

Abstract
A method for sampling and electrophoretic analysis of aqueous plugs segmented in a stream of immiscible oil is described. In the method, an aqueous buffer and oil stream flow parallel to each other to form a stable virtual wall in a microfabricated K-shaped fluidic element. As aqueous sample plugs in the oil stream make contact with the virtual wall coalescence occurs and sample is electrokinetically transferred to the aqueous stream. Using this virtual wall, two methods of injection for channel electrophoresis were developed. In the first, discrete sample zones flow past the inlet of an electrophoresis channel and a portion is injected by electroosmotic flow, termed the “discrete injector”. With this approach at least 800 plugs could be injected without interruption from a continuous segmented stream with 5.1% RSD in peak area. This method generated up to 1,050 theoretical plates; although analysis of the injector suggested that improvements may be possible. In a second method, aqueous plugs are sampled in a way that allows them to form a continuous stream that is directed to a microfluidic cross-style injector, termed the “desegmenting injector”. This method does not analyze each individual plug but instead allows periodic sampling of a high-frequency stream of plugs. Using this system at least 1000 injections could be performed sequentially with 5.8% RSD in peak area and 53,500 theoretical plates. This method was demonstrated to be useful for monitoring concentration changes from a sampling device with 10 s temporal resolution. Aqueous plugs in segmented flows have been applied to many different chemical manipulations including synthesis, assays, sampling processing and sampling. Nearly all such studies have used optical methods to analyze plug contents. This method offers a new way to analyze such samples and should enable new applications of segmented flow systems.
Keywords: Segmented Flow, Electrophoresis, Microfluidic, Virtual Wall
Introduction
Multiphase flows in microfluidic devices have recently received considerable interest because of their potential for improving sample manipulation and chemical reaction control on low volume systems 1–4. Segmented flow, in which sample plugs or droplets are carried by an immiscible fluid, is one type of multiphase system that has been extensively explored 5. Such plugs can be thought of as small test tubes in which chemical reactions and assays can be performed. Segmented flows have a number of useful properties including reduction of axial dispersion or mixing between adjacent plugs 6, rapid mixing within a plug due to induced recirculation 7, and large interfacial areas that contribute to high mass or energy transfer 8. Methods for generation 9,10, sorting 11, merging and mixing 12,13, fusion 14, freezing 15, splitting 16, and concentration enrichment of plugs 17 in the fL to nL volume range have been demonstrated. These elegant manipulations of droplets and plugs have enabled numerous applications including single cell assays and imaging 18, organismal analysis 19, bacterial cultivation 20, PCR 21,22, enzyme reactions 23, synthesis 13, high-throughput screening 24, and microdialysis sampling 25 where the properties of multiphase flows have led to substantial improvements.
Despite the considerable research effort aimed at manipulating sample plugs and applying them to technical problems, relatively few methods exist for sampling and analyzing contents of plugs which greatly limits their potential utility. Nearly all studies have relied on optical probing of plugs by fluorescence or absorbance. While many useful assays can be developed with this approach, frequently higher resolving power is necessary for complex mixtures or multi-analyte analysis. One approach to this problem that has been reported is collecting plugs off-line for MALDI-MS analysis 26. Another powerful approach to analyzing plugs would be electrophoresis. A previous seminal study has demonstrated an approach to use channel electrophoresis for analysis of individual pL droplets 27. This interesting method was proven useful for electrophoresis of individual droplets, but was not demonstrated for a continuous stream of plugs thus precluding applications requiring high throughput. Furthermore, the efficiency of the resulting separations was relatively low due to the use of PDMS as a substrate and a design that allowed oil to enter the electrophoresis channel.
In this work we describe two microfluidic devices, both based on a K-shaped element which can transfer small amounts from segmented plugs flowing through a larger channel to a continuous stream of aqueous buffer flowing through a smaller sample channel. These two different devices were developed for two different classes of applications in which aqueous plugs in an immiscible carrier stream would be used. In the first class, segmented plugs are generated as discrete, independent samples and analysis of each individual plug desired, for example, in high-throughput screening applications 22. In another class of applications, plugs are formed from a continuous sample stream, for example the effluent of a microdialysis probe 25, to preserve temporal resolution of concentration changes by preventing the axial dispersion of zones that would occur in parabolic flow to the analytical system. In such applications, if plugs are generated at a high rate (greater than 1 Hz) it is not generally necessary to analyze every plug; but, sampling the stream periodically or re-forming a continuous stream is sufficient. Appropriate to these different needs we have developed two methods for sampling segmented plugs for injection to on-chip electrophoresis. The “discrete” injector performs an injection from each plug, without carryover between plugs, for analysis of plugs as independent samples. The “desegmenting” injector samples plugs into closely-spaced zones in the continuously-flowing buffer stream, which is periodically injected for electrophoresis. The “desegmenting” system uses an electrokinetic gated cross injector, which provides higher efficiency in the CE separation than the discrete injector.
Experimental
Reagents
All reagents were used as received unless stated otherwise. All chemicals were purchased from Sigma Aldrich (St. Louis, MO) with the following exceptions. Naphthalene-2,3-dicarboxaldehyde (NDA) and 5-fluorescein isothiocyanate (5-FITC) were purchased from Invitrogen (Eugene, OR). Salts for preparing artificial cerebral spinal fluid (aCSF) were purchased from Fisher Scientific (Pittsburgh, PA).
Aqueous solutions were prepared using house deionized water that was further deionized to 18 MΩ resistivity using a Barnstead E-pure system (Series 1090, Barnstead Thermolyne Cooperation, Dubuque, IA). Artificial cerebral spinal fluid consisted of: 145 mM sodium chloride (NaCl), 2.68 mM potassium chloride (KCl), 1.01 mM magnesium sulfate (MgSO4), 1.22 mM calcium chloride (CaCl2), 1.55 mM sodium phosphate dibasic (Na2HPO4), and 0.45 mM sodium phosphate monobasic (NaH2PO4).
Microchip fabrication
The microfluidic devices used in this work had two depths of channel, a deeper channel for segmented flow and a shallower channel for electrophoresis, that overlapped as shown in Figure 1A. To prepare such chips, channels were made at a given depth in two different glass plates, which were then bonded together as shown in Figure 1B. The result was a microfluidic network capable of generating segmenting flow and coalescing the aqueous plugs into an electrophoresis stream. Channels were prepared using previously described photolithographic and HF etching methods 28. Briefly, 50,800 dpi photomasks were designed in AutoCAD and purchased from Fineline Imaging (Colorado Springs, CO). Photoresist-chromium coated slides (1 in × 3 in × 1.1 mm; Optical Density 2.8; Reflectivity 8%; Resist Type AZ 1500; Resist Thickness 5300Å; Bake time 30 min at 103 °C) were purchased from Telic (Valencia, CA). Masks were placed in contact with the photoresist-chromium slide using a quartz cover plate and exposed for 8 s using the 365 nm mercury line from a collimated flood exposure system (Optical Associates, Inc. Milpitas, CA) at a power of 45 mJ/cm2. Following exposure, slides were developed using AZ 726 MIF developer from AZ Electronic Materials USA Corp (Somerville, NJ). Chromium was then etched using CEP 200 micro-chrome etchant from HTA Enterprises (San Jose, CA). After both the electrophoresis channel manifold and the plug generation manifold were chromium etched for 2 min, they were HF etched to 10 μm and 80 μm depths, respectively. After etching, access holes were drilled using #79 diamond drill bits from Tartan Tool Company (Troy, MI) using a vertical drilling press from CPO Delta (South Pasadena, CA). The resulting glass slides were prepared for bonding by soaking them in “piranha solution” consisting of a 3:1 (V:V) mixture of H2SO4/H2O2 for 40 min and then “RCA” consisting of 5:1:1 (V/V/V) H2O/NH4OH/H2O2 at 60 °C for 40 min. After the acid and base baths the slides were thoroughly rinsed with water and then aligned under a microscope. After aligning the slides they were air dried for 2 hr at room temperature. At this point the chip could be picked up without loss of alignment. The aligned slides were placed in a Neytech Centurian Qex furnace (Pacific Combustion, Los Angeles, CA) for bonding at 610 °C. Reservoirs and Nanoports from Upchurch Scientific (Oak Harbor, OR) were applied to glass chips using the manufacturer’s protocol.
Figure 1.

A) Illustrates the cross section of the fabricated fluidic manifold at the K interface between the segmented flow and electrophoresis channels. The segmented flow channels are etched to 75 μm depths and 250 μm widths. The electrophoresis channels have 7.5 μm depths and 20 μm widths. B) The fabrication scheme for the fluidic manifold is illustrated. The segmented flow and electrophoresis channels are etched on separate substrates and are aligned and bonded. Perfluorodecalin (PFD) reservoir, and the amino acid (AA) reservoir are labeled. The T junction that connects these two reservoirs serves to break up the AA flow into plugs that are transported downstream in a continuous stream of perfluorodecalin. Arrows indicate direction of flow. After generation, these plugs are pumped into the segmented flow serpentine prior to coalescence at the K interface. Inset shows photomicrograph of the K-interface.
Surface Patterning of Octadecyltrichlorosilane
To achieve stable segmented flow and help form a virtual wall, the deeper channels were selectively derivatized with octadecyltrichlorosilane (OTCS) using previously described procedures 3. Figure 2 illustrates the patterning techniques and the final product. Briefly, the entire microfluidic manifold was initially filled with n-hexadecane by adding the solvent to all reservoirs and applying vacuum at the common waste reservoir. After air bubbles were removed from the manifold, the solvent in the plug generation reservoirs was replaced with freshly prepared 0.2% (V/V) OTCS in n-hexadecane so that a laminar interface of derivatization reagent and solvent was created as shown in Figure 2 yielding selective derivatization. The OTCS-hexadecane solution was flowed for 10 min at ~ 5 μL/min, after which the OTCS-hexadecane was removed from the open reservoirs using a pipette and replaced with n-hexadecane for rinsing. All channels were then sequentially rinsed with methanol, water and air for 5 min each. The process of changing solutions for rinsing was performed without interrupting flow. Since the vacuum at the common waste reservoir drives flow in all channels, rinsing these channels and changing the reservoir content does not significantly modify either laminar flow or flow rates within the channels.
Figure 2.

A) Illustrates the patterning of hydrophobic regions on a glass microfluidic channel using laminar flow. A vacuum was applied to the common waste reservoir on the manifold to flow octadecyltrichlorosilane (OTCS) solution through the segmented flow channel. Solvent was also simultaneously pumped through the electrophoresis channels to the K interface as shown in the inset. B) Micrograph showing the completed K fluid element and virtual wall formed by flowing perfluorodecalin through the large straight channel (right to left) and aqueous buffer through the V shaped channel (right to left).
Chip operation
Flow through the chips was driven by syringe pumps (Model 402, CMA Microdialysis, North Chelmsford, MA) loaded with 100 μL Hamilton syringes (Reno, NV) and connected via fused silica capillary (Polymicro, Phoenix, AZ) to the chip using Nanoport fittings (Upchurch Scientific, Oak Harbor, OR). The process of connecting the syringes with the Nanoports can create temporary pressure heads that will dissipate over the period of about 10 min. Hence, after connecting the syringes measurements were not started until a 10 min equilibration period. A stream of aqueous sample plugs was formed on the chip at the T-intersection by pumping aqueous samples and perfluorodecalin at 3 μL/min and 300 nL/min respectively 25. Under these conditions, plugs were formed at 1 Hz with a volume of 12 nL each. Sample solutions consisted of fluorescein or NDA-derivatized amino acids or FITC-derivatized amino acids dissolved in aCSF. Amino acid derivatives were prepared using previously described procedures 29–31. Another syringe pump was used to flow electrophoresis buffer into the chip consisting of 10 mM sodium tetraborate, and 0.9 mM hydroxypropyl-β-cyclodextran (HPβCD) at a pH of 10. Voltage for performing electrophoresis was applied using a CZE1000R power supply (Spellman, Hauppague, NY). Plug volume and temporal intervals were determined from micrographs collected sequentially using a Nikon stereoscopic microscope (Diaphot 300, Melville, NY). Sampling of plugs and electrophoresis injections were imaged using an inverted, epi-illumination microscope (RCM 8000, Nikon, Melville, NY) with a 10X objective. To collect electropherograms, chips were mounted on a confocal LIF detector with either 488 or 440 nm excitation as described previously 30,32. Igor Pro from Wavemetrics Inc. (Lake Oswego, OR) was utilized to analyze electropherograms, injection widths, separation efficiencies, and reproducibility.
Results and Discussion
We developed two methods to perform electrophoretic injection and separation of a stream of aqueous sample plugs segmented by oil. Both methods utilize a K-shaped microfluidic structure, shown in Figure 2B, to transfer a portion of aqueous plugs from the segmented flow into a stream of buffer. The K structure allows formation of a virtual wall, that is a stable interface of oil and buffer flowing side-by-side (see Figure 2B). To operate the system, the segmented flow and electrophoresis buffer (cross flow) are pumped through their respective channels as shown in Figure 3A. While oil is present in the K interface, a stable virtual wall is formed. When an aqueous plug in oil enters the interface, it contacts the aqueous buffer allowing coalescence or merging of the aqueous plug and electrophoresis buffer such that some of the aqueous plug is transferred or extracted to the electrophoresis buffer cross flow. A sequence of such events for a plug containing FITC-serine is shown in Figure 3B. In the discrete injector this transferred sample is carried past the inlet of the narrow electrophoresis channel by the cross flow so that a portion is pulled into that channel (see Figure 3B) by the electric field applied (see Figure 3A). The remaining plug and transferred portion are washed away by the segemented and cross flow respectively. In the desegmented injector, the same geometry is used but the plugs are delivered fast enough to the interface that they reform a constant stream in the downstream channel (labeled the electrophoresis channel in Figure 3B). This downstream channel, which we call a sampling channel in this case, allows the continuous analyte stream to be directed to cross-style electrokinetic gate injector downstream. We first describe the formation and operation of the K structure and virtual wall. We then cover the detailed operation and characterization of each injector.
Figure 3.

Illustration of coalescence and discrete injection using the K interface. A–B) The microfluidic scheme for the discrete injection scheme with a 10 cm electrophoresis channel, C) micrographs illustrating the approach and coalescence of plugs using a cross flow of 100 nL/min, and D) series electropherograms resulting from sampling and injection of plugs using the discrete injector and separation on a 5 cm long electrophoresis channel at 500 V/cm with 10 mM sodium tetraborate, pH 9.0 separation buffer. The fluorescent plugs consist of 1 μM serine derivatized with FITC.
K-fluidic Element and Virtual Wall Formation
A key to developing reproducible injections was forming a stable virtual wall in the K-interface. Virtual walls have previously been reported with application to extraction8,33–36 and membrane synthesis 37. The stability of a virtual wall depends on several variables including the hydrophobicity of surfaces, surface tension, viscosity, and microfluidic channel depth 3. It has previously been shown for example, that chemical patterning of a surface to define regions of hydrophilicity and hydrophobicity within a channel manifold can aid in forming a stable interface between flowing organic and water streams3. In such systems, non-polar solvents preferentially wet and flow over hydrophobic surfaces while aqueous samples are confined over the hydrophilic regions. It has also been shown that having channels of different depths, but with fluidic contact between them, can stabilize virtual walls 8,33.
In initial experiments we found that although both approaches used individually allowed stable virtual walls to be formed with continuous streams of oil and aqueous solution, it was necessary to use them in combination to generate a wall that was stable to segmented flows. Thus, the K fluidic element used in this work consists of a linear segmented flow channel (250 μm wide × 80 μm deep) and a V shaped “cross flow” channel (50 μm wide × 10 μm deep). The difference in fluidic resistance between the segmented flow channel and the electrophoresis channel reduced the movement of the non-aqueous carrier phase (which was typically flowing faster than the aqueous buffer stream) into the electrophoresis channel. To form the channels at different depths we etched the top of one substrate to one depth and the bottom of another to a different depth. These substrates were then aligned and bonded. Alignment differences of up to 15 μm were not observed to affect the fluid dynamics at the interface (n = 5 chips). The separation or sampling channel is located 65 μm from the interface to provide enough working distance so that small differences in alignment of the two layers would not produce a manifold where the electrophoresis channel contacted the segmented flow channel.
In addition to having a differential channel height, we also patterned the surface such that the oil (perfluorodecalin) channel was made hydrophobic by covalent attachment of octadecylsilane (C18) while the aqueous channel retained a native, hydrophilic glass surface. This surface pattern helped to prevent the perfluorodecalin from encroaching on the electrophoresis buffer channel and the electrophoresis buffer from pushing into the oil flow. Keeping the segmented flow channel hydrophobic also ensured that the oil wetted the surface and not the aqueous plugs thus preventing mass transfer between plugs. Keeping the aqueous channels hydrophilic helped ensure good electroosmotic flow and electrophoresis properties. The combination of both differential surface energies and channel heights yielded a stable interface between a segmented stream with flow rates from 3.5 to 5 μl/min (linear velocities of 0.29 to 0.42 cm/s) and an aqueous stream with flow rates of 40 to 220 nL/min (linear velocities of 0.03 to 0.42 cm/s). The relative flow rates of each stream at the K interface can be manipulated within these limits to control injection.
We only tested perfluorodecalin and electrophoresis buffer in these experiments; however a variety of other oils have been used in segmented flows 22,38,39. We expect that similar conditions could be used with such oils. Perfluorinated oils like perfluorodecalin have the advantage of low viscosity to allow low pressure pumping, wall wetting to hydrophobic channels with contact angles > 100°, and excellent resistance to extraction of analyte from aqueous plugs.
Discrete Injector
As described above and shown in Figure 3A and B, the K interface allows a series of samples can be injected without using relays or changing any parameters such as applied voltage. A series of sample plugs can be sampled and analyzed with 5.8% RSDs of peak area as illustrated by the sequence of traces in Figure 3C. At least 800 plugs (corresponding to ~130 min of operation) could be injected without interruption in this system.
Injection volume in the discrete injector is controlled by the electric field and temporal width of the plug. The temporal width of a plug is defined as the time it takes to traverse a fixed point in space and is governed by the flow velocity and plug spatial width. Thus, the injector performance is coupled to the size of sample plugs formed. To quantify how these factors affect the electrophoretic separation, we examined peak efficiency and area with respect to each of these variables as summarized in Figure 4.
Figure 4.

A) Effect of plug length on efficiency of peak for fluorescein dissolved in aCSF obtained using the discrete injection. The flow rate for the perfluorodecalin and separation buffer was 1 μL/min and 100 nL/min, respectively, whereas the sample flow rate was varied to generate different sized plugs and ranged from 30 nL/min-500 nL/min. The plug length was calculated 500 μm from the virtual wall of the fusion interface. The electrophoresis was performed with a field strength of 1 kV/cm and a separation buffer of 10 mM sodium tetraborate, 0.9 mM HPβ-CD at a pH of 9.7. B–C) The relationship between the field strength and cross flow for fluorescein plugs generated using a flow rate of 1 μL/min for the PFD and 300 nL/min for the sample. Cross flow was 100 nL/min. D–F) Peak area comparisons using the same segmentation parameters in B–C.
Decreasing temporal plug width improved efficiency as shown in Figure 4A by decreasing injection bandwidth. Although the oil flow rate may be increased to generate even more narrow injections, this strategy is limited in at least two ways. First, faster flows decrease the interval between sample plugs and leads to overlapping electropherograms if separation time is longer than the interval time. Secondly, higher flow rates can result in unstable virtual walls. We believe that using a different method for forming plugs that allows for plugs with shorter spatial width and longer intervals would be beneficial for improving efficiency.
Efficiency of a separation is expected to increase linearly with electric field as long as Joule heating is not a factor; however, in this system we found that greater electric field strengths had relatively little effect on efficiency (Figure 4B). This is likely because even though increasing electric field improves separation efficiency, it also increases the amount injected (see Figure 4E) leading to an extra column broadening effect that negates reduced broadening in the electrophoresis channel. Fields above 600 V/cm also tended to decrease reliability of the injections due to bubble formation in the interfacial zone.
In addition to field strength and plug length, the electrophoresis buffer cross flow rate has an effect on peak area and efficiency. The cross flow serves two functions: 1) it acts to counterbalance the pressure exerted by the segmented flow stream on the virtual wall, and 2) it washes analytes out of the coalescence interface and terminates the injection. As shown in Figure 4F, as cross flow is increased above 100 nL/min (linear velocities of 0.23 cm/s) the bulk motion of the solute outstrips the electrophoretic force in the direction of the electric field and results in undetectable analyte injection. As cross flow is decreased more analyte is injected, indicated by the increase in peak area, Figure 4F, while peak efficiency decreases (Figure 4C). The decrease of the efficiency with decreasing cross flows is a result of the longer residence time of the sample plug in the interface and longer injections. Thus, it is necessary to balance the goal of removing the analyte from the interface as soon as possible after a coalescence event, but still enabling sufficient time for electrokinetic injection into the electrophoresis channel.
To better understand the limits on separation efficiency imposed by the injector and its relationship to the band broadening in the electrophoresis channel, we examined peak variance, a measure of peak width, along the channel length, shown in Figure 5 40. For this experiment, sample plugs of 1 μM fluorescein were generated at 1 Hz and coalesced at a cross flow of 100 nL/min and separated at a field strength of 250 V/cm. The linear trend with a slope of 4.1 × 10−6 cm2/s, which is comparable to the diffusion coefficient of fluorescein 40, suggests diffusion is the dominant source of band broadening during transport down the electrophoresis channel. The large y-intercept of 0.001129 cm2 indicates that extra-column effects contribute 96.5% of the total band width over a 10 cm separation length. In view of the narrow detection window and strong effects of injector conditions on the efficiency, we conclude that the injector contributes this large effect and limits efficiency.
Figure 5.
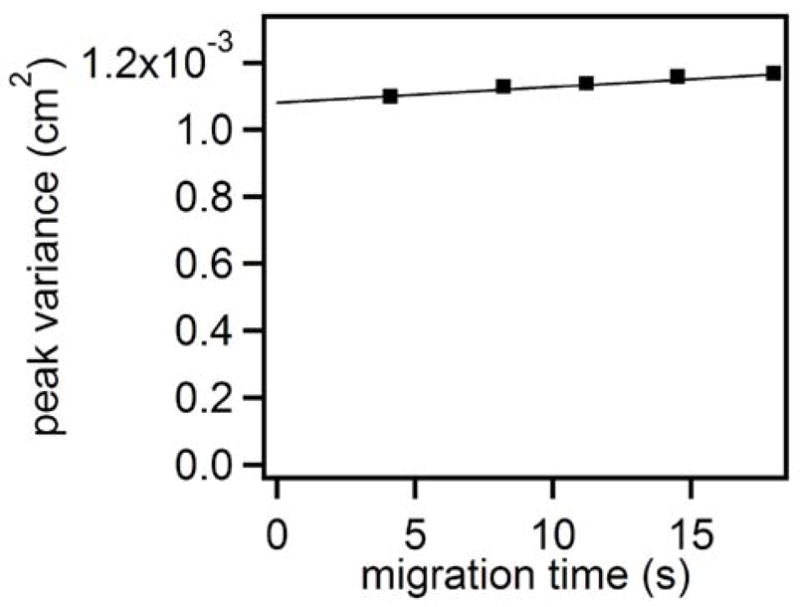
Effect of migration through a 10 cm channel on peak variance. Measurements of peak variance were recorded at 0.2, 0.4, 0.6, 0.8 and 1 cm corresponding to the migration times shown. Field strength applied across the electrophoresis channel was 250 V/cm with a plug frequency of 1.5 Hz and a separation buffer of 10 mM sodium borate, 0.9 mM HP-βCD at a pH of 10.0. Sample plug volumes were approximately 15 nL.
Based on broadening in the channel, we should be able to generate ~300,000 plates over the 10 cm separation distance at an applied electric field of 550 V/cm if the injection band width was infinitely narrow. Several improvements in this design are possible and may yield performance closer to this theoretical maximum. Methods of forming plugs that allow longer intervals with smaller plug sizes will aid efficiency by decreasing the plug width. Also, a method of manipulating voltage changes when sample is present at the interface would allow better control over the injection. In this way, lower voltage could be used to avoid over injection. Furthermore, if voltage could only be applied when the center, most concentrated, portion of the analyte zone was passing in front of the electrophoresis channel a higher efficiency injection could be performed with less effect on sensitivity. The trade-off of peak height and efficiency could also be decreased with methods of preconcentration from sample plugs because only a small fraction of the plug is injected.
Although the peak separation efficiency is low compared to conventional electrokinetic gated injection coupled to microchip CE 41, it is at least an order of magnitude greater than previously published plug electrophoresis 27,42 and unlike previous systems is compatible with a continuous stream of plugs. To illustrate separations capability, the discrete injection technique was used to sample from a segmented flow stream with samples containing fluorescently-labeled amino acids. Figure 6 demonstrate back to back coalescence, injection and separations of plugs containing 1 μM GABA, serine, and glutamic acid. These separations were performed at both 2.5 and 7.5 cm electrophoretic separation lengths, with plug intervals of 7 s and 12 s, respectively. These electropherograms illustrate the resolving capability of this separation technique. We envision this system to be applicable to analysis of cartridges of samples for applications requiring high-throughput.
Figure 6.

Serial plug coalescence, injection and electrophoresis using the discrete injector. NDA derivatized amines in 17 nL plugs, injected at 7 s intervals and separated at 2.5 cm (top) and injected at 12 s intervals and separated at 7.5 cm (bottom). The separation buffer was 10 mM sodium tetraborate, 0.9 mM HPβ-CD at a pH of 9.7 and the field strength of 550 V/cm.
Desegmenting injector
The desegmenting injection method uses the same K-interface used for the discrete injector, but cross flow velocity is reduced relative to oil flow velocity. In this way, new aqueous plugs are brought to the interface before the previous plug is washed out resulting in a continuous, secondary aqueous stream forming from the sample plugs in the sampling channel (See Figure 7A). This secondary analyte stream is directed to a microfluidic cross for electrokinetic injection 30. A sequence of images illustrating coalescence and transfer to cross injector is shown in Figure 7A. With this approach, reliable and stable injections can be achieved as illustrated in Figure 7B. Using this system, it was possible to coalesce over 10,000 plugs at the K interface and make over 1,000 serial electrokinetic injections without interruption (~83 min of operation) and with peak height RSD of 5.1%. Compared with the discrete injector, an advantage of this approach is that injection is controlled independently of plug generation allowing optimization of the injection independent of the method of plug generation. On the other hand, plugs do not remain distinct prior to injection. Thus, this method is suited for periodic sampling of a stream of plugs rather than analysis of discrete samples.
Figure 7.

A) Illustration of desegmenting injection. Top drawing shows the lay-out of channels with addition of sampling channel and cross-style injector. Drawing illustrates a sample plug coalescing and portion being transferred to sampling channel for injection and separation. Below are two sequential fluorescence micrographs showing coalescence of a fluorescein plug to the virtual wall illustrating “residence time”. Times (t) indicate time since the plug arrived at the interface. The plugs were pumped past the coalescence interface at 5 μL/min, with a linear velocity of 0.42 cm/s. The cross flow separation buffer was pumped at a rate of 40 nL/min, with a linear velocity of 0.03 cm/s. B) Sequential fluorescein injections performed by sampling from a K interface with a cross flow of 40 nL/min and a 1 Hz segmented flow stream. C) Effect of cross flow rate on residence time in the sampling channel.
For tests of this system, aqueous sample plugs were generated at 1 Hz in a T junction, pumped through a serpentine, and coalesced to a virtual wall within the K fluidic element. Under this condition, we explored how the cross-flow rate affected the resulting secondary sample stream. As the cross flow rate is increased, the residence time of a given flow segment in the interface decreased as shown in Figure 7C. The residence times provide an estimate of the plug frequency that is necessary to generate a continuous analyte stream into the electrokinetic gated injector. For example, a residence time of ~1 s at 40 nL/min suggests that plugs need to arrive at the interface at ≥ 1 Hz to avoid generating a discontinuous sample stream. As the cross flow rate increases, the frequency of plug formation would have to be increased. If discontinuities occur in the secondary sample stream, it is not possible to continuously inject sample unless the injection is synchronized to match the arrival of a sample plug at the cross. Thus, for a given plug stream, the cross-flow should be kept within a fixed range to achieve reliable sampling and injection.
In this system, we observed that a minimum electrophoresis separation length of 15 cm for 20 μm deep channels was required to prevent flow splitting into the electrophoresis channel. Such splitting causes parabolic flow in the electrophoresis channel and must be minimized to achieve good separations. With this condition, fluorescein plugs at a concentration of 10 μM and a frequency of 0.9 Hz were coalesced to a virtual wall (within the K fluidic element), transferred to the electrokinetic gated injector, and electrophoresed at a field strength of 525 V/cm. Sequential injections of 10 ms produced efficiencies at 10 cm of 53,500 ± 6,400 (n = 30), shown in Figure 6B. The efficiencies for these separations are limited by the injection band width and applied field strength. As with the discrete injector, bubble formation above 600 V/cm prevented use of higher fields for the separation.
As suggested above, the main application of this approach to injection is sampling a stream of plugs. For example, we have recently demonstrated that temporal resolution in microdialysis sampling can be improved by segmenting dialysate prior to transport from the sampling site to the analysis system 25. In such a case, the segmentation rate may be arbitrarily high, then sampled downstream by electrophoresis or other analytical methods at the temporal resolution desired. (Frequently it is not possible to segment at a rate that exactly matches the electrophoretic rate. This approach eliminates the need to match these rates and still allows the utility of preventing axial dispersion during sampling.) To demonstrate the potential for monitoring rapid concentration changes, a concentration step change was generated by filling a capillary with two known analyte solutions separated by an oil plug to mimic a sampling system that had collected a rapid change of analytes. This solution was pumped into the T junction, broken up into a segmented flow stream at 2.1 Hz, pumped into the K interface, injected at 5 s intervals with a tinj of 300 ms and separated at 525 V/cm. The resulting electropherograms, shown in Figure 8, illustrates that the change from high to low concentration of amino acids could be recorded over 10 s, or two electropherograms and cross flows of 40 nL/min and 0.03 cm/s linear velocities. The 10 s temporal resolution limit is likely due to mixing between plugs that occurs between the point at which the plugs are coalesced and the point at which the analyte is injected into the CE channel. During sampling, peak area RSDs were 5.2% RSD (n = 20) for a fixed concentration. These results demonstrate the suitability of this system for monitoring concentration changes in a sample stream.
Figure 8.

A 250 μm id capillary was utilized with a valco union and a 100 μL Hamilton gas tight syringe to initially aspirate the sample, oil plug, and blank solution into the capillary. After the cartridge was formed it was screwed into a nanoport and pumped into the segmentation channel and analyzed using the desegmenting gated injector. Electropherograms recorded using a 525 V/cm field strength with 10 mM borate and 0.9 mM HPβ-CD as the separation buffer. Separation distances were 5 cm and injection widths were 200 ms.
Conclusions
The 3-dimensional microfluidic manifold with differing channel depths and patterned surface chemistry described here can be used to generate a stable interface between a segmented flow stream and aqueous buffer. The stable interface enabled reproducible sampling of plugs that passed through the interface and transfer to an aqueous stream. Such sampling may be used for a variety of applications. We used this sampling method in developing two types of injectors for electrophoretic analysis of streams of plugs. The discrete injection method enables serial electrophoretic analysis of individual droplets or plugs with ~1,050 theoretical plates and 5% RSDs. Analysis of the injection method suggests potential routes to improving efficiency including use of different plug formation method and voltage control at the injector. The desegmenting method performs a fluidic “digital to analog conversion” by transforming a stream of plugs into a continuous aqueous stream that is then periodically injected using a conventional electokinetic scheme. This method produced higher efficiencies of ~52,000 plates with comparable RSDs. This method is suited for periodic analysis of high frequency plug streams. Both methods showed good stability for analysis of up to 2 hrs. This approach advances the state-of-the-art for analyzing sample plugs confined in oil streams. They also provide a new approach to manipulating samples prior to electrophoresis. These new injection methods could have many applications including high-throughput screening, measuring products of in-plug reactions, monitoring reactions, process analytical technology, and microdialysis sampling.
Acknowledgments
We thank Kendra Reid and John Dishinger for technical help and useful discussions. This work was supported by NIH grants R37 EB003320 and P41 EB002030-120002.
References
- 1.Gunther A, Jhunjhunwala M, Thalmann M, Schmidt MA, Jensen KF. Langmuir. 2005;21:1547–1555. doi: 10.1021/la0482406. [DOI] [PubMed] [Google Scholar]
- 2.Gunther A, Jensen KF. Lab Chip. 2006;6:1487–1503. doi: 10.1039/b609851g. [DOI] [PubMed] [Google Scholar]
- 3.Zhao B, Moore JS, Beebe DJ. Science. 2001;291:1023–1026. doi: 10.1126/science.291.5506.1023. [DOI] [PubMed] [Google Scholar]
- 4.Teh SY, Lin R, Hung LH, Lee AP. Lab Chip. 2008;8:198–220. doi: 10.1039/b715524g. [DOI] [PubMed] [Google Scholar]
- 5.Song H, Chen DL, Ismagilov RF. Angew Chem Int Ed. 2006;45:7336–7356. doi: 10.1002/anie.200601554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gunther A, Khan SA, Thalmann M, Trachsel F, Schmidt MA, Jensen KF. Lab Chip. 2004;4:278. doi: 10.1039/b403982c. [DOI] [PubMed] [Google Scholar]
- 7.Tice JD, Song H, Lyon AD, Ismagilov RF. Langmuir. 2003;19:9127–9133. [Google Scholar]
- 8.Aota A, Nonaka M, Hibara A, Kitamori T. Ang Chemi Int Ed. 2007;46:878–880. doi: 10.1002/anie.200600122. [DOI] [PubMed] [Google Scholar]
- 9.Nisisako T, Torii T, Higuchi T. Lab Chip. 2002;2:24–26. doi: 10.1039/b108740c. [DOI] [PubMed] [Google Scholar]
- 10.Garstecki P, Fuerstman MJ, Stone HA, Whitesides GM. Lab Chip. 2006;6:437–446. doi: 10.1039/b510841a. [DOI] [PubMed] [Google Scholar]
- 11.Chabert M, Viovy JL. Proc Nat Acad Sci. 2008;105:3191–3196. doi: 10.1073/pnas.0708321105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rhee M, Burns MA. Langmuir. 2008;24:590–601. doi: 10.1021/la702575j. [DOI] [PubMed] [Google Scholar]
- 13.Khan SA, Gunther A, Schmidt MA, Jensen KF. Langmuir. 2004;20:8604–8611. doi: 10.1021/la0499012. [DOI] [PubMed] [Google Scholar]
- 14.Lorenz RM, Edgar JS, Jeffries GDM, Zhao Y, Mcgloin D, Chiu DT. Anal Chem. 2007;79:224–228. doi: 10.1021/ac061586w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sgro AE, Allen PB, Chiu DT. Anal Chem. 2007;13:4845–4851. doi: 10.1021/ac062458a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ody CP, Baroud CN, Langre E. J Colloid and Interface Science. 2007;308:231–238. doi: 10.1016/j.jcis.2006.12.018. [DOI] [PubMed] [Google Scholar]
- 17.He M, Sun C, Chiu DT. Anal Chem. 2004;76:1222–1227. doi: 10.1021/ac035196a. [DOI] [PubMed] [Google Scholar]
- 18.He M, Edgar JS, Jeffries GDM, Lorenz RM, Shelby JP, Chiu DT. Anal Chem. 2005;77:1539–1544. doi: 10.1021/ac0480850. [DOI] [PubMed] [Google Scholar]
- 19.Funfak A, Brosing A, Brand M, Kohler JM. Lab Chip. 2007;7:1132–1138. doi: 10.1039/b701116d. [DOI] [PubMed] [Google Scholar]
- 20.Martin K, Henkel T, Baier V, Grodrian A, Schon T, Martin R, Kohler JM, Metze J. Lab Chip. 2003;3:202–207. doi: 10.1039/b301258c. [DOI] [PubMed] [Google Scholar]
- 21.Dorfman KD, Chabert M, Codarbox JH, Rousseau G, Cremoux P, Viovy JL. Anal Chem. 2005;77:3700–3704. doi: 10.1021/ac050031i. [DOI] [PubMed] [Google Scholar]
- 22.Chabert M, Dorfman KD, deCremoux P, Roeraade J, Viovy JL. Anal Chem. 2006;78:7722–7728. doi: 10.1021/ac061205e. [DOI] [PubMed] [Google Scholar]
- 23.Song H, Ismagilov RF. J Am Chem Soc. 2003;125:14613–14615. doi: 10.1021/ja0354566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumaresan P, Yang CJ, Cronier SA, Blazej RG, Mathies RA. Anal Chem. 2008;80:3522–3529. doi: 10.1021/ac800327d. [DOI] [PubMed] [Google Scholar]
- 25.Wang M, Roman GT, Schultz K, Jennings C, Kennedy RT. Anal Chem. 2008 doi: 10.1021/ac801317t. ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hatakeyama T, Chen DL, Ismagilov RF. J Am Chem Soc. 2006;128:2518–2519. doi: 10.1021/ja057720w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edgar JS, Pabbati CP, Lorenz RM, He M, Fiorini GS, Chiu DT. Anal Chem. 2006;78:6948–6954. doi: 10.1021/ac0613131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roper MG, Shackman JG, Dahlgren GM, Kennedy RT. Anal Chem. 2003;75:4711–4717. doi: 10.1021/ac0346813. [DOI] [PubMed] [Google Scholar]
- 29.Roman GT, Hlaus T, Bass KJ, Seelhammer TG, Culbertson CT. Anal Chem. 2005;77:1414–1422. doi: 10.1021/ac048811z. [DOI] [PubMed] [Google Scholar]
- 30.Sandlin ZD, Shou M, Shackman JG, Kennedy RT. Anal Chem. 2005;77:7702–7708. doi: 10.1021/ac051044z. [DOI] [PubMed] [Google Scholar]
- 31.Shou M, Ferrario CR, Schultz KN, Robinson TE, Kennedy RT. Anal Chem. 2006;78:6717–6725. doi: 10.1021/ac0608218. [DOI] [PubMed] [Google Scholar]
- 32.Qian WJ, Aspinwall CA, Battiste MA, Kennedy RT. Anal Chem. 2000;72:711–717. doi: 10.1021/ac991085t. [DOI] [PubMed] [Google Scholar]
- 33.Aota A, Hibara A, Kitamori T. Anal Chem. 2007;79:3919–3924. doi: 10.1021/ac070031d. [DOI] [PubMed] [Google Scholar]
- 34.Xiao H, Liang D, Liu GC, Guo M, Zing WL, Cheng J. Lab Chip. 2006;6:1067–1072. doi: 10.1039/b600374e. [DOI] [PubMed] [Google Scholar]
- 35.Dong L, Jiang H. App Phys Lett. 2007;91:041109. [Google Scholar]
- 36.Hibara A, Iwayama S, Matsuoka S, Ueno M, Kikutani Y, Tokeshi M, Kitamori T. Anal Chem. 2005;77:943–947. doi: 10.1021/ac0490088. [DOI] [PubMed] [Google Scholar]
- 37.Atencia J, Beebe DJ. Nature. 2005;437:648. doi: 10.1038/nature04163. [DOI] [PubMed] [Google Scholar]
- 38.Zheng B, Tice JD, Ismagilov RF. Anal Chem. 2004;76:4977–4982. doi: 10.1021/ac0495743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gadd JC, Kuyper CL, Fujimoto BS, Allen RW, Chiu DT. Anal Chem. 2008;80:3450–3457. doi: 10.1021/ac8000385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Culbertson CT, Jacobson SC, Ramsey JM. Talanta. 2002;56:365–373. doi: 10.1016/s0039-9140(01)00602-6. [DOI] [PubMed] [Google Scholar]
- 41.Culbertson CT, Jacobson SC, Ramsey JM. Anal Chem. 2000;72:5814–5819. doi: 10.1021/ac0006268. [DOI] [PubMed] [Google Scholar]
- 42.Burns MA, Johnson BN, Brahmasandra SN, Handique K, Webster JR, Krishnan M, Sammarco TS, Man PM, Jones D, Heldsinger DC, Mastrangelo CH, Burke DT. Science. 1998;282:484–487. doi: 10.1126/science.282.5388.484. [DOI] [PubMed] [Google Scholar]