

Abstract
Mitochondrial DNA (mtDNA) mutations contribute to the pathology of a number of age-related disorders, including Parkinson disease(1, 2), muscle-wasting(3), and the metastatic potential of cancers(4). The impact of mitochondrial DNA mutations on a wide variety of human diseases has made it increasingly important to understand the mechanisms that drive mitochondrial mutagenesis. In order to provide new insight into the etiology and natural history of mtDNA mutations, we have developed an assay that can detect mitochondrial mutations in a variety of tissues and experimental settings(5, 6). This methodology, termed the Random Mutation Capture assay, relies on single-molecule amplification to detect rare mutations among millions of wild-type bases(7), and can be used to analyze mitochondrial mutagenesis to a single base pair level in mammals.
Keywords: Mitochondria, Mitochondrial DNA, Mutations, Mutagenesis, qPCR, Mutation detection
1. Introduction
Our understanding of mitochondrial mutagenesis has been hampered by the lack of highly sensitive mutation detection assays(8). In contrast, many assays are available to detect somatic mutations in nuclear DNA(9-11). Some of the most informative assays employ transgenic mouse models that contain reporter genes such as LacZ(12) or EYFP(13), which allow investigators to carefully track the behaviour of mutations in a time dependent manner. Unfortunately, we currently lack the ability to manipulate the sequence of the mitochondrial genome at will, preventing us from inserting mutation reporters or selectable markers into the mitochondrial genome. Resistance to chloramphenicol can be used to score mtDNA mutations in the 16SrRNA subunit(14), but the requirement for proliferating cells limits the use of this assay to cell types that can be expanded in culture. In addition, it is important to point out that, due to the multiplicity of mtDNA molecules within a cell, resistance mutations need to be clonally expanded before they confer resistance to the cell as a whole. The need for clonal expansion adds a layer of complexity to the data analysis that makes it difficult to define critical parameters such as the mutation rate and spectrum of mtDNA, which are necessary to fully understand the role of mitochondrial mutagenesis in human disease. In order to define these parameters, we need to be able to detect spontaneous mutations, which are rare events that occur randomly throughout genome. The most straightforward way to detect mutations is by sequencing. Unfortunately, mutations that arise de novo are not easily detected by sequencing, since they are only present in a single copy of mtDNA, whereas a mutation must be present in as much as 10-25% of a population of molecules to be detected by sequencing(15). One way to get around this problem, and still use sequencing as a detection method, would be to sequence each mtDNA molecule individually. Several assays have been designed to do this, all of which separate, and then amplify individual DNA molecules before sequencing(16-19). Importantly though, the amplification of single DNA molecules prior to sequencing may allow spurious mutations to be generated during amplification. This may be one of the reasons why there is no consensus in the literature on the mutation frequency of mitochondrial DNA in mammalian tissues. The frequencies measured depend strongly on the background of the assay used(20). This discrepancy suggests that the spontaneous mutation frequency of mtDNA is either below, or very close to the detection limit of these technologies. In order to resolve this controversy, we have adapted a highly sensitive single-molecule sequencing method, termed the random mutation capture (RMC) assay, to measure the spontaneous mutation frequency of mammalian mtDNA(6).
2. Concept of the RMC assay
Although the RMC-assay was initially conceived to quantify mutations in the nuclear genome(7), it was designed to use DNA as its source material, so that, in principle, the methodology could be applied to any organism or DNA desired, with minimal changes to the protocol. Here, we describe how we adapted the protocol to measure mutations in the mitochondrial genome.
The assay revolves around a restriction enzyme, TaqI, which is used to discriminate between WT DNA and rare DNA molecules that contain a mutation in the TaqI restriction site. By digesting the mitochondrial genome with TaqI, the vast majority of mtDNA molecules will be cleaved at a known restriction site (figure 1). However, a small number of molecules will be resistant to TaqI cleavage due to a mutation in the restriction site. The key idea behind the assay, is that these molecules can be quantified with real time PCR, using primers that flank the restriction site. Thus, PCR can be used to screen a large number of mtDNA molecules for the presence of rare, mutated copies. The number of molecules screened is determined with a second PCR reaction, which amplifies a region that is adjacent to, but unaffected by the TaqI restriction site. To increase the number of molecules that can be screened, PCR amplification is carried out in a 96-well format. One example of how this information can be used to calculate the mutation frequency is presented in figure 2. The use of a 96-well plate also allows mutant molecules to be quantified at a single-molecule level. These molecules are then sequenced to verify the presence of a mutation in the TaqI restriction site. Thus, like other assays that use brute force to sequence many single molecules at once, the RMC assay is ultimately a single-molecule sequencing approach. The key difference being that, instead of PCR amplifying and sequencing every DNA molecule present in a sample, the RMC-assay inserts a simple DNA digestion step prior to PCR amplification, in order to remove WT molecules from further analysis. This ensures that only molecules known to contain a mutation need to be sequenced, greatly speeding up the process of mutation discovery and reducing costs. In addition, because genotypic selecting for mutant molecules occurs prior to PCR amplification, mutation discovery is disconnected from the fidelity of the PCR amplification, which greatly increases the sensitivity of the assay.
Figure 1. Concept of the Random Mutation Capture assay.
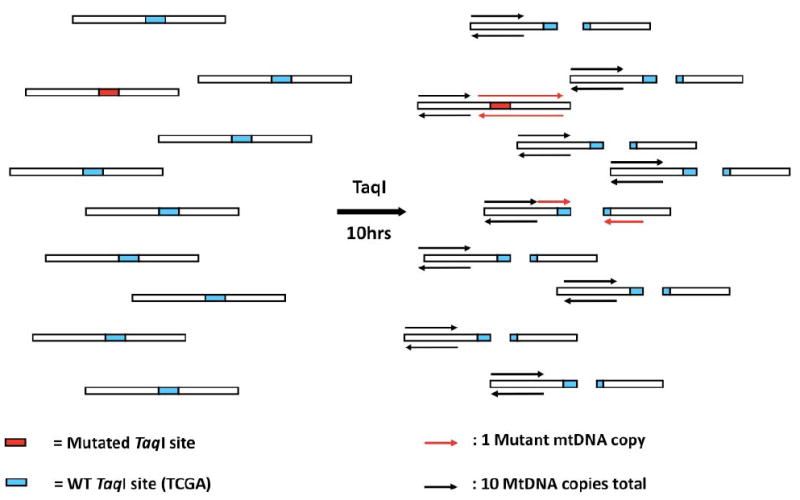
The RMC-assay exploits the ability of TaqI, a restriction enzyme, to discriminate between DNA molecules with either a WT, or a mutant TaqI restriction site. After mtDNA digestion with TaqI, a PCR reaction is attempted across a TaqI restriction site (red arrows). This PCR reaction will amplify only DNA molecules that contain a mutation in the restriction site (red box), that rendered it resistant to cleavage. Amplicons with a WT restriction site (green box) are no longer a template for PCR amplification. These mutant molecules are then quantified by qPCR. A second qPCR reaction, adjacent to the restriction site, quantifes every DNA molecule in a sample. The ratio of mutant molecules to the total number of molecules is a direct measurement of the mutant frequency, which can be used to calculate the mutation frequency per base pair.
Figure 2. Schematic of the Random Mutation Capture assay.
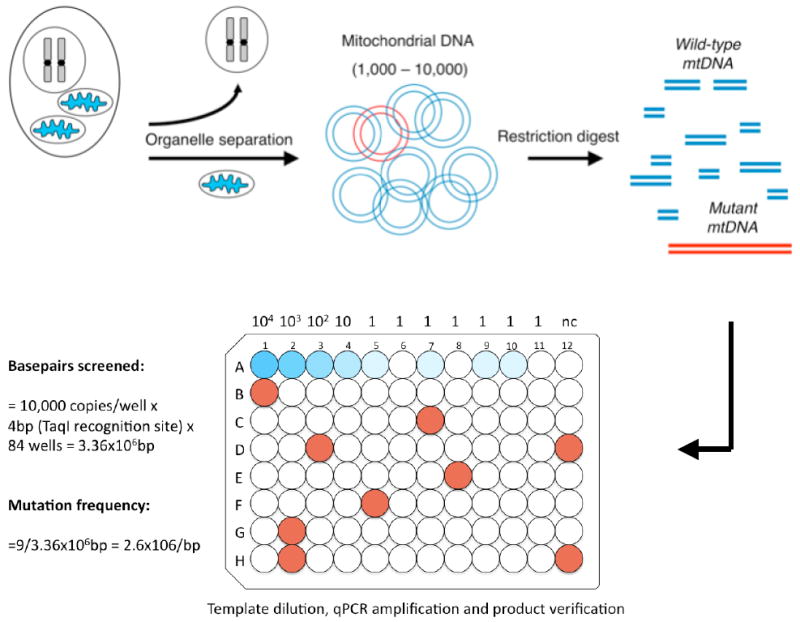
The RMC-assay consists of 4 steps, organelle separation, DNA extraction, DNA digestion, and qPCR amplification. In order to calculate the mutation frequency, mtDNA is displayed in a 96-well format. In the example presented above, 10,000 molecules of the mitochondrial genome are inserted into rows B-H. In each of these wells, a PCR reaction is attempted across the TaqI restriction site. Here, 9 wells, displayed in red contained an amplified molecule. Sequencing of each of these PCR reactions confirmed that a mutation was present in the TaqI restriction site. Serial dilutions, from 10,000 copies to 1, in row A, are used to confirm the copy number present in each well, and provide an important control for PCR efficiency. At an estimated 1 copy per well (wells A5-A11), some wells do, and some do not contain an amplified DNA molecule. The mutation frequency can be calculated as follows; If 10,000 copies of mtDNA are screened per well, that is equivalent to a screen of 40,000 basepairs per well, since the TaqI site is 4bps long, and a mutation in any one of these basepairs will render it resistant to cleavage. 84 wells were screened in this experiment, which amounts to 3.36×106bps screened. 9 Mutants were found, yielding a mutation frequency of 2.6×10-6.
3. Description of the Random Mutation Capture assay
The following sections will describe the methodologies used to perform the random mutation capture assay. The assay consists of 4 basic steps, which are depicted in figure 2. The first two steps, organelle separation and DNA extraction will allow you to isolate mtDNA with a limited amount of nuclear DNA contamination. Then, mtDNA is digested with TaqI restriction enzyme, and qPCR-amplification is utilized to measure the mutation frequency. Each of these steps is described in detail below.
3.1 Organelle separation
An important requirement for the RMC-assay is to start with a purified preparation of mtDNA. Nuclear DNA (nDNA) contamination can result in incomplete digestion of mtDNA by restriction enzymes and increases the variation of PCR efficiency. Moreover, a number of mitochondrial pseudogenes are present in the nucleus, that are subject to PCR amplification by mtDNA specific primers. Some of these pseudogenes contain single base substitutions in the TaqI restriction site, which may be erroneously scored as mutations. One way to limit nDNA contamination is by isolating mitochondria prior to DNA extraction, so that nuclear DNA is excluded from further analysis. The best mitochondrial preparations are derived from fresh tissue. Therefore, it is recommended to use fresh tissue, or cell cultures, whenever possible, although flash frozen tissue can be used. If tissue is to be harvested from mice, organs need to be extracted as quickly as possible, and placed on ice immediately to halt tissue degradation.
3.1.2 Extracting mitochondria from tissues
Several methods can be used to lyse cells and harvest mitochondria, and a number of kits are commercially available (Pierce, cat. #89801, and #89874). Most methods use differential centrifugation to separate mitochondria from nuclei. This section will describe how differential centrifugation can be carried out with a homemade kit, and is based on a previous publication in Methods(21). The process starts by disrupting cell membranes with a Dounce homogenizer, and gently spinning down a pellet that contains nuclei and cell debris. Mitochondria are present in the supernatant, and are recovered by centrifugation at a higher speed.
To lyse the cells, tissues need to be homogenized in buffers that keep mitochondria intact. Specific buffers are recommended for different tissues. For liver and kidney, this buffer contains 0.32M sucrose, 1mM EDTA, and 10mM Tris-HCl, pH 7.4. For brain and heart tissue, the buffer consists of 0.075M sucrose, 0.225M sorbitol, 1mM EGTA, 0.1% fatty acid free bovine serum albumin (BSA), and 10mM Tris-HCl, pH 7.4. Use 4ml of homogenization buffer per gram of liver, 5ml/g of kidney or brain, and 10 ml/g of heart. Prior to homogenization, samples should be washed in ice cold 1×PBS, or the appropriate homogenization buffer, to remove blood and connective tissue. Make sure to keep the tissue cold, and use a scalpel or a razor blade to cut the tissue into smaller pieces and to remove blood clots inside the tissue. Then, use a hand driven, glass pestle, to homogenize the tissue with approximately 10 firm strokes, until large tissue pieces are dispersed, and allow the cells to swell for 2 minutes before performing 10 additional strokes with the pestle to complete the homogenization process. When monitoring the process under a microscope, homogenization should be stopped when 70% of the cells are lysed. Transfer the homogenates to a 10ml tube, and centrifuge at 1000g for 10 minutes at 4°C to pellet nuclei and large cell debris. Collect the supernatant in 1.5ml Eppendorf tubes and centrifuge for 15 minutes at 13,000 rpm, at 4°C to obtain a mitochondrial pellet. Contamination by lysosomes and peroxisomes in the pellet can be decreased by 50% by centrifuging at 4,000rpm instead of 13,000rpm. However, this will reduce the mitochondrial yield. Remove the supernatant, and resuspend the pellets in a single tube with 750μl ice-cold homogenization buffer. Spin the mitochondrial fraction down again for 10 minutes at 13,000 rpm, at 4°C. Remove the supernatant, and resuspend the pellet in 650μl lysis buffer containing 10mM Tris-HCl, 0.15M NaCl, and 0.05M EDTA.
3.2 MtDNA extraction
Add Proteinase K to the lysis buffer at a final concentration of 0.2mg/ml, and add SDS to a final concentration of 0.5%. Incubate the samples at 55°C for 30 minutes to lyse the mitochondria. RNaseA can be added to hydrolyze the RNA at a final concentration of 30ng/ml. DNA can then be isolated by phenol-chloroform extraction, followed by ethanol precipitation(22). Add phenol-chloroform-isoamyl alcohol (25:24:1 by volume) in a 1:1 ratio with the lysis reaction, mix thoroughly by shaking, and centrifuge for 2 minutes at 13,000rpm. Gently remove the aqueous phase from the top of the solution, without disturbing the interphase. Cut off the end of your pipette tip to decrease the force of pipetting if the interphase is unstable. Mix the aqueous solution again with phenol-chloroform-isoamyl alcohol in a 1:1 ratio, and repeat the extraction. Perform one final extraction with chloroform only, in a 1:1 ratio, to remove any traces of phenol. Add a 1:20 volume to volume of 3M sodium acetate to the final extract, and 2μl of glycogen in a 10μg/ml concentration, to facilitate DNA precipitation. Add ice-cold 100% ethanol to the solution in a 2.5:1 volume to volume ratio, and mix thoroughly by shaking. Store samples for 15 minutes at -80°C, or for 30 minutes at -20°C. Then, centrifuge samples at 13,000rpm for 30 minutes at 4°C to pellet mtDNA. Rince the surface of the pellet with 70% ethanol, and centrifuge again for 5 minutes at 4°C. Remove ethanol and air-dry samples for 15 minutes before resuspending the pellet in 10mM Tris-HCl. At this point, it is possible to determine the amount of nuclear DNA contamination. The most precise way to do this, is to perform real-time PCR using a nuclear and a mitochondrial target. A number of validated primersets to amplify nuclear DNA are available at http://www.realtimeprimers.org, and two primersets that we recommend can be found in Table 1. If nDNA contamination is high, and results in incomplete digestion of mtDNA with TaqI or disruption of efficient DNA amplifiction, we recommend using a sucrose gradient purification after differential centrifugation to further isolate the mitochondria. An excellent protocol for sucrose gradient purification of mitochondria can be found in a paper by Yoon et al(23).
Table 1.
Primers
| Mouse RMC-assay |
| mControl forward: TCGGCGTAAAACGTGTCAAC |
| mControl reverse: CCGCCAAGTCCTTTGAGTTT |
| mTaq634 forward: ACTCAAAGGACTTGGCGGTA |
| mTaq634 reverse: AGCCCATTTCTTCCCATTTC |
| Mouse nuclear DNA contamination |
| nDNA forward: ATGGAAAGCCTGCCATCATG |
| nDNA reverse: TCCTTGTTGTTCAGCATCAC |
| mtDNA forward: CCTATCACCCTTGCCATCAT |
| mtDNA reverse: GAGGCTGTTGCTTGTGTGAC |
| Human RMC-assay |
| hcontrol forward: ACAGTTTATGTAGCTTACCTCC |
| hcontrol reverse: TTGCTGCGTGCTTGATGCTTG |
| hTaq1216 forward: AACTGCTCGCCAGAACACTAC |
| hTaq1216 reverse: GGGCTACACCTTGACCTAAC |
| Human nuclear DNA contamination |
| nDNA forward: CCCCATGAAACTCCTTCTTT |
| nDNA reverse: GAAAACTGAGGAAATCGGGT |
| mtDNA forward: CATAGGAGGCTTCATTCACTG |
| mtDNA reverse: CAGGTTTATGGAGGGTTCTTC |
3.3 TaqI digestion
Although we prefer to use TaqI for DNA digestion and mutation detection, other restriction enzymes may also be explored and might be equally efficacious. The use of different enzymes could yield valuable information about differences in mutation rate between loci in various genetic contexts. However, the efficiency of the RMC-assay hinges almost entirely on complete digestion of the mitochondrial genome by the chosen restriction enzyme. Therefore, not every enzyme is suitable for this kind of application and only highly robust restriction enzymes should used.
TaqI was selected by our laboratory for four reasons. Firstly, TaqI has proven to be a very robust enzyme that is unaffected by several types of DNA damage, including DNA ethylation(7) and oxidation(6). Secondly, the restriction site of TaqI (TCGA) contains all base pairs, allowing each nucleotide to be screened for mutaganesis. Thirdly, TaqI is a thermostable enzyme, derived from the same host as Taq polymerase, the most prevalent enzyme in real time PCR reactions. This consistency allows for limited TaqI digestion to continue during the PCR reaction, which could help digest residual WT DNA copies during PCR amplification. And fourthly, TaqI can be purchased from NEB in either a 1x (cat.# RO149L) or a 5x concentration (cat.# RO149M). Higher concentrations of TaqI are used during digestion to inhibit star-activity due to high glycerol content.
DNA digestion is carried out in 100μl reactions that contain 100 units of TaqI, 100mM NaCl, 10mM Tris-HCl, 1×BSA, and 10mM MgCl2. This buffer should have a pH of 8.4 at 25°C, to compensate for the decrease in pH that occurs during prolonged incubation at 65°C, the optimal temperature for TaqI digestion. Add 100 units of TaqI to the reaction mixture every hour to replenish any enzyme that has been deactivated due to extended incubation. Incomplete digestion by TaqI will result in PCR amplification of DNA molecules that do not contain a mutation in the TaqI restriction site. Thus, extra care should be taken to make sure DNA digestion is complete. Mix the contents of the reaction buffer thoroughly each time after adding TaqI enzyme, and make sure that condensation is removed from the cap of the tube. Subsequent steps include PCR amplification of single DNA molecules, experiments that are easily contaminated with minute amounts of airborne DNA. Therefore, in order to avoid DNA cross-contamination between samples or other reagents, all tubes should be spun down before they are opened, and waterbaths should be avoided for incubations.
Although TaqI is a very robust restriction enzyme, we have found that some TaqI sites cannot be fully digested. We suspect that resistance to digestion is the result of unusual structures or base pairs that are present in mtDNA. For instance, TaqI prefers to digest double stranded DNA. Hence, the D-loop, which contains a triple stranded structure, cannot be completely digested. DNA replication can also interfere with TaqI digestion by generating single stranded structures and ribosubstituted intermediates. Therefore, some residual WT DNA is expected to be present, even after extended incubation periods with TaqI, and PCR products should always be sequenced or digested with TaqI and analyzed by gel electrophoresis to confirm the presence of mtDNA mutations.
3.4 qPCR amplification
To calculate the mutation frequency of a sample, only two data points are required: the total number of molecules present in the sample, and the number of molecules that are mutant. To determine these numbers, the RMC-assay uses two primer pairs. One primer pair flanks a TaqI restriction site, and is used to quantify DNA molecules that contain a mutation. The second primer pair does not flank a TaqI restriction site, and quantifies every mtDNA molecule present in the sample. Together these two primer pairs can be used to calculate the mutation frequency.
The design of these primers should be considered very carefully, and PCR conditions should be extensively optimized before experiments are started. Ideally, both primers are approximately 20 base pairs long, with an annealing temperature of 60°C. To make sure that they behave in a nearly identical fashion in terms of annealing temperature and amplification profiles, several online programs can help with the design, including Invitrogen’s Vector NTI software (www.invitrogen.com/Applications/Cloning/Vector-Design-Software/Vector-NTI-Software.html), Primer3 (http://primer3.sourceforge.net), and less elaborate programs such as the Operon Oligo design toolkit s (https://www.operon.com/oligos/toolkit.php). Amplicons should not exceed a maximum length of 350 base pairs, with a minimum length of 150 base pairs.
Since single DNA molecules will be amplified for mutation detection, it is very important to keep DNA contamination to a minimum. Therefore, reagents should be aliquoted extensively and discarded after use. A dedicated set of pipettors should be used for PCR experiments, and calibrated every three months. Always use barrier tips when pipetting, and change gloves whenever tubes are handled that contain DNA preparations. Pipetting steps should be kept to a minimum, and in order to decrease variation among samples, pipetting volumes should not drop below 5μl. Ideally, PCR reactions are set up in a PCR hood, away from regular workbenches where gels are run, and DNA contamination is expected. Regardless of the precautions taken though, contamination of primers, water, or PCR reagents, can always occur, most notably from previous PCR reactions were target molecules are amplified into trillions of copies. Therefore, in order to further avoid DNA contamination, we recommend using real time PCR reagents that contain dUTP in place of dTTP (Stratagene, SYBR Green Brilliant Mastermix, cat. #600548 and #929548). Using these reagents will ensure that all PCR products contain dUMP instead of dTMP, which minimizes the threat of PCR products contaminating future experiments, since all PCR reactions can be pre-incubated with uracil DNA glycosylase (UDG), to destroy dUMP-containing templates.
PCR amplification is carried out in 25μl reactions, containing 12.5μl 2x SYBR Green Brilliant Mastermix, 0.2μl UDG (NEB, cat. #0280S), 2μl of 10pM/μl forward and reverse primers, and 3.3μl H2O. Here is the PCR program we normally use:

The protocol shown above contains only one step that is uncommon to standard real time PCR programs, a 37°C incubation period, which allows UDG to destroy contaminating PCR products. In addition, we prefer the time allocated for DNA melting, primer annealing and primer extension to be significantly longer than required for a 250bp product, to allow every DNA template to be amplified during each cycle. This decreases the variation between samples, and increases the yield of PCR products with SYBR-green, a potent inhibitor of PCR amplification. We prefer 45 cycles for DNA amplification, since single DNA molecules become detectable around cycle 34.
3.4.2 Preliminary PCR experiments
Since amplification of mutant molecules occurs at a single-molecule level, preliminary experiments need to be performed to quantify these molecules. This knowledge is then used to determine at what dilution single molecules are expected to be present. In the final experiment, an average of 0.2 molecules should be present in each well of a 96-well plate. This dilution ensures that only 1/10 wells will contain a single, amplifiable copy of mutant DNA, and only 1/400 wells will contain 2 molecules. Quantifying DNA copy number can be done by a stepwise, endpoint dilution of mtDNA, followed by PCR amplification of each step with primers that amplify mutant DNA molecules. This will provide an approximation of the number of mutant molecules present in a sample. A follow-up experiment determines the exact copy number by amplifying ≈0.5 molecules in 30 individual reactions. The absolute number of molecules can then be calculated according to the formula:
where p(0) is the fraction of the reactions that did not contain an amplifiable DNA molecule. For example, if a 1/100 dilution results in 10 PCR products out of 30 reactions, then each well contained −ln 0.666 = 0.4 copies of mutant mtDNA. By taking the dilution factor into account, the amount of molecules present in the source material can be calculated. The same procedure can be applied to measure the total number of mtDNA molecules present in the sample with the second primer set.
3.4.3 Plate setup
In the example presented in figure 2, preliminary experiments determined that 10,000 copies of WT mtDNA were present per 0.2 copies of mutant DNA. Therefore, 10,000 copies were distributed into each well of rows B-H, so that 840,000 copies of mtDNA were screened for mutations in the entire plate. Row A is used as a control for PCR efficiency and mtDNA copy number. To control for PCR efficiency, dilute mtDNA down in a stepwise manner until single-molecule level is reached. A linear dilution of DNA should result in a linear increase in C(t) values, the cycle number at which DNA amplification crosses the detection threshold. To control for mtDNA copy number, aliquot the dilution at which mtDNA is thought to be present at 1 copy over wells A5-A11. If DNA copy number was calculated correctly, several wells should, and several wells should not contain a PCR product. PCR products should cross the detection threshold at the proper C(t) value. Well A12 can be used as a negative control.
3.4.4 Mutation verificaton
Proper dilution of DNA templates will result in PCR amplificaton of single DNA molecules resistant to TaqI cleavage. To verify the presence of DNA mutations in these molecules, each PCR product is sequenced. Although the presence of dUMP in the PCR product is compatible with high quality sequencing, residual activity of uracil DNA glycosylase in the PCR reaction will eventually destroy the PCR product. Therefore, in order to improve sequencing results, it is advised to store PCR reactions immediately at -20°C after completion of each experiment. PCR products can be purified by standard columns (Qiagen, Qiaquick PCR purification kit, #28104) prior to sequencing. As an alternative to sequencing, digestion of the PCR product with TaqI restriction enzyme can be used to verify the presence of a mutation. Since most real time PCR buffers use Taq-polymerase as the replicative enzyme, 1μl of 1x TaqI can be directly added to 10μl of a 25μl PCR reaction, and incubated for 10 minutes at 65°C, before resolving the reaction by size on a gel.
4. MtDNA deletions
Although the majority of the mutations recovered by the RMC-assay are single base pair substitutions, any type of mutation that destroys the TaqI restriction site can be detected, including insertions and deletions. However, since point mutations affect TaqI sites more frequently than other types of mutations, complex rearrangements such as mtDNA deletions are rarely detected. However, by relocating the primers, the detection prevalence of the RMC-assay can be skewed from point mutations to DNA deletions.
The key is to flank multiple TaqI sites with a single primer pair. For a PCR-product to form under those conditions, all TaqI sites need to be mutated simultaneously on a single mtDNA molecule. The frequency of such an event occurring becomes increasingly small when multiple TaqI sites are flanked. For instance, if a single TaqI restriction site is mutated at a frequency of ≈1×10-5, 2 sites will be mutated at a frequency of (1×10-5)2. Similarly, three sites will be mutated at a frequency of (1×10-5)3, and so forth. Thus, by increasing the number of restriction sites flanked by a primer pair, it becomes increasingly unlikely to get a PCR product as a result of individual point mutations. Instead, you will skew your detection prevalence towards mutation events that remove all TaqI sites simultaneously, such as DNA deletions. To measure the frequency of these deletions, the same procedure can be followed as described in section 3.
The most important aspect for the design of this assay, are the primers. It will be important to allow ample spacing between the annealing sites of the primers and the TaqI restriction sites. The smaller the distance, the less likely it will be that the deletion that removed the TaqI restriction sites will also keep the primer landing site intact. On the other hand, this buffer can also not be too large, because the efficiency of real-time PCR decreases with increasing amplicon size. Therefore, a fine balance needs to be struck, and we recommend spacing your forward and reverse primers no further than 400 base pairs away from the nearest TaqI restriction sites. This will keep PCR products from exceeding a maximum of 800 bases.
The spacing between the primers and the TaqI restrictions sites has important ramifications for the interpretation of your data. For instance, because the distance you allow between the primers and the TaqI restriction sites is directly related to the amount of mtDNA deletions you can detect, the deletion frequencies you get are only reproducible with the same primers and identical PCR conditions. This makes it very difficult to extrapolate your findings to other regions in the genome.
It is also important to realize that you are screening a very limited part of the genome. By locating the forward primer 400 base pairs away from the nearest TaqI restriction site, you are limiting your screen to 1/40th of the mitochondrial genome. Most likely, there are many deletions that remove all the TaqI restriction sites that separate your primers, however, only those deletions that have a breakpoint near your forward primer will be detected. And of those deletions that satisfy this condition, you will only detect the ones that contain a second breakpoint that is near the reverse primer. Thus, in reality, you will only detect 1/1600 (1/40 × 1/40) of all possible deletions that affect the TaqI restriction sites. Together, these considerations limit the extent to which you can generalize your results. In our view, the RMC-assay is an excellent tool to measure mtDNA deletions. It provides precise information on the burden of deletions at any given site, making it very easy to determine relative changes in the deletion rate as a result of genotype, treatment, or age. In addition, mtDNA deletions can be measured anywhere in the genome, and the size of the deletion that is detected can be adjusted by flanking TaqI sites that are closer, or further apart. However, by its nature, it is an assay that gives relative values and it is important to realize that it cannot be used to provide absolute numbers for an overall deletion rate of the entire mitochondrial genome. To do this, other methods are much more suitable, such as a single molecule analysis described elsewhere in this issue(20). This method is especially useful in the analysis of single cells.
5. Closing remarks
The RMC-assay has several benefits that make it ideal for mutation detection. Perhaps the most important advantage of the assay is its sensitivity. One mutation can be detected among 109 WT bases(7). In addition, the assay can be applied to any tissue, of any organism, at any age, which makes it an ideal assay when addressing either the role of age, environment, or transgenic constructs on mitochondrial mutagenesis. Finally, the methodology is very cost-effective, especially when it is compared to large scale sequencing efforts. One drawback of the assay is, that the target where mutations are scored, the TaqI restriction site, is 4 bps long. Consequently, the mutation frequency at one restriction site may not accurately predict the rate of mutagenesis elsewhere in the genome. Therefore, multiple sites should always be surveyed in parallel. However, as long as the same restriction site is compared between different ages or genotypes, unequivocal evidence for relative increases or decreases in mutation rate can be obtained.
Figure 3. By flanking multiple TaqI restriction sites with a primer pair, the detection prevalence of the RMC-assay can be skewed from point mutations towards DNA deletions.
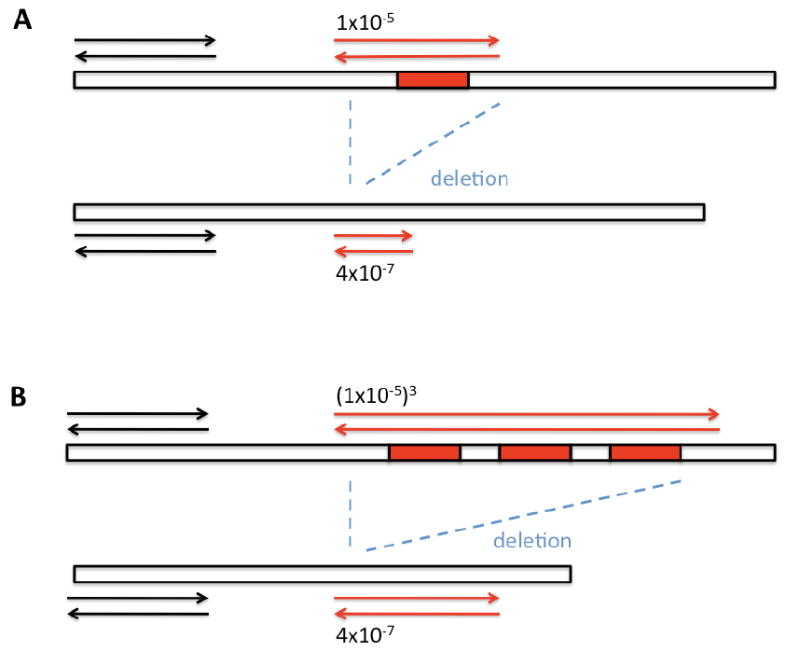
In this example, two primer pairs are used to detect mtDNA mutations with the RMC-assay. In panel A, mtDNA mutations are detected with primers (red arrows) that flank a single TaqI restriction site (red box). Most of the mutations recovered from this reaction are single base subsititions, which occur at a frequency of 1×10-5. Deletions are also present, but detected infrequently, because they are 25-fold less prevalent than mtDNA point mutations. In panel B, a similar reaction is attempted. However, this time, mutations are scored with a primer pair that flanks 3 TaqI restriction sites. As a result, the expected frequency of a PCR-product due to mtDNA point mutations drops exponentially to 1×10-15. In contrast, mtDNA deletions occur at a much higher rate, and hence, every mutation detected with these primer pairs will be a deletion.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bender A, et al. Nat Genet. 2006 May;38:515. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 2.Kraytsberg Y, et al. Nat Genet. 2006 May;38:518. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- 3.Wanagat J, Cao Z, Pathare P, Aiken JM. Faseb J. 2001 Feb;15:322. doi: 10.1096/fj.00-0320com. [DOI] [PubMed] [Google Scholar]
- 4.Ishikawa K, et al. Science. 2008 May 2;320:661. doi: 10.1126/science.1156906. [DOI] [PubMed] [Google Scholar]
- 5.Vermulst M, et al. Nat Genet. 2008 Mar 2; [Google Scholar]
- 6.Vermulst M, et al. Nat Genet. 2007 Apr;39:540. doi: 10.1038/ng1988. [DOI] [PubMed] [Google Scholar]
- 7.Bielas JH, Loeb LA. Nat Methods. 2005 Apr;2:285. doi: 10.1038/nmeth751. [DOI] [PubMed] [Google Scholar]
- 8.Jacobs HT. Aging Cell. 2003 Feb;2:11. doi: 10.1046/j.1474-9728.2003.00032.x. [DOI] [PubMed] [Google Scholar]
- 9.Parsons BL, Heflich RH. Mutat Res. 1997 Oct;387:97. doi: 10.1016/s1383-5742(97)00026-4. [DOI] [PubMed] [Google Scholar]
- 10.Dean SW, et al. Mutagenesis. 1999 Jan;14:141. doi: 10.1093/mutage/14.1.141. [DOI] [PubMed] [Google Scholar]
- 11.Weinstock DM, Richardson CA, Elliott B, Jasin M. DNA Repair (Amst) 2006 Sep 8;5:1065. doi: 10.1016/j.dnarep.2006.05.028. [DOI] [PubMed] [Google Scholar]
- 12.Garcia AM, et al. Methods Mol Biol. 2007;371:267. doi: 10.1007/978-1-59745-361-5_20. [DOI] [PubMed] [Google Scholar]
- 13.Hendricks CA, et al. Proc Natl Acad Sci U S A. 2003 May 27;100:6325. doi: 10.1073/pnas.1232231100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Minowa O, et al. Proc Natl Acad Sci U S A. 2000 Apr 11;97:4156. doi: 10.1073/pnas.050404497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones S, et al. Proc Natl Acad Sci U S A. 2008 Mar 18;105:4283. doi: 10.1073/pnas.0712345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin MT, Simon DK, Ahn CH, Kim LM, Beal MF. Hum Mol Genet. 2002 Jan 15;11:133. doi: 10.1093/hmg/11.2.133. [DOI] [PubMed] [Google Scholar]
- 17.Monnat RJ, Jr, Loeb LA. Proc Natl Acad Sci U S A. 1985 May;82:2895. doi: 10.1073/pnas.82.9.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang D, et al. Genomics. 2000 Oct 15;69:151. doi: 10.1006/geno.2000.6333. [DOI] [PubMed] [Google Scholar]
- 19.Kraytsberg Y, Khrapko K. Expert Rev Mol Diagn. 2005 Sep;5:809. doi: 10.1586/14737159.5.5.809. [DOI] [PubMed] [Google Scholar]
- 20.K Y, M P, N A, Khrapko K. Methods. 2008 [Google Scholar]
- 21.Fernandez-Vizarra E, Lopez-Perez MJ, Enriquez JA. Methods. 2002 Apr;26:292. doi: 10.1016/S1046-2023(02)00034-8. [DOI] [PubMed] [Google Scholar]
- 22.Ausubel F, et al. In: Current protocols in molecular biology. Chanda VB, editor. Vol. 1 John Wiley & Sons; 2002. [Google Scholar]
- 23.Yoon YG, Koob MD. Nucleic Acids Res. 2003 Mar 1;31:1407. doi: 10.1093/nar/gkg228. [DOI] [PMC free article] [PubMed] [Google Scholar]