activate the caspase cascade, NF-κB and MAPK pathways (left) although there is variation in the specific MAP kinase intermediates that are used to activate p38MAPK, ERK1/2 and JNK. In contrast, only TNF-R1 and IL-1β-R1 enhance both neutral and acidic sphingomyelinase activity, although each receptors uses distinct mechanisms to do so (right). The ceramide pathways enhance MAPK pathways, and particularly JNK activation.
TNFα binding to the trimeric TNF-R1 induces receptor activation and recruitment of several cytoplasmic accessory proteins. The intracellular portion of the TNF-R1 contains a death domain (DD) region that binds TNF-R associated death domain (TRADD)/Fas associated death domain (FADD) [55, 56], TRAF [46, 57] and the receptor interacting protein (RIP) domain. These adaptor proteins then activate signaling cascades that modulate the majority of cytokine receptor cellular processes, including activation of stress proteins and modulation of cellular proliferation, differentiation, survival and apoptosis.
As shown in Fig. 2, the principal pathways activated by the TNF-R1 are members of the MAPK and the NF-κB signaling pathways. Following TNF-R1 activation, TRADD and FADD recruit and bind TRAF proteins. TRAF2 is important for the activation of NF-κB. After binding to the DD of receptor-coupled adapter proteins, TRAF2 becomes accessible to NIK (NF-κB-inducing kinase) [46, 58]. NIK association with TRAF2 leads to IκK phosphorylation, resulting in phosphorylation and degradation of IκB, the inhibitory subunit of NF-κB [58]. NF-κB is then released from the complex with IκB, translocates to the nucleus and activates transcription of several inflammatory and proliferation factors that play a critical role in myogenesis.
The MAPK family is composed of five groups of kinases; ERK-1/2, stress-associated protein kinase (SAPK) p38MAPK, SAPK JNK, ERK-3/4 and ERK-5 [59]. ERK-1/2, p38MAPK and JNK are considered the classical MAPKs and have been well described, while relatively little is known about ERK-3/4 and ERK-5. The diverse functions of MAPKs are reflected in the variety of stimuli that activate this kinase family. However, it is generally accepted that ERK-1/2 are activated by growth factors, while p38MAPK and JNK are induced by proinflammatory cytokines as well as stress stimuli, including oxidative stress and ionizing radiation. Although JNK and p38MAPK are the primary MAPKs activated by cytokine receptors, ERK-1/2 was also shown to be induced by TNF-R1 activation [58]. The specific MAPK pathway that is activated depends on the assortment of adapter proteins that bind TNF-Rs. Activation of p38MAPK and JNK is mediated by the association of TRAF2 and RIP with the TNF-R1, and this action is augmented by SMase activation. TNF-R1 activates both N-SMase and A-SMase, utilizing different scaffolding complexes associated with either Factor Associated with N-SMase (FAN), the N-SMase activation domain (NSD) of the receptor, or FADD.
Unlike TRAF2 and RIP that bind the DDs, FAN utilizes the NSD for binding to the TNF-R1 (Fig. 2). After binding to NSD, FAN recruits and activates N-SMase [53, 60]. Association with FAN brings N-SMase into proximity with the plasma membrane. N-SMase then catalyzes conversion of membrane-associated sphingomyelin to ceramide, resulting in enhanced phosphorylation and activation of ERK-1/2, p38MAPK and JNK [61]. Although the activation of SAPKs by N-SMase has been reported [49, 58, 61], the details of how this occurs are not yet clear. An emerging concept indicates that N-SMase-dependent activation of SAPKs is mediated indirectly via ceramide. This view is consistent with the recent results showing that ceramide can bind directly to upstream effector kinases of JNK and induce its activation [62]. Consequently, mutations and deletions in either FAN or the NSD domains block the ability of TNF-R1 to activate N-SMase, to generate ceramide and to activate the MAPK family of kinases [60, 63].
IL-1 receptor type 1 (IL-1R1) and the decoy receptor (IL-1R2) are members of the rapidly growing IL-1R/Toll-like receptor (TLR) superfamily that play a critical role in host defense and tissue remodeling following injury (Table 2) [64]. Whereas IL-1R1 signaling is required for IL-1β activity [48], the IL-1R2 serves as a decoy that inhibits IL-1β signaling and is primarily expressed on hematopoietic cells [65, 66]. Ligation of IL-1R1 with IL-1β induces the formation of receptor and IL-1R accessory protein (IL-1R AcP) complex [64]. This association increases the receptor ligand-binding affinity and recruits IL-1R associating kinase (IRAK) to the complex [67]. IRAK is phosphorylated following binding and can then associate with myeloid differentiation factor (MyD88) [68] to transduce the majority of the IL-1R signals.
Similar to the TNF-R1, IL-1R1 transduces most of its biological actions through four protein kinase cascades, NF-κB, ERK-1/2, p38MAPK and JNK. NF-κB is activated following complex formation of MyD88 and IRAK. Upon binding to MyD88, IRAK becomes hyperphosphorylated and is then released into the cytosol where it interacts with TRAF6. The TRAF6/IRAK complex formation is critical for NF-κB activation [47] and results in phosphorylation and subsequent degradation of IκB [64, 69]. Loss of IκB unmasks the nuclear localization sequence and releases NF-κB, which then translocates to the nucleus to drive transcription of numerous genes. In addition to activating NF-κB, TRAF6 also induces the phosphorylation and activation of p38MAPK and JNK, although the details of this activation have not yet been elucidated [46, 64, 69]. The most likely mechanism for activation of SAPKs via TRAF6 is formation of a complex that brings TRAF6 into close proximity with the upstream effector kinases.
A mechanism for maximal activation of NF-κB and the MAPK family of kinases exists, which is dependent on the SMases. IL-1β initiates the MAPK cascade [48] resulting in ERK-1/2 and JNK phosphorylation [46, 64, 69]. These pathways can be enhanced by A-SMase and N-SMase activation (Fig. 2). Although IL-1β induces both A-Smase and N-SMase [70, 71], the specific consequences of this activation have yet to be defined.
4. Proinflammatory cytokine-induced IGF resistance: Molecular mechanisms
Cells interpret and respond to a complex local environment that contains numerous molecular signals from a wide variety of effector cells. Most cells are grown and maintained in medium containing serum or growth supplements that contain members of the IGF family. In addition, most cells secrete factors into the culture medium in which they are maintained, and these may include proinflammatory cytokines and the IGFs. We now know that distinct intracellular signaling cascades can be activated by a single ligand, and the signaling networks become even more complex and difficult to unravel when cytokines and growth factors are considered together (see Fig. 1 and Fig 2). As a result, only a few investigators have attempted to model the interaction between proinflammatory cytokines and growth factors, even though cells are constantly bathed in a milieu of extracellular fluid that contains a variety of hormones and cytokines. This early work has shown that multiple external ligands signal through a molecular network. Recent attempts aimed at modeling these intracellular signaling networks have been successful. For example, a model has been developed that used 7,980 intracellular signals as inputs to 1440 apoptotic outputs that occurred in response to an extracellular death signal (TNFα) combined with one of two extracellular survival signals, insulin or epidermal growth factor [72]. The resulting mathematical model accurately predicted cell survival or death, depending upon the concentration of ligand and the very important interaction between life-and-death proteins.
The intracellular signaling components for both proinflammatory cytokine- and IGF receptor-activated pathways are present in most cell types, and are sometimes even shared (see ERK-1/2 for both IGF-I and proinflammatory cytokine in Fig. 1 and Fig. 2). This situation creates the potential for signal amplification or resistance. However, in the case of insulin, TNF-R activated pathways are well known to engage in intracellular crosstalk and reduce the availability of substrates used by insulin receptor activated pathways, resulting in resistance. At least two proinflammatory cytokine-activated pathways interact with IGF-IR signaling to impair the induction of PI3-K dependent events. Evidence supporting the notion of proinflammatory cytokine-induced IGF resistance is presented below.
Cancer cells are not only an important clinical target, but also they have been extensively used as a model to delineate the function of proinflammatory cytokines and growth factors in regulating cell proliferation and survival. These cells are usually immortalized and have short doubling times, so they provide a robust proliferative response and typically do not undergo spontaneous differentiation. Importantly, many cancer cells retain their responsiveness to both proinflammatory cytokines and growth factors. Cytokine immunotherapy and IGF-I receptor inhibition are two anti-cancer treatments that have been validated in animal models and are in clinical trials for the treatment of certain types of human cancers [73, 74]. Activation of intracellular signaling pathways by the IGF-IR drives cell cycle progression, and over the past several years, intracellular signaling cascades activated by proinflammatory cytokine receptors have been shown to interact with those of the IGF-IR to attenuate the proliferation induced by IGF-I.
Breast, prostate, lung, colon and bladder cancer growth have been intimately tied to activation of the IGF-IR [20]. Constituitive activation of IGF-IR activated signaling pathways, PI3-K and Akt (Fig. 1), has been described in many cancers, and the catalytic p110 subunit of PI3-K is chronically activated in several immortalized cell lines [75]. These findings appear to be supported by both animal and human studies. In laboratory animals, IGF has been shown to promote tumor growth and metastasis [20, 76, 77]. Consistent with these findings, human patients whose IGF-I levels are in the upper quartile of the normal range have an increased incidence of several cancers [20]. The inverse association has been made as well, whereby low IGF-I concentrations are associated with diminished tumor growth and metastasis [20, 76].
The case that proinflammatory cytokines restrain the growth of cancer cells, such as leukemia [78], melanoma [79], breast [80], rhabdomyosarcoma [81], salivary gland [82], sarcoma [83] and ovarian adenocarcinoma [84], has been confirmed in vitro. However, in vivo administration of systemic proinflammatory cytokines is not practical because the acute systemic inflammatory response that develops can cause severe sickness or, in extreme cases, death. None-the-less, local administration of cytokines has significant therapeutic potential [73]. Interestingly, the effects of locally administered proinflammatory cytokines and inhibition of IGF-I activity have strikingly similar results on the growth and proliferation of cancer cells, yet a link between these two approaches to treat cancer has only recently been made.
Historically, the effects of cytokines have typically been interpreted to be a direct cellular effect. However, as previously discussed, most in vitro studies (and certainly all in vivo experiments) that were designed to unravel proinflammatory cytokine action were actually performed in the presence of serum or growth supplements containing growth factors, such as insulin or IGF-I. As the signaling cascades activated by proinflammatory cytokines began to unravel, it became clear that no pathway was present that directly interfered with cell cycle progression. Thus, using carefully designed serum-free culture, experiments were designed based on the assumption that proinflammatory cytokines may act indirectly by interfering with the proliferative actions of serum-derived factors such as IGF-I (Fig. 3). Further, many cancer cells synthesize significant amounts IGF-I or IGF-II, so cytokines may act to blunt autocrine proliferation caused by endogenously produced IGF. Several experiments have now been reported that demonstrate the anti-proliferative effects of proinflammatory cytokines on cells grown in serum-free culture systems were only apparent when cells were treated with exogenous growth factor [10, 85, 86]. In fact, TNF-α, IL-1β, or IL-6 alone have no effect on cell proliferation in experiments carried out under serum-free conditions [10], but the cytokines inhibited cell proliferation and activation of cell cycle proteins in IGF-I-treated cells [85, 86].
Fig. 3. Proinflammatory cytokines inhibit proliferation of MCF-7 breast cancer cells.

Proliferation of MCF-7 cells by IGF-I is an Akt-dependent event. We have shown that both TNFα and IL-1β induce IGF resistance of these cells. As shown in the Western blots, IGF-I strikingly activates, whereas TNFα clearly decreases, the ability of IGF-I to activate the cyclin loop; i.e., the IGF-I-induced increase in cyclin A, hyperphosphorylated RB (ppRB) and E2F-1 are all blocked by TNFα. Data shown with permission J. Biol. Chem. 279 (2004): 7438–7446.
IGF binding to its receptor results in autophosphorylation on tyrosine residues of the IGF-IR, followed by recruitment of insulin receptor substrate (IRS) adapter proteins and activation of downstream signaling pathways (Fig. 1). In human MCF-7 breast cancer cells, cytokines do not inhibit proliferation by themselves, but they do attenuate the mitogenic effects of IGF-I [10]. This effect occurs downstream of the receptor, as autophosphorylation of the β chains of the IGF-IR is unchanged. Instead, both TNF-α and IL-1β exert an inhibitory interaction by reducing tyrosine phosphorylation/activation of IRS-1 [10]. The diminished phosphorylation of IRS-1 reduces its ability to function as a docking molecule and effectively blunts the activation of downstream effector pathways, including PI3-K and Akt. These data establish that proinflammatory cytokines act in human cancer cells by blocking IGF-IR activated intracellular signaling events.
Downstream of PI3-K and Akt, the impact of cytokine interference on IGF-I action is reflected as an inhibition of IGF-induced activation of the cell cycle machinery (Fig. 3, inset). TNFα alone does not activate or inhibit cyclin A, retinoblastoma protein (Rb) or E2F-1. However, IGF-I induced upregulation of cyclin A and E2F-1 is completely blocked by TNFα. Similarly, IGF-I-induced Cdk2 activity, as measured by phosphorylation of its substrate, Rb, is also blocked by TNFα. These effects occur as a result of proinflammatory cytokine inhibition of PI3-K activation and blockade of the induction stimulus for the formation of Cdk2/cyclin A complexes [85, 86]. One of the 22 ways in which proinflammatory cytokines exert their suppressive effects on breast cancer cells is by inhibition of intracellular signaling constituents that become activated by IGF. Even 100 ng/ml of IGF-I is unable to overcome the inhibitory effect of proinflammatory cytokines because the effect of TNFα and IL-1β on MCF-7 cells is a classic case of receptor crosstalk, causing IGF resistance rather than diminished sensitivity.
This anti-proliferative effect of proinflammatory cytokines occurs because activation of the PI3-K pathway is a limiting factor for the proliferation of MCF-7 breast cancer cells. However, not all cells are similarly sensitive to the anti-proliferative effects of these cytokines. An example can be found in the case of skeletal muscle myoblasts, which appear to be dependent on IGF-induced MAPK signaling for proliferation [87, 88]. Instead of inhibiting proliferation, both TNFα and IGF-I activate MAPK/ERK-1/2 signaling and enhance myoblast growth [89].
Skeletal muscle, which constitutes the largest tissue in the body, is now known to synthesize and respond to a plethora of inflammatory mediators. Local or systemic inflammation ultimately affects muscle function and is associated with decreased muscle mass. For instance, elevated levels of proinflammatory cytokines, particularly TNFα and IL-1β, are implicated in muscle wasting that occurs during sarcopenia of normal aging [90] and in chronic diseases such as AIDS and cancer. A review of several recent studies demonstrates that prolonged inflammation may also contribute to myopathies associated with work-related musculoskeletal disorders [91]. These pathophysiological actions of proinflammatory cytokines are, at least in part, mediated by decreasing expression and activity of growth-promoting hormones such as GH and IGF-I. In chronic disease and aging, a decrease in IGF-I activity coincides with prolonged elevated concentrations of TNFα, IL-1β and IL-6 [92–97]. Coupled with the observation that nutritional supplementation and GH administration fail to reverse the loss of muscle mass in cachectic animals and humans [92–94, 96, 97], these data suggest that host immune factors are indirectly involved in eliciting wasting syndromes by inducing IGF resistance. Studies in skeletal muscle progenitor cells have confirmed and extended this concept. Physiological concentrations (0.1 – 1 ng/ml) of exogenous TNFα or IL-1β significantly suppress differentiation of muscle progenitor cells by disrupting IGF-IR signaling and consequently preventing IGF-I-induced protein synthesis and myogenesis [41–43, 49]. Promising recent results demonstrate that TNFα- and IL-1β-induced hormone resistance involves common signaling intermediates, including ceramide and JNK [43, 49]. Collectively, these novel data lend support for the biological significance of immune-endocrine interactions in the physiology of skeletal muscle development.
Muscle dystrophies, aging [90] and chronic diseases such as AIDS [98–102] and cancer [93, 103], are all accompanied by a loss of muscle mass and diminished regenerative capacity of muscle cells. Wasting symptoms in these conditions are often indistinguishable, despite major differences in etiology [97]. Furthermore, despite using pharmacological doses of IGF-I and growth hormone to treat such wasting symptoms, the treatments frequently fail to sustain prolonged beneficial effects. For example, administration of GH and IGF-I to AIDS patients with cachexia does not result in increased protein synthesis or muscle mass despite elevated IGF-I levels [104], whereas a significant improvement in protein synthesis and muscle mass is observed in AIDS patients without muscle wasting and with comparable IGF-I levels [104, 105]. Similar findings have been reported in patients with pediatric HIV in which the severity of infection is correlated with increased resistance to the growth promoting properties of IGF-I, GH and insulin. Initial administration of GH to young or adult patients increased protein synthesis and lean body mass while decreasing fat mass [101, 104, 106, 107]. However, prolonged treatment did not show a significant increase in lean body mass, despite elevated IGF-I levels [101, 104, 106, 107]. Hormone (IGF) resistance, by definition, cannot be overcome by a simple elevation of ligand. These data support the hypothesis that hormone resistance is involved in the loss of muscle mass. They also suggest that humoral host factors such as proinflammatory cytokines are involved.
Proinflammatory cytokines are now known to regulate muscle development, injury and repair [108]. This concept was very recently demonstrated in young dystrophic mice in which injections of a soluble TNF receptor antagonist, etanercept, or depletion of neutrophils, reduced muscle necrosis [109]. Similarly, TNFα and IL-1β inhibited myoblast differentiation [110] and fusion [111], which may occur by reducing expression (Fig. 4 A) of muscle specific transcription factors promoted by IGF-I, myogenin and MyoD [41–43, 49, 112, 113]. Results from skeletal muscle indicate that the inhibitory actions of cytokines on skeletal muscle development are mediated indirectly by inducing changes in growth factor expression and activity. For example, in skeletal muscle, as well as in myoblasts, TNFα decreased IGF-I mRNA by 80%, and this effect was blocked by a TNFβ antagonist [114]. Such results indicate that the actions of proinflammatory cytokines in muscle can be mediated by targeting growth factor expression, but they do not explain the findings from clinical studies in which supplementation with growth hormone did not significantly ameliorate wasting in AIDS patients, despite elevated IGF-I levels [104]. This is consistent with the observations that whereas circulating IGF-I concentrations are not altered consistently in cachectic patients, there is a prominent decrease in the ability of IGF-I to activate protein synthesis that coincides with wasting [102, 104].
Fig. 4. Proinflammatory cytokines impair differentiation of C2C12 myoblasts only in the presence of IGF-I.

IGF-I increases both differentiation (myogenin expression; A and B) and hypertrophy (myosin heavy chain expression, MHC; C) of muscle cells. IGF-I increases differentiation in both a PI3-K- and Akt-dependent manner (not shown). TNFα and IL-1β depress IGF-I activity (both differentiation; A, and hypertrophy; C) by inducing IGF resistance. This resistance is associated with depressed activation of IRS-1. We have defined a pathway required for a low concentration of TNFα, 1 ng/ml, to inhibit IGF-I-induced myogenesis as follows: TNFα → N-SMase/A-SMase/ceramide synthase → ceramide → JNK → ↓ IGF effect by showing first that: A) inhibition of ceramide synthesis (de novo with FB1; A-SMase with D609 or N-SMase with GSH) blocks cytokine inhibition of IGF activity. Second, B) addition of N-SMase or a ceramide-derivative, C2-cer, mimic action of the cytokine. Third, B and C) the IGF resistance-inducing actions of N-SMase, C2-cer and TNFα are all blocked by a JNK inhibitor. Data shown with permission Endocrinology 145 (2004) : 4592–4602 and 147 (2006) : 4363–4373.
Several reports have demonstrated the ability of cytokines to induce a state of IGF resistance without affecting the expression of IGF-I or IGF-IRs [42, 115]. Indeed, physiologically relevant concentrations (0.1–1.0 ng/ml) of TNFα or IL-1β inhibit the ability of IGF-I to induce global protein synthesis and expression of muscle specific transcription factors, myogenin and MyoD, but have no effect in absence of IGF-I [41–43] (Fig. 4, inset). TNFα also blocks IGF-I-induced differentiation as assessed by expression of myosin heavy chain (MHC), a well-established marker of differentiation [41, 49]. We recently reported that TNFα does not affect the ability of IGF-I to bind to and activate the IGF-IR, but rather it induces IGF resistance by diminishing signaling downstream of IGF-IR in both myoblasts [42] and breast tumor epithelial cells [10]. These results are consistent with previous findings that TNFα decreased IGF-I function despite normal serum levels of IGF-I and normal binding of IGF-I to IGF-IR [115]. Both TNFα and IL-1β can inhibit IGF-I-induced tyrosine phosphorylation of IRS-1 and IRS-2 [10, 41–43]. This inhibition could block events (protein synthesis and differentiation) that are dependent on downstream signaling pathways involving PI3-K, Akt and mTOR (Fig. 1). The ability of proinflammatory cytokines to inhibit early IGF-I-induced signaling events such as IRS-1 tyrosine phosphorylation, as well as late events such as myogenin and MHC expression, (Fig. 4, inset) indicates that proinflammatory cytokines suppress myogenesis by directly blocking IGF-I signaling. This is an important concept that may help explain why nutrient and IGF-I supplementation does not yield consistent successful clinical results.
Promising results now indicate that common intermediary factors such as ceramide and JNK that lie downstream of several cytokine receptors play an important role in IGF resistance in skeletal muscle progenitors. Activation of TNFα and IL-1β receptors induces intracellular expression of the sphingosine-based lipid second messenger, ceramide [54, 116–118] via three major pathways; the de novo ceramide synthesis [118, 119] and the two SMase dependent pathways; A-SMase and N-SMase [54, 60, 117, 120]. Ceramide is a potent second messenger that mediates cytokine actions in a variety of cell types, including embryonic kidney cells [121], adipocytes [122] and HMN1 motor neurons [123], and it does so, in part, by inhibiting growth factor signaling. These results are consistent with findings in muscle cells. Ceramide was shown to inhibit insulin-induced Akt phosphorylation in C2C12 myotubes [124] and block PDGF-induced Akt phosphorylation and [3H]-thymidine incorporation in smooth muscle cells [125]. The possibility that ceramide synthesis is required for cytokine inhibition of IGF-I biological activity was more recently tested using pharmacological inhibitors for each of the three ceramide generating pathways, GSH as an inhibitor of N-SMase, D609 as an inhibitor of A-SMase and fumonisin B1 as an inhibitor of de novo ceramide synthesis [43]. The results from these studies established that TNFα and IL-1β do not affect myogenin expression when added alone, but they completely block the stimulatory action of IGF-I (Fig. 4 A). All three inhibitors of ceramide synthesis suppressed the ability of TNFα and IL-1β to inhibit IGF-I-induced global protein synthesis and restore IGF-I-induced expression of muscle specific differentiation factors, myogenin (Fig. 4 A) and MyoD. Furthermore, we were able to show that exogenous N-SMase and a cell permeable ceramide analog (C2-cer) mimicked the effect of TNFα and IL-1β and inhibited IGF-I-induced myogenin expression by myoblasts (Fig. 4B). These results demonstrated that ceramide is necessary for cytokine-induced IGF-resistance in skeletal muscle myoblasts.
JNK is another important protein that has been implicated in disrupting skeletal muscle development and has recently gained significant clinical attention because it was found to be closely associated with the pathophysiology of many neurodegenerative, metabolic and inflammatory disorders [126]. The potent inhibitory activity of JNK can be attributed in part to its ability to induce resistance to several hormones including insulin, glucocorticoids and IGF. JNK is activated in vivo and in vitro in response to various stress stimuli, including proinflammatory cytokines and ceramide [59, 61, 62]. Recent results have attributed the catabolic actions of JNK in muscle tissue to its ability to suppress IGF-I expression [9]. Consistent with these findings, a pharmacological inhibitor of JNK, SP600125, completely blocked the ability of TNFα to suppress GH-dependent mRNA expression of IGF-I [114]. JNK is also thought to act in muscle cells by directly blocking components of growth factor signal transduction pathways. For example, JNK can associate with IRS-1 and inhibit activation of substrates downstream of the IGF-IR [49]. We recently tested the possibility that JNK mediates cytokine inhibition of myogenesis. Using a novel cell-permeable peptide inhibitor of JNK (I-JNK), we found that JNK is required for TNFα to inhibit IGF-I-induced expression of myogenin (Fig. 4 B) and myosin heavy chain (MHC) (Fig 4 C). This JNK inhibitor completely blocked the ability of TNFα to induce serine phosphorylation of IRS-1 and restored tyrosine phosphorylation of IRS-1 in response to IGF-I [49]. These data demonstrated that JNK is a key downstream intermediate by which proinflammatory cytokines induce IGF-resistance.
5. IGF-I Antagonizes Proinflammatory Cytokines in the Brain
Despite considerable information regarding intracellular signaling and the separate actions of both hormones and cytokines in vitro, including the expanding field of receptor crosstalk, relatively little is known about their interaction within the brain. Clearly, central inflammation plays a critical role in protecting the brain from infection and aides in repair of damaged neuronal tissue [127, 128]. However, proinflammatory cytokines can also promote neurodegeneration, excitotoxicity and accentuate ischemic injury [129–131]. In these latter cases, targeting inflammation is a feasible means to protect the brain [132]. Another important facet of inflammation in the central nervous system is that proinflammatory cytokines modulate brain functions independent of neurotoxicity, resulting in altered behavior [133, 134], pain sensitivity (allodynia and hyperalgesia) [135, 136] and cognition [129, 137]; all of which worsen if the inflammation becomes chronic.
One very important aspect of proinflammatory cytokine action on behavior that does not involve neurotoxicity is the induction of sickness behaviors. Sickness behavior is the incidence of one or a combination of several symptoms including general malaise, lack of energy/fatigue, loss of interest in social interaction, reduction in appetite and changes in sleep patterns. Despite the induction of sickness behaviors by central and peripheral proinflammatory cytokines that has been known for nearly two decades [138], the molecular circuits that mediate these events within the brain are only now being defined. Peripheral or central administration of the proinflammatory cytokines, IL-1β and TNFα, in rodents induces sickness behaviors that dissipate within 24 h [14]. Common quantitative criteria used for rodents treated with proinflammatory cytokines include depressed food intake, weight loss, immobility, decreased motivation for naturalistic behaviors such as exploration aimed at either novel animals or objects and decreased rearing. These experimentally-induced behavioral changes mimic sickness behavior of humans treated with proinflammatory cytokines [138, 139]. Interestingly, behavioral changes following chronic elevations in proinflammatory cytokines can culminate in the development of major depressive disorders [140].
There is a clear interaction between IGF-I and proinflammatory cytokines on sickness behavior. In the brain, IGF-I induces resistance (or decreases sensitivity) to TNFα, whereas in breast cancer cells and myoblasts TNFα clearly induces IGF-I resistance, as discussed above. IGF-I, given directly into the lateral ventricles of the brain (i.c.v.) at 1 µg, significantly attenuates sickness behavior induced by i.c.v. injection of lipopolysaccharide (LPS) [141]. Since LPS induces synthesis of proinflammatory cytokines in the brain, this finding indicates that IGF-I directly interacts with the activity of centrally-produced cytokines. We subsequently confirmed this hypothesis by showing that IGF-I given i.c.v. completely blocks sickness behavior induced by central TNFα [142] but only partially opposes sickness behavior induced by IL-1β. In the latter example, IL-1β at 2 ng/mouse completely reduces motivation for social behavior (exploration of a con-specific novel juvenile, Fig. 5A left) and results in almost complete immobility of mice (immobile for ~180 out of 240 seconds of observation, Fig. 5A right). IGF-I blocks only 50 %, of IL-1β’s effect on both indices.
Fig. 5. Peripheral and central IGF-I reduces sickness behavior: a case of pro-inflammatory cytokine resistance?

A) IGF-I action involves a central component as IGF-I given i.c.v. impairs sickness behavior, as assessed by social exploration and duration of immobility, in response to centrally administered IL-1β. The mechanism for this central effect of IGF-I is unknown. Significant differences between bars are indicated by brackets (p<0.05). B) Peripheral administration of IGF-I reduces sickness behavior induced by peripheral administration of LPS in wild-type db/+ mice. However, db/db mice have elevated circulating and tissue proinflammatory cytokines and are resistant to IGF-I. C) Critical and essential challenges for the future are to define the largely unknown mechanism(s) by which cytokines induce and by which IGF-I abrogates sickness behaviors; whether sickness is a case of cytokine-induced IGF resistance and if IGF-I depresses sickness by triggering pro-inflammatory cytokine resistance. D) The identities of all the cellular targets in the central nervous system (or in the periphery) that are responsible for sickness behavior have not been determined. Proinflammatory cytokines may act directly on neurons or on glia. E) Similarly, IGF-I may act directly to prevent proinflammatory cytokine signaling (not shown) or to depress the central cytokine loop by acting like IL-10. Data shown with permission Brain, Behavior, and Immunity 20 (2006): 57–63 and PNAS 102 (2005): 15184–15189.
We recently reported that the protective ability of central IGF-I in blocking sickness behavior caused by central TNFα occurrs similarly in wild type and TNF-R2 knockout mice [143]. This finding indicates that IGF-I somehow interacts with central TNF-R1 receptors. When given systemically instead of centrally, IGF-I also antagonizes the behavioral effects of LPS. For example, when administered to db/+ mice treated with LPS, systemic IGF-I diminishes sickness behavior by approximately 50% (Fig. 5B left) [33]. However, homozygous db/db mice are resistant to IGF-I. These mice are well known to be deficient in the leptin receptor, leading to obesity and chronic inflammation [144]. In these mice, an acute systemic injection of LPS also induces an exaggerated response to LPS (greater decrease in social exploration), but IGF-I has no effect (Fig. 5B right). Two important and distinct points can be derived from these data. First, IGF-I inhibits sickness behaviors induced by acute injections of proinflammatory cytokines, which could be due to IGF-I-induced resistance or decreased sensitivity to TNFα. Second, chronic inflammation, as found in obese db/db mice, decreases the ability of IGF-I to block sickness behaviors. This could occur by reducing sensitivity to TNFα (e.g., by promoting shedding of antagonistic soluble TNF receptors) or by inducing TNF resistance. To adequately address these issues, future experiments will need to answer two questions: What are the mechanisms of action of cytokines in sickness behavior? And, how does IGF-I act to antagonize sickness behaviors induced by proinflammatory cytokines?
Behavioral changes that result from i.c.v. administration of proinflammatory cytokines TNFα [145, 146] and IL-1β [142, 147] or LPS [147, 148] are similar to those following i.p. administration of proinflammatory cytokines IL-1β [147] or LPS [147]. The main difference is that 100–500 fold lower concentrations of cytokines cause the same behavioral changes when given i.c.v. rather than i.p. These similarities occur because induction of sickness by peripheral cytokines is dependent on central cytokine expression [14, 139]. TNFα and IL-1β activate the MAPK kinase pathway, JNK, p38MAPK and Erk-1/2, as well as NF-κB (Fig. 2). IL-1β induces sickness behavior via activation of the IL-1R1 [147] and requires activation of NF-κB [149]. TNFα acts within the hypothalamus to depress food intake [150], an effect that requires nitric oxide synthesis associated with elevations in JNK and p38MAPK. Thus, each of the MAPK kinase pathway has been implicated in behavioral changes, but their respective roles are likely to be different for distinct cytokines.
The signaling pathways that are responsible for sickness behaviors induced by TNFα and IL-1β are likely to differ. IL-1β strongly activates c-Fos in neuronal but also in glial cells, and this action parallels the induction of sickness behavior [149]. TNF-R1 is strongly expressed in neurons, whereas TNF-R2 is primarily localized to glia [151]. It could therefore be that TNFα induces sickness behavior by activating only one receptor isoform on different cell types. Since human TNFα binds only to the murine TNF-R1 in mice to induce sickness behaviors [145, 146], TNFα could induce sickness behavior by direct activation of neurons. If so, this would be in striking contrast to IL-1β, which appears to act via glia. Following influenza A viral infection, p38MAPK activation in glia is associated with TNFα expression, but JNK activation in neurons is associated with neuronal apoptosis [152]. These findings indicate that the MAPK pathways have different roles within the brain, and these roles require distinct signaling pathways that differ according to the phenotype of the cell. This separation may explain the differing ability of IGF-I to reverse sickness behaviors caused by TNFα versus IL-1β.
Some, but not all, of the behavioral effects induced by TNFα are blocked by IL-1ra [146], indicating that full-blown sickness behavior requires activation of a cytokine network loop (Fig. 5E) and possibly both neuronal and glia activation by proinflammatory cytokines. This possibility is further supported by the ability of both TNF-R1 and TNF-R2 to mediate depressive-like responses, as determined by decreased immobility of both TNF-R1 and R2 knockout mice in the forced swim test [153]. Clearly, a single proinflammatory cytokine can induce a variety of sickness behaviors independent of the actions of the other. TNFα drives LPS-induced sickness behavior in IL-1R1 knockout (KO) mice but not in wild-type mice [147]. Indeed, TNFα sensitivity is enhanced in IL-1R1 KO mice. This indicates a compensatory response within the brain when the action of one cytokine is missing. IL-1β is active in IL-6 KO mice [154], but IL-6 KO mice are less sensitive to IL-1β than are wild-type mice [148]. The decrease in IL-1β sensitivity may result from depressed proinflammatory cytokine production within the brain (decreased cytokine loop Fig. 5E) since IL-6 plays an initiating role in central production of IL-1β and TNFα [155]. These results point to a complicated interplay between the various proinflammatory cytokines within the brain during induction of sickness behavior. Clearly, additional experiments are necessary to clarify the molecular mechanisms involved in cytokine-induced sickness behavior.
There are several possible mechanisms, both direct and indirect, by which IGF-I could reduce sickness behavior. IGF-I may act on the same cells as proinflammatory cytokines and, by doing so, interact via receptor crosstalk within those cells. By this proposed mechanism, IGF-I would directly act to depress proinflammatory cytokine activity (Fig. 2). In support of this idea, IGF-II secreted by microglial cells inhibits TNF-activation of JNK in cultured oligodendrocytes [156]. IGF-I also stimulates dephosphorylation of IκB in astrocytes, thus preventing its degradation and diminishing NF-κB nuclear translocation [157]. Similarly, IGF-I depresses TNFα-induced NF-κB activation in human colonic adenocarcinoma cells [158]. These direct intracellular signaling abilities of IGF-I have the potential to reduce sickness behaviors, and these actions are equivalent to pharmacological inhibition of brain NF-κB to impair IL-1β-induced sickness behavior [149]. Although there is only circumstantial information regarding the mechanism by which IGF-I causes reduced behavioral responses to TNFα, these findings provide a sound basis for future experiments aimed at deciphering the molecular mechanisms by which IGF-I inhibits sickness behaviors.
IGF-I may also induce proinflammatory cytokine resistance by enhancing the production of anti-inflammatory cytokines. IGF-I stimulates IL-10 secretion by T-cells in the inflamed pancreas [159, 160]. If such an effect occurred within the brain, this phenomenon might well attenuate proinflammatory cytokine-induced sickness behaviors because central administration of IL-10 tempers the behavioral effects of LPS [161]. IL-10 is the prototypical anti-inflammatory cytokine that acts to diminish proinflammatory cytokine secretion and receptor expression [162, 163]. If this were the case, IGF-I would act by an indirect mechanism to abrogate sickness behavior via the induction of IL-10 secretion. Recent studies have also identified IGF-I as having potent anti-depressant-like properties in rodent models. Either exogenously administered IGF-I [23] or administration of an inhibitor of IGF-I binding proteins (which increases endogenous IGF-I concentrations) [24] effectively impairs anxiety- and depressive-like behaviors in mice.
Perhaps the most promising non-pharmaceutical approach to abrogate sickness behavior is the simplest: exercise. Exercise protects the brain from ischemic damage, an effect dependent on exercise-induced increases in central IGF-I levels [164]. It is tempting to speculate that exercise would diminish sickness behavior by its ability to increase central IGF-I. However, the behavioral benefits of exercise and IGF-I, independent of other health benefits, still await characterization. Collectively, these data reinforce the model that the balance of proinflammatory cytokines in the brain in relation to anti-inflammatory agents, such as IGF-I, is critical for determining the extent and perhaps recovery from sickness (Fig. 6). Furthermore, the data establish that IGF-I impairs proinflammatory cytokine-induced sickness behaviors by a mechanism that involves the induction of TNFα-resistance or a reduction in TNFα sensitivity.
6. Conclusion
Interactions between the immune and endocrine system have historically been viewed from the perspective that hormones regulate immune function. More data have now accumulated that further define the bi-directional interaction between the two systems. Proinflammatory cytokines are able to induce a state of hormone resistance. This review has provided several examples that demonstrate, at the cellular and behavioral levels, how proinflammatory cytokines interact with the IGF growth factors. The balance between proinflammatory cytokines and hormones is an important natural component of the body’s homeostatic process. The balance of this interaction is likely essential for the maintenance of health and well being (Fig. 6), especially when one considers the number of clinical conditions associated with chronic inflammation such as type 2 diabetes, rheumatoid arthritis, inflammatory bowel disease, HIV/AIDS, cardiovascular disease, cerebrovascular disease and even normal aging. Therefore, a comprehensive understanding of the interaction between the immune and endocrine systems is paramount to our ability to develop new, more effective means to improve overall quality of life.
Acknowledgements
This research was supported by grants from National Institutes of Health to K.W.K. (AI 50442, MH 51569, AG 029573), R.D. (MH 071349 and MH 079829) and R.W.J. (AG 023580 and AG 16710) and from the USDA to R.H.M. (2004-35206-14144)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hehlgans T, Pfeffer K. The intriguing biology of the tumour necrosis factor/tumour necrosis factor receptor superfamily: players, rules and the games. Immunology. 2005;115:1–20. doi: 10.1111/j.1365-2567.2005.02143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Valverde AM, Benito M, Lorenzo M. The brown adipose cell: a model for understanding the molecular mechanisms of insulin resistance. Acta Physiol Scand. 2005;183:59–73. doi: 10.1111/j.1365-201X.2004.01384.x. [DOI] [PubMed] [Google Scholar]
- 3.Frystyk J. Free insulin-like growth factors -- measurements and relationships to growth hormone secretion and glucose homeostasis. Growth Horm IGF Res. 2004;14:337–375. doi: 10.1016/j.ghir.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 4.Kim JH, Park HH, Lee CE. IGF-1 potentiation of IL-4-induced CD23/Fc(epsilon)RII expression in human B cells. Mol Cells. 2003;15:307–312. [PubMed] [Google Scholar]
- 5.Bernabei P, Bosticardo M, Losana G, Regis G, Di Paola F, De Angelis S, Giovarelli M, Novelli F. IGF-1 down-regulates IFN-gamma R2 chain surface expression and desensitizes IFN-gamma/STAT-1 signaling in human T lymphocytes. Blood. 2003;102:2933–2939. doi: 10.1182/blood-2003-01-0100. [DOI] [PubMed] [Google Scholar]
- 6.Arkins SR, Jonhson W, Minshall C, Dantzer R, Kelley KW. Immunophysiology: The interaction of hormone, lymphohemopoietic cytokines and the neuroimmune axis. In: McEwen BS, editor. Coping with the Environment: Neural and Endocrine Mechanisms, Handbook of Physiology. Vol. 4. New York: Oxford University Press; 2001. pp. 469–495. [Google Scholar]
- 7.Avitsur R, Kavelaars A, Heijnen C, Sheridan JF. Social stress and the regulation of tumor necrosis factor-alpha secretion. Brain Behav Immun. 2005;19:311–317. doi: 10.1016/j.bbi.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 8.Silverman MN, Pearce BD, Biron CA, Miller AH. Immune modulation of the hypothalamic-pituitary-adrenal (HPA) axis during viral infection. Viral Immunol. 2005;18:41–78. doi: 10.1089/vim.2005.18.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lang CH, Hong-Brown L, Frost RA. Cytokine inhibition of JAK-STAT signaling: a new mechanism of growth hormone resistance. Pediatr Nephrol. 2005;20:306–312. doi: 10.1007/s00467-004-1607-9. [DOI] [PubMed] [Google Scholar]
- 10.Shen WH, Zhou JH, Broussard SR, Freund GG, Dantzer R, Kelley KW. Proinflammatory cytokines block growth of breast cancer cells by impairing signals from a growth factor receptor. Cancer Res. 2002;62:4746–4756. [PubMed] [Google Scholar]
- 11.Mocellin S, Rossi CR, Pilati P, Nitti D. Tumor necrosis factor, cancer and anticancer therapy. Cytokine Growth Factor Rev. 2005;16:35–53. doi: 10.1016/j.cytogfr.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 12.Wajant H, Gerspach J, Pfizenmaier K. Tumor therapeutics by design: targeting and activation of death receptors. Cytokine Growth Factor Rev. 2005;16:55–76. doi: 10.1016/j.cytogfr.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 13.Spate U, Schulze PC. Proinflammatory cytokines and skeletal muscle. Curr Opin Clin Nutr Metab Care. 2004;7:265–269. doi: 10.1097/00075197-200405000-00005. [DOI] [PubMed] [Google Scholar]
- 14.Kelley KW, Bluthe RM, Dantzer R, Zhou JH, Shen WH, Johnson RW, Broussard SR. Cytokine-induced sickness behavior. Brain Behav Immun. 2003;17 Suppl 1:S112–S118. doi: 10.1016/s0889-1591(02)00077-6. [DOI] [PubMed] [Google Scholar]
- 15.Schiepers OJ, Wichers MC, Maes M. Cytokines and major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:201–217. doi: 10.1016/j.pnpbp.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 16.McCusker RH, Strle K, Broussard SR, Dantzer R, Bluthé RM, Kelley KW. Crosstalk between insulin-like growth factors and pro-inflammatory cytokines. In: Ader R, Dantzer R, Glaser R, Heijnen C, Irwin M, Padgett D, Sheridan J, editors. Psychoneuroimmunology. Vol. 1. New York: Academic Press; 2006. pp. 171–191. [Google Scholar]
- 17.Lupu F, Terwilliger JD, Lee K, Segre GV, Efstratiadis A. Roles of growth hormone and insulin-like growth factor 1 in mouse postnatal growth. Dev Biol. 2001;229:141–162. doi: 10.1006/dbio.2000.9975. [DOI] [PubMed] [Google Scholar]
- 18.Sutter NB, Bustamante CD, Chase K, Gray MM, Zhao K, Zhu L, Padhukasahasram B, Karlins E, Davis S, Jones PG, Quignon P, Johnson GS, Parker HG, Fretwell N, Mosher DS, Lawler DF, Satyaraj E, Nordborg M, Lark KG, Wayne RK, Ostrander EA. A single IGF1 allele is a major determinant of small size in dogs. Science. 2007;316:112–115. doi: 10.1126/science.1137045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kenyon C. The plasticity of aging: insights from long-lived mutants. Cell. 2005;120:449–460. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 20.LeRoith D, Helman L. The new kid on the block(ade) of the IGF-1 receptor. Cancer Cell. 2004;5:201–202. doi: 10.1016/s1535-6108(04)00054-6. [DOI] [PubMed] [Google Scholar]
- 21.Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, McDowell EP, Lazo-Kallanian S, Williams IR, Sears C, Armstrong SA, Passegue E, DePinho RA, Gilliland DG. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325–339. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 22.Panowski SH, Wolff S, Aguilaniu H, Durieux J, Dillin A. PHA-4/Foxa mediates diet-restriction-induced longevity of C. elegans. Nature. 2007 doi: 10.1038/nature05837. [DOI] [PubMed] [Google Scholar]
- 23.Hoshaw BA, Malberg JE, Lucki I. Central administration of IGF-I and BDNF leads to long-lasting antidepressant-like effects. Brain Res. 2005;1037:204–208. doi: 10.1016/j.brainres.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 24.Malberg JE, Platt B, Rizzo SJ, Ring RH, Lucki I, Schechter LE, Rosenzweig-Lipson S. Increasing the levels of insulin-like growth factor-I by an IGF binding protein inhibitor produces anxiolytic and antidepressant-like effects. Neuropsychopharmacology. 2007 doi: 10.1038/sj.npp.1301358. [DOI] [PubMed] [Google Scholar]
- 25.Sun LY, Evans MS, Hsieh J, Panici J, Bartke A. Increased neurogenesis in dentate gyrus of long-lived Ames dwarf mice. Endocrinology. 2005;146:1138–1144. doi: 10.1210/en.2004-1115. [DOI] [PubMed] [Google Scholar]
- 26.Savage MO, Camacho-Hubner C, Dunger DB. Therapeutic applications of the insulin-like growth factors. Growth Horm IGF Res. 2004;14:301–308. doi: 10.1016/j.ghir.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 27.Bahr C, Groner B. The insulin like growth factor-1 receptor (IGF-1R) as a drug target: novel approaches to cancer therapy. Growth Horm IGF Res. 2004;14:287–295. doi: 10.1016/j.ghir.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 28.Yakar S, Leroith D, Brodt P. The role of the growth hormone/insulin-like growth factor axis in tumor growth and progression: Lessons from animal models. Cytokine Growth Factor Rev. 2005;16:407–420. doi: 10.1016/j.cytogfr.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 29.Dohm GL, Elton CW, Raju MS, Mooney ND, DiMarchi R, Pories WJ, Flickinger EG, Atkinson SM, Jr, Caro JF. IGF-I--stimulated glucose transport in human skeletal muscle and IGF-I resistance in obesity and NIDDM. Diabetes. 1990;39:1028–1032. doi: 10.2337/diab.39.9.1028. [DOI] [PubMed] [Google Scholar]
- 30.Grzelkowska-Kowalczyk K, Wieteska W. The impairment of IGF-I-stimulated protein synthesis and activation of protein kinase B, p70(S6k), MAP kinase, and p90(rsk) in mouse C2C12 myogenic cells exposed to high glucose and high insulin. Pol J Vet Sci. 2005;8:241–250. [PubMed] [Google Scholar]
- 31.Lang CH, Kumar V, Liu X, Frost RA, Vary TC. IGF-I induced phosphorylation of S6K1 and 4E-BP1 in heart is impaired by acute alcohol intoxication. Alcohol Clin Exp Res. 2003;27:485–494. doi: 10.1097/01.ALC.0000057061.28704.AC. [DOI] [PubMed] [Google Scholar]
- 32.Lang CH, Pruznak AM, Deshpande N, Palopoli MM, Frost RA, Vary TC. Alcohol intoxication impairs phosphorylation of S6K1 and S6 in skeletal muscle independently of ethanol metabolism. Alcohol Clin Exp Res. 2004;28:1758–1767. doi: 10.1097/01.alc.0000145787.66405.59. [DOI] [PubMed] [Google Scholar]
- 33.Johnson DR, O'Connor JC, Dantzer R, Freund GG. Inhibition of vagally mediated immune-to-brain signaling by vanadyl sulfate speeds recovery from sickness. Proc Natl Acad Sci U S A. 2005;102:15184–15189. doi: 10.1073/pnas.0507191102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pratipanawatr T, Pratipanawatr W, Rosen C, Berria R, Bajaj M, Cusi K, Mandarino L, Kashyap S, Belfort R, DeFronzo RA. Effect of IGF-I on FFA and glucose metabolism in control and type 2 diabetic subjects. Am J Physiol Endocrinol Metab. 2002;282:E1360–E1368. doi: 10.1152/ajpendo.00335.2001. [DOI] [PubMed] [Google Scholar]
- 35.Ren J, Sowers JR, Walsh MF, Brown RA. Reduced contractile response to insulin and IGF-I in ventricular myocytes from genetically obese Zucker rats. Am J Physiol Heart Circ Physiol. 2000;279:H1708–H1714. doi: 10.1152/ajpheart.2000.279.4.H1708. [DOI] [PubMed] [Google Scholar]
- 36.Lebl J, Pruhova S, Zapletalova J, Pechova M. IGF-I resistance and Turner's syndrome. J Pediatr Endocrinol Metab. 2001;14:37–41. doi: 10.1515/jpem.2001.14.1.37. [DOI] [PubMed] [Google Scholar]
- 37.Tappy L, Fujita-Yamaguchi Y, LeBon TR, Boden G. Antibodies to insulin-like growth factor I receptors in diabetes and other disorders. Diabetes. 1988;37:1708–1714. doi: 10.2337/diab.37.12.1708. [DOI] [PubMed] [Google Scholar]
- 38.Thompson K, Dempsher DP, Bier DM, Tollefsen SE. Low prevalence of autoantibodies to the insulin-like growth factor I receptor in children with short stature. Pediatr Res. 1992;32:455–459. doi: 10.1203/00006450-199210000-00016. [DOI] [PubMed] [Google Scholar]
- 39.Dupont J, Holzenberger M. IGF type 1 receptor: a cell cycle progression factor that regulates aging. Cell Cycle. 2003;2:270–272. [PubMed] [Google Scholar]
- 40.Boldt S, Kolch W. Targeting MAPK signalling: Prometheus' fire or Pandora's box? Curr Pharm Des. 2004;10:1885–1905. doi: 10.2174/1381612043384420. [DOI] [PubMed] [Google Scholar]
- 41.Broussard SR, McCusker RH, Novakofski JE, Strle K, Shen WH, Johnson RW, Dantzer R, Kelley KW. IL-1beta impairs insulin-like growth factor i-induced differentiation and downstream activation signals of the insulin-like growth factor i receptor in myoblasts. J Immunol. 2004;172:7713–7720. doi: 10.4049/jimmunol.172.12.7713. [DOI] [PubMed] [Google Scholar]
- 42.Broussard SR, McCusker RH, Novakofski JE, Strle K, Shen WH, Johnson RW, Freund GG, Dantzer R, Kelley KW. Cytokine-hormone interactions: tumor necrosis factor alpha impairs biologic activity and downstream activation signals of the insulin-like growth factor I receptor in myoblasts. Endocrinology. 2003;144:2988–2996. doi: 10.1210/en.2003-0087. [DOI] [PubMed] [Google Scholar]
- 43.Strle K, Broussard SR, McCusker RH, Shen WH, Johnson RW, Freund GG, Dantzer R, Kelley KW. Proinflammatory cytokine impairment of insulin-like growth factor I-induced protein synthesis in skeletal muscle myoblasts requires ceramide. Endocrinology. 2004;145:4592–4602. doi: 10.1210/en.2003-1749. [DOI] [PubMed] [Google Scholar]
- 44.Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel DV. TRAF6 is a signal transducer for interleukin-1. Nature. 1996;383:443–446. doi: 10.1038/383443a0. [DOI] [PubMed] [Google Scholar]
- 45.Muzio M, Ni J, Feng P, Dixit VM. IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science. 1997;278:1612–1615. doi: 10.1126/science.278.5343.1612. [DOI] [PubMed] [Google Scholar]
- 46.Bradley JR, Pober JS. Tumor necrosis factor receptor-associated factors (TRAFs) Oncogene. 2001;20:6482–6491. doi: 10.1038/sj.onc.1204788. [DOI] [PubMed] [Google Scholar]
- 47.Janssens S, Beyaert R. Functional diversity and regulation of different interleukin-1 receptor-associated kinase (IRAK) family members. Mol Cell. 2003;11:293–302. doi: 10.1016/s1097-2765(03)00053-4. [DOI] [PubMed] [Google Scholar]
- 48.O'Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Sci STKE. 2000;2000:RE1. doi: 10.1126/stke.442000re1. [DOI] [PubMed] [Google Scholar]
- 49.Strle K, Broussard SR, McCusker RH, Shen WH, LeCleir JM, Johnson RW, Freund GG, Dantzer R, Kelley KW. C-jun N-terminal kinase mediates tumor necrosis factor-alpha suppression of differentiation in myoblasts. Endocrinology. 2006;147:4363–4373. doi: 10.1210/en.2005-1541. [DOI] [PubMed] [Google Scholar]
- 50.Schutze S, Machleidt T, Kronke M. The role of diacylglycerol and ceramide in tumor necrosis factor and interleukin-1 signal transduction. J Leukoc Biol. 1994;56:533–541. doi: 10.1002/jlb.56.5.533. [DOI] [PubMed] [Google Scholar]
- 51.Andrieu-Abadie N, Levade T. Sphingomyelin hydrolysis during apoptosis. Biochim Biophys Acta. 2002;1585:126–134. doi: 10.1016/s1388-1981(02)00332-3. [DOI] [PubMed] [Google Scholar]
- 52.Obeid LM, Hannun YA. Ceramide: a stress signal and mediator of growth suppression and apoptosis. J Cell Biochem. 1995;58:191–198. doi: 10.1002/jcb.240580208. [DOI] [PubMed] [Google Scholar]
- 53.Adam-Klages S, Schwandner R, Adam D, Kreder D, Bernardo K, Kronke M. Distinct adapter proteins mediate acid versus neutral sphingomyelinase activation through the p55 receptor for tumor necrosis factor. J Leukoc Biol. 1998;63:678–682. doi: 10.1002/jlb.63.6.678. [DOI] [PubMed] [Google Scholar]
- 54.Mathias S, Younes A, Kan CC, Orlow I, Joseph C, Kolesnick RN. Activation of the sphingomyelin signaling pathway in intact EL4 cells and in a cell-free system by IL-1 beta. Science. 1993;259:519–522. doi: 10.1126/science.8424175. [DOI] [PubMed] [Google Scholar]
- 55.Chinnaiyan AM, O'Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 56.Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell. 1995;81:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 57.Wajant H, Henkler F, Scheurich P. The TNF-receptor-associated factor family: scaffold molecules for cytokine receptors, kinases and their regulators. Cell Signal. 2001;13:389–400. doi: 10.1016/s0898-6568(01)00160-7. [DOI] [PubMed] [Google Scholar]
- 58.MacEwan DJ. TNF receptor subtype signalling: differences and cellular consequences. Cell Signal. 2002;14:477–492. doi: 10.1016/s0898-6568(01)00262-5. [DOI] [PubMed] [Google Scholar]
- 59.Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68:320–344. doi: 10.1128/MMBR.68.2.320-344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Adam-Klages S, Adam D, Wiegmann K, Struve S, Kolanus W, Schneider-Mergener J, Kronke M. FAN, a novel WD-repeat protein, couples the p55 TNF-receptor to neutral sphingomyelinase. Cell. 1996;86:937–947. doi: 10.1016/s0092-8674(00)80169-5. [DOI] [PubMed] [Google Scholar]
- 61.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 62.Huwiler A, Xin C, Brust AK, Briner VA, Pfeilschifter J. Differential binding of ceramide to MEKK1 in glomerular endothelial and mesangial cells. Biochim Biophys Acta. 2004;1636:159–168. doi: 10.1016/j.bbalip.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 63.Adam D, Wiegmann K, Adam-Klages S, Ruff A, Kronke M. A novel cytoplasmic domain of the p55 tumor necrosis factor receptor initiates the neutral sphingomyelinase pathway. J Biol Chem. 1996;271:14617–14622. doi: 10.1074/jbc.271.24.14617. [DOI] [PubMed] [Google Scholar]
- 64.Dunne A, O'Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Sci STKE. 2003;2003:re3. doi: 10.1126/stke.2003.171.re3. [DOI] [PubMed] [Google Scholar]
- 65.Colotta F, Re F, Muzio M, Bertini R, Polentarutti N, Sironi M, Giri JG, Dower SK, Sims JE, Mantovani A. Interleukin-1 type II receptor: a decoy target for IL-1 that is regulated by IL-4. Science. 1993;261:472–475. doi: 10.1126/science.8332913. [DOI] [PubMed] [Google Scholar]
- 66.McMahan CJ, Slack JL, Mosley B, Cosman D, Lupton SD, Brunton LL, Grubin CE, Wignall JM, Jenkins NA, Brannan CI, et al. A novel IL-1 receptor, cloned from B cells by mammalian expression, is expressed in many cell types. Embo J. 1991;10:2821–2832. doi: 10.1002/j.1460-2075.1991.tb07831.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Greenfeder SA, Nunes P, Kwee L, Labow M, Chizzonite RA, Ju G. Molecular cloning and characterization of a second subunit of the interleukin 1 receptor complex. J Biol Chem. 1995;270:13757–13765. doi: 10.1074/jbc.270.23.13757. [DOI] [PubMed] [Google Scholar]
- 68.Bonnert TP, Garka KE, Parnet P, Sonoda G, Testa JR, Sims JE. The cloning and characterization of human MyD88: a member of an IL-1 receptor related family. FEBS Lett. 1997;402:81–84. doi: 10.1016/s0014-5793(96)01506-2. [DOI] [PubMed] [Google Scholar]
- 69.Daun JM, Fenton MJ. Interleukin-1/Toll receptor family members: receptor structure and signal transduction pathways. J Interferon Cytokine Res. 2000;20:843–855. doi: 10.1089/10799900050163217. [DOI] [PubMed] [Google Scholar]
- 70.Hofmeister R, Wiegmann K, Korherr C, Bernardo K, Kronke M, Falk W. Activation of acid sphingomyelinase by interleukin-1 (IL-1) requires the IL-1 receptor accessory protein. J Biol Chem. 1997;272:27730–27736. doi: 10.1074/jbc.272.44.27730. [DOI] [PubMed] [Google Scholar]
- 71.Nalivaeva NN, Rybakina EG, Pivanovich I, Kozinets IA, Shanin SN, Bartfai T. Activation of neutral sphingomyelinase by IL-1beta requires the type 1 interleukin 1 receptor. Cytokine. 2000;12:229–232. doi: 10.1006/cyto.1999.0547. [DOI] [PubMed] [Google Scholar]
- 72.Janes KA, Albeck JG, Gaudet S, Sorger PK, Lauffenburger DA, Yaffe MB. A systems model of signaling identifies a molecular basis set for cytokine-induced apoptosis. Science. 2005;310:1646–1653. doi: 10.1126/science.1116598. [DOI] [PubMed] [Google Scholar]
- 73.Dranoff G. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer. 2004;4:11–22. doi: 10.1038/nrc1252. [DOI] [PubMed] [Google Scholar]
- 74.Khandwala HM, McCutcheon IE, Flyvbjerg A, Friend KE. The effects of insulin-like growth factors on tumorigenesis and neoplastic growth. Endocr Rev. 2000;21:215–244. doi: 10.1210/edrv.21.3.0399. [DOI] [PubMed] [Google Scholar]
- 75.Martin GS. Cell signaling and cancer. Cancer Cell. 2003;4:167–174. doi: 10.1016/s1535-6108(03)00216-2. [DOI] [PubMed] [Google Scholar]
- 76.Baserga R, Peruzzi F, Reiss K. The IGF-1 receptor in cancer biology. Int J Cancer. 2003;107:873–877. doi: 10.1002/ijc.11487. [DOI] [PubMed] [Google Scholar]
- 77.Wu Y, Cui K, Miyoshi K, Hennighausen L, Green JE, Setser J, LeRoith D, Yakar S. Reduced circulating insulin-like growth factor I levels delay the onset of chemically and genetically induced mammary tumors. Cancer Res. 2003;63:4384–4388. [PubMed] [Google Scholar]
- 78.Mudipalli A, Li Z, Hromchak R, Bloch A. NF-kappaB (p65/RelA) as a regulator of TNFalpha-mediated ML-1 cell differentiation. Leukemia. 2001;15:808–813. doi: 10.1038/sj.leu.2402083. [DOI] [PubMed] [Google Scholar]
- 79.Lazar-Molnar E, Hegyesi H, Toth S, Falus A. Autocrine and paracrine regulation by cytokines and growth factors in melanoma. Cytokine. 2000;12:547–554. doi: 10.1006/cyto.1999.0614. [DOI] [PubMed] [Google Scholar]
- 80.Rozen F, Zhang J, Pollak M. Antiproliferative action of tumor necrosis factor-alpha on MCF-7 breastcancer cells is associated with increased insulin-like growth factor binding protein-3 accumulation. Int J Oncol. 1998;13:865–869. doi: 10.3892/ijo.13.4.865. [DOI] [PubMed] [Google Scholar]
- 81.Storz P, Doppler H, Horn-Muller J, Muller G, Pfizenmaier K. TNF down-regulation of receptor tyrosine kinase-dependent mitogenic signal pathways as an important step in cytostasis induction and commitment to apoptosis of Kym-1 rhabdomyosarcoma cells. Cell Death Differ. 2000;7:955–965. doi: 10.1038/sj.cdd.4400732. [DOI] [PubMed] [Google Scholar]
- 82.Katz J, Nasatzky E, Werner H, Le Roith D, Shemer J. Tumor necrosis factor alpha and interferon gamma--induced cell growth arrest is mediated via insulin-like growth factor binding protein-3. Growth Horm IGF Res. 1999;9:174–178. doi: 10.1054/ghir.1999.0101. [DOI] [PubMed] [Google Scholar]
- 83.Shalita-Chesner M, Katz J, Shemer J, Werner H. Regulation of insulin-like growth factor-I receptor gene expression by tumor necrosis factor-alpha and interferon-gamma. Mol Cell Endocrinol. 2001;176:1–12. doi: 10.1016/s0303-7207(01)00484-1. [DOI] [PubMed] [Google Scholar]
- 84.Simonitsch I, Krupitza G. Autocrine self-elimination of cultured ovarian cancer cells by tumour necrosis factor alpha (TNF-alpha) Br J Cancer. 1998;78:862–870. doi: 10.1038/bjc.1998.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shen WH, Jackson ST, Broussard SR, McCusker RH, Strle K, Freund GG, Johnson RW, Dantzer R, Kelley KW. IL-1beta suppresses prolonged Akt activation and expression of E2F-1 and cyclin A in breast cancer cells. J Immunol. 2004;172:7272–7281. doi: 10.4049/jimmunol.172.12.7272. [DOI] [PubMed] [Google Scholar]
- 86.Shen WH, Yin Y, Broussard SR, McCusker RH, Freund GG, Dantzer R, Kelley KW. Tumor necrosis factor alpha inhibits cyclin A expression and retinoblastoma hyperphosphorylation triggered by insulin-like growth factor-I induction of new E2F-1 synthesis. J Biol Chem. 2004;279:7438–7446. doi: 10.1074/jbc.M310264200. [DOI] [PubMed] [Google Scholar]
- 87.Czifra G, Toth IB, Marincsak R, Juhasz I, Kovacs I, Acs P, Kovacs L, Blumberg PM, Biro T. Insulin-like growth factor-I-coupled mitogenic signaling in primary cultured human skeletal muscle cells and in C2C12 myoblasts. A central role of protein kinase Cdelta. Cell Signal. 2006;18:1461–1472. doi: 10.1016/j.cellsig.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 88.Coolican SA, Samuel DS, Ewton DZ, McWade FJ, Florini JR. The mitogenic and myogenic actions of insulin-like growth factors utilize distinct signaling pathways. J Biol Chem. 1997;272:6653–6662. doi: 10.1074/jbc.272.10.6653. [DOI] [PubMed] [Google Scholar]
- 89.Foulstone EJ, Huser C, Crown AL, Holly JM, Stewart CE. Differential signalling mechanisms predisposing primary human skeletal muscle cells to altered proliferation and differentiation: roles of IGF-I and TNFalpha. Exp Cell Res. 2004;294:223–235. doi: 10.1016/j.yexcr.2003.10.034. [DOI] [PubMed] [Google Scholar]
- 90.Roth SM, Metter EJ, Ling S, Ferrucci L. Inflammatory factors in age-related muscle wasting. Curr Opin Rheumatol. 2006;18:625–630. doi: 10.1097/01.bor.0000245722.10136.6d. [DOI] [PubMed] [Google Scholar]
- 91.Barbe MF, Barr AE. Inflammation and the pathophysiology of work-related musculoskeletal disorders. Brain Behav Immun. 2006;20:423–429. doi: 10.1016/j.bbi.2006.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Argiles JM, Meijsing SH, Pallares-Trujillo J, Guirao X, Lopez-Soriano FJ. Cancer cachexia: a therapeutic approach. Med Res Rev. 2001;21:83–101. doi: 10.1002/1098-1128(200101)21:1<83::aid-med4>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 93.Baracos VE. Regulation of skeletal-muscle-protein turnover in cancer-associated cachexia. Nutrition. 2000;16:1015–1018. doi: 10.1016/s0899-9007(00)00407-x. [DOI] [PubMed] [Google Scholar]
- 94.Baracos VE. Management of muscle wasting in cancer-associated cachexia: understanding gained from experimental studies. Cancer. 2001;92:1669–1677. doi: 10.1002/1097-0142(20010915)92:6+<1669::aid-cncr1495>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 95.Grounds MD. Reasons for the degeneration of ageing skeletal muscle: a central role for IGF-1 signalling. Biogerontology. 2002;3:19–24. doi: 10.1023/a:1015234709314. [DOI] [PubMed] [Google Scholar]
- 96.Moldawer LL, Copeland EM., 3rd Proinflammatory cytokines, nutritional support, and the cachexia syndrome: interactions and therapeutic options. Cancer. 1997;79:1828–1839. [PubMed] [Google Scholar]
- 97.Tracey KJ. Lethal weight loss: the focus shifts to signal transduction. Sci STKE. 2002;2002:PE21. doi: 10.1126/stke.2002.130.pe21. [DOI] [PubMed] [Google Scholar]
- 98.Baronzio G, Zambelli A, Comi D, Barlocco A, Baronzio A, Marchesi P, Gramaglia A, Castiglioni E, Mafezzoni A, Beviglia E, Crespi F, Cargnel AI, Pravettoni G. Proinflammatory and regulatory cytokine levels in AIDS cachexia. In Vivo. 1999;13:499–502. [PubMed] [Google Scholar]
- 99.Chang HR, Bistrian B. The role of cytokines in the catabolic consequences of infection and injury. JPEN J Parenter Enteral Nutr. 1998;22:156–166. doi: 10.1177/0148607198022003156. [DOI] [PubMed] [Google Scholar]
- 100.Chang HR, Dulloo AG, Bistrian BR. Role of cytokines in AIDS wasting. Nutrition. 1998;14:853–863. doi: 10.1016/S0899-9007(98)00108-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jain S, Golde DW, Bailey R, Geffner ME. Insulin-like growth factor-I resistance. Endocr Rev. 1998;19:625–646. doi: 10.1210/edrv.19.5.0348. [DOI] [PubMed] [Google Scholar]
- 102.Kelley KW. From hormones to immunity: the physiology of immunology. Brain Behav Immun. 2004;18:95–113. doi: 10.1016/j.bbi.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 103.Crown AL, Cottle K, Lightman SL, Falk S, Mohamed-Ali V, Armstrong L, Millar AB, Holly JM. What is the role of the insulin-like growth factor system in the pathophysiology of cancer cachexia, and how is it regulated? Clin Endocrinol (Oxf) 2002;56:723–733. doi: 10.1046/j.1365-2265.2002.01540.x. [DOI] [PubMed] [Google Scholar]
- 104.McNurlan MA, Garlick PJ, Steigbigel RT, DeCristofaro KA, Frost RA, Lang CH, Johnson RW, Santasier AM, Cabahug CJ, Fuhrer J, Gelato MC. Responsiveness of muscle protein synthesis to growth hormone administration in HIV-infected individuals declines with severity of disease. J Clin Invest. 1997;100:2125–2132. doi: 10.1172/JCI119747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Garlick PJ, McNurlan MA, Bark T, Lang CH, Gelato MC. Hormonal regulation of protein metabolism in relation to nutrition and disease. J Nutr. 1998;128:356S–359S. doi: 10.1093/jn/128.2.356S. [DOI] [PubMed] [Google Scholar]
- 106.Krentz AJ, Koster FT, Crist DM, Finn K, Johnson LZ, Boyle PJ, Schade DS. Anthropometric, metabolic, and immunological effects of recombinant human growth hormone in AIDS and AIDS-related complex. J Acquir Immune Defic Syndr. 1993;6:245–251. [PubMed] [Google Scholar]
- 107.Mulligan K, Grunfeld C, Hellerstein MK, Neese RA, Schambelan M. Anabolic effects of recombinant human growth hormone in patients with wasting associated with human immunodeficiency virus infection. J Clin Endocrinol Metab. 1993;77:956–962. doi: 10.1210/jcem.77.4.8408471. [DOI] [PubMed] [Google Scholar]
- 108.Tidball JG. Inflammatory processes in muscle injury and repair. Am J Physiol Regul Integr Comp Physiol. 2005;288:R345–R353. doi: 10.1152/ajpregu.00454.2004. [DOI] [PubMed] [Google Scholar]
- 109.Hodgetts S, Radley H, Davies M, Grounds MD. Reduced necrosis of dystrophic muscle by depletion of host neutrophils, or blocking TNFalpha function with Etanercept in mdx mice. Neuromuscul Disord. 2006;16:591–602. doi: 10.1016/j.nmd.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 110.Coletti D, Yang E, Marazzi G, Sassoon D. TNFalpha inhibits skeletal myogenesis through a PW1-dependent pathway by recruitment of caspase pathways. Embo J. 2002;21:631–642. doi: 10.1093/emboj/21.4.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ji SQ, Neustrom S, Willis GM, Spurlock ME. Proinflammatory cytokines regulate myogenic cell proliferation and fusion but have no impact on myotube protein metabolism or stress protein expression. J Interferon Cytokine Res. 1998;18:879–888. doi: 10.1089/jir.1998.18.879. [DOI] [PubMed] [Google Scholar]
- 112.Harrington MA, Daub R, Song A, Stasek J, Garcia JG. Interleukin 1 alpha mediated inhibition of myogenic terminal differentiation: increased sensitivity of Ha-ras transformed cultures. Cell Growth Differ. 1992;3:241–248. [PubMed] [Google Scholar]
- 113.Szalay K, Razga Z, Duda E. TNF inhibits myogenesis and downregulates the expression of myogenic regulatory factors myoD and myogenin. Eur J Cell Biol. 1997;74:391–398. [PubMed] [Google Scholar]
- 114.Frost RA, Nystrom GJ, Lang CH. Tumor necrosis factor-alpha decreases insulin-like growth factor-I messenger ribonucleic acid expression in C2C12 myoblasts via a Jun N-terminal kinase pathway. Endocrinology. 2003;144:1770–1779. doi: 10.1210/en.2002-220808. [DOI] [PubMed] [Google Scholar]
- 115.Frost RA, Lang CH, Gelato MC. Transient exposure of human myoblasts to tumor necrosis factor-alpha inhibits serum and insulin-like growth factor-I stimulated protein synthesis. Endocrinology. 1997;138:4153–4159. doi: 10.1210/endo.138.10.5450. [DOI] [PubMed] [Google Scholar]
- 116.Levade T, Jaffrezou JP. Signalling sphingomyelinases: which, where, how and why? Biochim Biophys Acta. 1999;1438:1–17. doi: 10.1016/s1388-1981(99)00038-4. [DOI] [PubMed] [Google Scholar]
- 117.Rybakina EG, Nalivaeva NN, Pivanovich YU, Shanin SN, Kozinets A, Korneva EA. The role of neutral sphingomyelinase in interleukin-1beta signal transduction in mouse cerebral cortex cells. Neurosci Behav Physiol. 2001;31:439–444. doi: 10.1023/a:1010448930994. [DOI] [PubMed] [Google Scholar]
- 118.Xu J, Yeh CH, Chen S, He L, Sensi SL, Canzoniero LM, Choi DW, Hsu CY. Involvement of de novo ceramide biosynthesis in tumor necrosis factor-alpha/cycloheximide-induced cerebral endothelial cell death. J Biol Chem. 1998;273:16521–16526. doi: 10.1074/jbc.273.26.16521. [DOI] [PubMed] [Google Scholar]
- 119.Merrill AH, Jr, Jones DD. An update of the enzymology and regulation of sphingomyelin metabolism. Biochim Biophys Acta. 1990;1044:1–12. doi: 10.1016/0005-2760(90)90211-f. [DOI] [PubMed] [Google Scholar]
- 120.Wiegmann K, Schutze S, Machleidt T, Witte D, Kronke M. Functional dichotomy of neutral and acidic sphingomyelinases in tumor necrosis factor signaling. Cell. 1994;78:1005–1015. doi: 10.1016/0092-8674(94)90275-5. [DOI] [PubMed] [Google Scholar]
- 121.Bourbon NA, Yun J, Berkey D, Wang Y, Kester M. Inhibitory actions of ceramide upon PKC-epsilon/ERK interactions. Am J Physiol Cel Physiol. 2001;280:C1403–C1411. doi: 10.1152/ajpcell.2001.280.6.C1403. [DOI] [PubMed] [Google Scholar]
- 122.Summers SA, Garza LA, Zhou H, Birnbaum MJ. Regulation of insulin-stimulated glucose transporter GLUT4 translocation and Akt kinase activity by ceramide. Mol Cell Biol. 1998;18:5457–5464. doi: 10.1128/mcb.18.9.5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhou H, Summers SA, Birnbaum MJ, Pittman RN. Inhibition of Akt kinase by cell-permeable ceramide and its implications for ceramide-induced apoptosis. J Biol Chem. 1998;273:16568–16575. doi: 10.1074/jbc.273.26.16568. [DOI] [PubMed] [Google Scholar]
- 124.Chavez JA, Knotts TA, Wang LP, Li G, Dobrowsky RT, Florant GL, Summers SA. A role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acids. J Biol Chem. 2003;278:10297–10303. doi: 10.1074/jbc.M212307200. [DOI] [PubMed] [Google Scholar]
- 125.Bourbon NA, Sandirasegarane L, Kester M. Ceramide-induced inhibition of Akt is mediated through protein kinase Czeta: implications for growth arrest. J Biol Chem. 2002;277:3286–3292. doi: 10.1074/jbc.M110541200. [DOI] [PubMed] [Google Scholar]
- 126.Manning AM, Davis RJ. Targeting JNK for therapeutic benefit: from junk to gold? Nat Rev Drug Discov. 2003;2:554–565. doi: 10.1038/nrd1132. [DOI] [PubMed] [Google Scholar]
- 127.Hauwel M, Furon E, Canova C, Griffiths M, Neal J, Gasque P. Innate (inherent) control of brain infection, brain inflammation and brain repair: the role of microglia, astrocytes, "protective" glial stem cells and stromal ependymal cells. Brain Res Brain Res Rev. 2005;48:220–233. doi: 10.1016/j.brainresrev.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 128.Sofroniew MV. Reactive astrocytes in neural repair and protection. Neuroscientist. 2005;11:400–407. doi: 10.1177/1073858405278321. [DOI] [PubMed] [Google Scholar]
- 129.Leonard BE, Myint A. Inflammation and depression: is there a causal connection with dementia? Neurotox Res. 2006;10:149–160. doi: 10.1007/BF03033243. [DOI] [PubMed] [Google Scholar]
- 130.Lucas SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. Br J Pharmacol. 2006;147 Suppl 1:S232–S240. doi: 10.1038/sj.bjp.0706400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zipp F, Aktas O. The brain as a target of inflammation: common pathways link inflammatory and neurodegenerative diseases. Trends Neurosci. 2006;29:518–527. doi: 10.1016/j.tins.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 132.Gilgun-Sherki Y, Melamed E, Offen D. Anti-inflammatory drugs in the treatment of neurodegenerative diseases: current state. Curr Pharm Des. 2006;12:3509–3519. doi: 10.2174/138161206778343091. [DOI] [PubMed] [Google Scholar]
- 133.Avitsur R, Padgett DA, Sheridan JF. Social interactions, stress, and immunity. Neurol Clin. 2006;24:483–491. doi: 10.1016/j.ncl.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 134.Zalcman SS, Siegel A. The neurobiology of aggression and rage: role of cytokines. Brain Behav Immun. 2006;20:507–514. doi: 10.1016/j.bbi.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 135.Minami M, Katayama T, Satoh M. Brain cytokines and chemokines: roles in ischemic injury and pain. J Pharmacol Sci. 2006;100:461–470. doi: 10.1254/jphs.crj06005x. [DOI] [PubMed] [Google Scholar]
- 136.Myers RR, Campana WM, Shubayev VI. The role of neuroinflammation in neuropathic pain: mechanisms and therapeutic targets. Drug Discov Today. 2006;11:8–20. doi: 10.1016/S1359-6446(05)03637-8. [DOI] [PubMed] [Google Scholar]
- 137.Godbout JP, Johnson RW. Age and neuroinflammation: a lifetime of psychoneuroimmune consequences. Neurol Clin. 2006;24:521–538. doi: 10.1016/j.ncl.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 138.Dantzer R, Kelley KW. Twenty years of research on cytokine-induced sickness behavior. Brain Behav Immun. 2007;21:153–160. doi: 10.1016/j.bbi.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Dantzer R. Cytokine, sickness behavior, and depression. Neurol Clin. 2006;24:441–460. doi: 10.1016/j.ncl.2006.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Dunn AJ, Swiergiel AH, de Beaurepaire R. Cytokines as mediators of depression: what can we learn from animal studies? Neurosci Biobehav Rev. 2005;29:891–909. doi: 10.1016/j.neubiorev.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 141.Dantzer R, Gheusi G, Johnson RW, Kelley KW. Central administration of insulin-like growth factor-1 inhibits lipopolysaccharide-induced sickness behavior in mice. Neuroreport. 1999;10:289–292. doi: 10.1097/00001756-199902050-00015. [DOI] [PubMed] [Google Scholar]
- 142.Bluthe RM, Kelley KW, Dantzer R. Effects of insulin-like growth factor-I on cytokine-induced sickness behavior in mice. Brain Behav Immun. 2006;20:57–63. doi: 10.1016/j.bbi.2005.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Palin K, Bluthe RM, McCusker RH, Moos F, Dantzer R, Kelley KW. TNFalpha-induced suckness behavior in mice with functional 55 kD TNF receptors is blocked by central IGF-I. J Neuroimmunol. 2007;187:55–60. doi: 10.1016/j.jneuroim.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.O'Connor JC, Satpathy A, Hartman ME, Horvath EM, Kelley KW, Dantzer R, Johnson RW, Freund GG. IL-1beta-mediated innate immunity is amplified in the db/db mouse model of type 2 diabetes. J Immunol. 2005;174:4991–4997. doi: 10.4049/jimmunol.174.8.4991. [DOI] [PubMed] [Google Scholar]
- 145.Bluthe RM, Dantzer R, Kelley KW. Interleukin-1 mediates behavioural but not metabolic effects of tumor necrosis factor alpha in mice. Eur J Pharmacol. 1991;209:281–283. doi: 10.1016/0014-2999(91)90184-r. [DOI] [PubMed] [Google Scholar]
- 146.Bluthe RM, Pawlowski M, Suarez S, Parnet P, Pittman Q, Kelley KW, Dantzer R. Synergy between tumor necrosis factor alpha and interleukin-1 in the induction of sickness behavior in mice. Psychoneuroendocrinology. 1994;19:197–207. doi: 10.1016/0306-4530(94)90009-4. [DOI] [PubMed] [Google Scholar]
- 147.Bluthe RM, Laye S, Michaud B, Combe C, Dantzer R, Parnet P. Role of interleukin-1beta and tumour necrosis factor-alpha in lipopolysaccharide-induced sickness behaviour: a study with interleukin-1 type I receptor-deficient mice. Eur J Neurosci. 2000;12:4447–4456. [PubMed] [Google Scholar]
- 148.Bluthe RM, Michaud B, Poli V, Dantzer R. Role of IL-6 in cytokine-induced sickness behavior: a study with IL-6 deficient mice. Physiol Behav. 2000;70:367–373. doi: 10.1016/s0031-9384(00)00269-9. [DOI] [PubMed] [Google Scholar]
- 149.Nadjar A, Bluthe RM, May MJ, Dantzer R, Parnet P. Inactivation of the cerebral NFkappaB pathway inhibits interleukin-1beta-induced sickness behavior and c-Fos expression in various brain nuclei. Neuropsychopharmacology. 2005;30:1492–1499. doi: 10.1038/sj.npp.1300755. [DOI] [PubMed] [Google Scholar]
- 150.Moraes JC, Amaral ME, Picardi PK, Calegari VC, Romanatto T, Bermudez-Echeverry M, Chiavegatto S, Saad MJ, Velloso LA. Inducible-NOS but not neuronal-NOS participate in the acute effect of TNF-alpha on hypothalamic insulin-dependent inhibition of food intake. FEBS Lett. 2006;580:4625–4631. doi: 10.1016/j.febslet.2006.07.042. [DOI] [PubMed] [Google Scholar]
- 151.Bette M, Kaut O, Schafer MK, Weihe E. Constitutive expression of p55TNFR mRNA and mitogen-specific up-regulation of TNF alpha and p75TNFR mRNA in mouse brain. J Comp Neurol. 2003;465:417–430. doi: 10.1002/cne.10877. [DOI] [PubMed] [Google Scholar]
- 152.Mori I, Goshima F, Koshizuka T, Koide N, Sugiyama T, Yoshida T, Yokochi T, Nishiyama Y, Kimura Y. Differential activation of the c-Jun N-terminal kinase/stress-activated protein kinase and p38 mitogen-activated protein kinase signal transduction pathways in the mouse brain upon infection with neurovirulent influenza A virus. J Gen Virol. 2003;84:2401–2408. doi: 10.1099/vir.0.19188-0. [DOI] [PubMed] [Google Scholar]
- 153.Simen BB, Duman CH, Simen AA, Duman RS. TNFalpha signaling in depression and anxiety: behavioral consequences of individual receptor targeting. Biol Psychiatry. 2006;59:775–785. doi: 10.1016/j.biopsych.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 154.Swiergiel AH, Dunn AJ. Feeding, exploratory, anxiety- and depression-related behaviors are not altered in interleukin-6-deficient male mice. Behav Brain Res. 2006;171:94–108. doi: 10.1016/j.bbr.2006.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sparkman NL, Buchanan JB, Heyen JR, Chen J, Beverly JL, Johnson RW. Interleukin-6 facilitates lipopolysaccharide-induced disruption in working memory and expression of other proinflammatory cytokines in hippocampal neuronal cell layers. J Neurosci. 2006;26:10709–10716. doi: 10.1523/JNEUROSCI.3376-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Nicolas V, Prewett A, Bettica P, Mohan S, Finkelman RD, Baylink DJ, Farley JR. Age-related decreases in insulin-like growth factor-I and transforming growth factor-beta in femoral cortical bone from both men and women: implications for bone loss with aging. J Clin Endocrinol Metab. 1994;78:1011–1016. doi: 10.1210/jcem.78.5.8175953. [DOI] [PubMed] [Google Scholar]
- 157.Pons S, Torres-Aleman I. Insulin-like growth factor-I stimulates dephosphorylation of ikappa B through the serine phosphatase calcineurin (protein phosphatase 2B) J Biol Chem. 2000;275:38620–38625. doi: 10.1074/jbc.M004531200. [DOI] [PubMed] [Google Scholar]
- 158.Vallee S, Fouchier F, Bremond P, Briand C, Marvaldi J, Champion S. Insulin-like growth factor-1 downregulates nuclear factor kappa B activation and upregulates interleukin-8 gene expression induced by tumor necrosis factor alpha. Biochem Biophys Res Commun. 2003;305:831–839. doi: 10.1016/s0006-291x(03)00866-0. [DOI] [PubMed] [Google Scholar]
- 159.Kooijman R, Coppens A. Insulin-like growth factor-I stimulates IL-10 production in human T cells. J Leukoc Biol. 2004;76:862–867. doi: 10.1189/jlb.0404248. [DOI] [PubMed] [Google Scholar]
- 160.Warzecha Z, Dembinski A, Ceranowicz P, Konturek SJ, Tomaszewska R, Stachura J, Konturek PC. IGF-1 stimulates production of interleukin-10 and inhibits development of caerulein-induced pancreatitis. J Physiol Pharmacol. 2003;54:575–590. [PubMed] [Google Scholar]
- 161.Bluthe RM, Castanon N, Pousset F, Bristow A, Ball C, Lestage J, Michaud B, Kelley KW, Dantzer R. Central injection of IL-10 antagonizes the behavioural effects of lipopolysaccharide in rats. Psychoneuroendocrinology. 1999;24:301–311. doi: 10.1016/s0306-4530(98)00077-8. [DOI] [PubMed] [Google Scholar]
- 162.Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 163.Strle K, Zhou JH, Shen WH, Broussard SR, Johnson RW, Freund GG, Dantzer R, Kelley KW. Interleukin-10 in the brain. Crit Rev Immunol. 2001;21:427–449. [PubMed] [Google Scholar]
- 164.Carro E, Trejo JL, Busiguina S, Torres-Aleman I. Circulating insulin-like growth factor I mediates the protective effects of physical exercise against brain insults of different etiology and anatomy. J Neurosci. 2001;21:5678–5684. doi: 10.1523/JNEUROSCI.21-15-05678.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Samuel DS, Ewton DZ, Coolican SA, Petley TD, McWade FJ, Florini JR. Raf-1 activation stimulates proliferation and inhibits IGF-stimulated differentiation in L6A1 myoblasts. Horm Metab Res. 1999;31:55–64. doi: 10.1055/s-2007-978699. [DOI] [PubMed] [Google Scholar]
- 166.Landin-Wilhelmsen K, Lundberg PA, Lappas G, Wilhelmsen L. Insulin-like growth factor I levels in healthy adults. Horm Res. 2004;62 Suppl 1:8–16. doi: 10.1159/000080753. [DOI] [PubMed] [Google Scholar]
- 167.Street ME, Ziveri MA, Spaggiari C, Viani I, Volta C, Grzincich GL, Virdis R, Bernasconi S. Inflammation is a modulator of the insulin-like growth factor (IGF)/IGF-binding protein system inducing reduced bioactivity of IGFs in cystic fibrosis. Eur J Endocrinol. 2006;154:47–52. doi: 10.1530/eje.1.02064. [DOI] [PubMed] [Google Scholar]
- 168.Arosio M, Ronchi CL, Beck-Peccoz P, Gebbia C, Giavoli C, Cappiello V, Conte D, Peracchi M. Effects of modified sham feeding on ghrelin levels in healthy human subjects. J Clin Endocrinol Metab. 2004;89:5101–5104. doi: 10.1210/jc.2003-032222. [DOI] [PubMed] [Google Scholar]