

Abstract
Dominant negative inhibition is most commonly seen when a mutant subunit of a multisubunit protein is coexpressed with the wild-type protein so that assembly of a functional oligomer is impaired. By analogy, it should be possible to interfere with the functional assembly of a monomeric enzyme by interfering with the folding pathway. Experiments in vitro by others suggested that fragments of a monomeric enzyme might be exploited for this purpose. We report here dominant negative inhibition of bacterial cell growth by expression of fragments of a tRNA synthetase. Inhibition is fragment-specific, as not all fragments cause inhibition. An inhibitory fragment characterized in more detail forms a specific complex with the intact enzyme in vivo, leading to enzyme inactivation. This fragment also associated stoichiometrically with the full-length enzyme in vitro after denaturation and refolding, and the resulting complex was catalytically inactive. Inhibition therefore appears to arise from an interruption in the folding pathway of the wild-type enzyme, thus suggesting a new strategy to design dominant negative inhibitors of monomeric enzymes.
Dominant negative inhibition is a phenomenon in which the function of a wild-type gene product is impaired by a coexpressed mutant variant of the same gene product (1). These mutants are powerful tools for studying the function of wild-type genes where gene disruption techniques are not available (2–4) and are potential therapeutic agents (5, 6). Naturally occurring trans-dominant mutants have been implicated in human disease, including tumorigenesis (7). In normal cells, alternatively spliced variant proteins that lack portions of full-length proteins have been demonstrated to act in a dominant negative manner (8, 9). In most cases, such mutants interfere with the functional assembly of oligomeric wild-type proteins and result in formation of non-functional protein complexes (2–4, 7). Previously reported dominant negative mutants of monomeric enzymes presumably have distinct mechanisms for inhibition, such as substrate sequestering (1, 10).
Most proteins are comprised of more than one structural domain and these domains assemble together to form the native structure. With this perspective, the assembly of the native structure of a monomeric and of an oligomeric protein share features in common. Earlier work of Hall and Frieden (11) suggested that fragments of dihydrofolate reductase interfered with refolding in vitro of the wild-type enzyme. These considerations suggested the plausibility of dominant negative inhibition of cell growth being achieved by expression of fragments of an essential monomeric enzyme. For this purpose, we chose one of the tRNA synthetases, enzymes that are essential for protein synthesis.
Isoleucyl-tRNA synthetase catalyzes the attachment of isoleucine to its cognate tRNAs and its activity is essential for cell growth (12). In Escherichia coli, the enzyme is a monomer of 939 amino acids and is composed of a N-terminal catalytic domain and a C-terminal domain. Although no crystal structure is available for Ile-tRNA synthetase, limited homology with Met-tRNA synthetase, the structure of which has been determined (13), has permitted mapping of the structural domains and elements of secondary structure in Ile-tRNA synthetase (14). The N-terminal region of Ile-tRNA synthetase is predicted to have the nucleotide binding domain, a common structural motif among 10 aminoacyl-tRNA synthetases (14). The C-terminal domain contains an anticodon binding region and a zinc binding site, both of which are necessary for aminoacylation activity (12, 15–17). Previous work demonstrated that active Ile-tRNA synthetase can be reconstituted from two or three fragments of this enzyme in vivo (18, 19). Thus, the noncovalent interactions between disconnected polypeptide fragments of Ile-tRNA synthetase are sufficiently strong that tertiary and quartenary structure may exist in the absence of a continuous polypeptide backbone. We report here that C-terminal enzyme fragments exert a dominant negative effect on the growth of host cells. To study the underlying mechanism of inhibition, we have carried out in vitro experiments with purified Ile-tRNA synthetase and an inhibitory fragment. The results suggest that C-terminal fragments of Ile-tRNA synthetase inhibit the native enzyme by interfering with its folding.
MATERIALS AND METHODS
Growth Inhibition by Ile-tRNA Synthetase Fragments.
Plasmids pKS246 (ColE1 ori, Apr) encoding the C-terminal fragment (residues 585–939) of isoleucyl-tRNA synthetase, and pKS171 (p15A ori, Tcr) encoding the N-terminal fragment (residues 1–583), were introduced into E. coli strain JM109 [F′(traD36proAB+lacIqlacZΔM15)endA1 recA1 hsdR17 (rk−, mk+) supE44 thi − 1 gyrA96 relA1 Δ(lac-proAB)] individually or in combination (18). Vector controls were pTZ19R (ColE1 ori, Apr) or pKS42 (p15A ori, Tcr). The growth of these cells was scored by replica plating on Luria–Bertani plates with the appropriate antibiotics at 30°C, 37°C, and 42°C. Isopropyl β-d-thiogalactopyranoside (0.1 mM) was added to some plates to increase the expression of the plasmid encoding fragments. For measuring growth curves in liquid medium, saturated cultures were diluted 1:250 into prewarmed Luria–Bertani medium (42°C) containing ampicillin and isopropyl β-d-thiogalactopyranoside (0.4 mM). The optical densities of the cultures at 600 nm were measured at various time points.
Purification of Ile-tRNA Synthetase and Fragments.
Purification of wild-type Ile-tRNA synthetase from E. coli cells harboring pKS21 was carried out by methods described previously (16). Fragments of Ile-tRNA synthetase were expressed from the pKS series of plasmids (18). Plasmids encoding the individual fragments were introduced into E. coli strain JM109. Expression of fragment 585–939 in 1L cultures was carried out by overnight induction with 0.2 mM isopropyl β-d-thiogalactopyranoside. E. coli cells expressing the fragment were harvested, resuspended in lysis buffer [50 mM potassium phosphate, pH 7.5/0.1 M NaCl/50 mM 2-mercaptoethanol/0.1% phenylmethylsulfonyl fluoride] and lysed in a French pressure cell. After centrifugation of the cell lysates (13,000 × g, 30 min), protein was precipitated with 65% ammonium sulfate. The pellet was dialyzed in 25 mM Tris (pH 7.5), 1 mM 2-mercaptoethanol, and 0.1% phenylmethylsulfonyl fluoride. The dialysate was then applied to an fast protein liquid chromatography Mono Q ion-exchange column (1 × 10 cm). Mono Q fractions containing fragment (as judged by A280 and SDS/PAGE) were applied to a Superose-12 column under denaturing conditions. The Superose 12 column was developed in 50 mM potassium phosphate (pH 7.5), 0.1 M NaCl, 50 mM 2-mercaptoethanol, 0.1% phenylmethylsulfonyl fluoride, and 6 M guanidinium HCl. Absorbance peaks from the Superose-12 column were analyzed by SDS/PAGE, and fractions containing Ile-tRNA synthetase fragments were pooled and concentrated on a Centriplus-30 device (Amicon). Dialysis of the fragment against guanidine-free buffer confirmed that fragment 585–939 is a soluble, monomeric polypeptide.
Western blot analysis was carried out during the fragment preparation to ensure complete separation of wild-type Ile-tRNA synthetase. Samples were analyzed by SDS/9% polyacrylamide gels. The separated proteins in the gel were transferred to polyvinylidene difluoride membrane by semidry transfer using a Milliblot SDE electroblotting system (Millipore). The membranes were blocked overnight in Tris-buffered saline-Tween buffer, probed with rabbit anti-E. coli Ile-tRNA synthetase antiserum (15), and detected with the Amersham Enhanced Chemiluminescence system.
Denaturation and Refolding.
Purified E. coli Ile-tRNA synthetase, in the presence or absence of polypeptide fragments, was dissolved in 50 mM sodium phosphate buffer (pH 7.0) containing 6 M guanidine·HCl, 0.5 mM dithiothreitol, 30 μM ZnCl2, and 10 μM EDTA. After 30 min, refolding of Ile-tRNA synthetase was accomplished by rapid dilution of the sample into 50 mM sodium phosphate buffer (pH 7.0) containing 1 mM ATP, 1 mM MgCl2, 0.5 mM dithiothreitol, 30 μM ZnCl2, and 10 μM EDTA (20). After a 2-hr incubation period, aminoacylation activity in the sample was measured using purified tRNAIle (Subriden RNA, Rolling Bay, WI) and 3H-isoleucine (Amersham) as described previously (16). Assays were carried out at 37°C in 20 mM Hepes (pH 7.5), 0.1 mM EDTA, 0.15 M NH4Cl, 100 μg/ml BSA, 2.0 mM ATP, 4.0 mM MgCl2, and 20 μM 3H-Ile.
To characterize the complex between full-length Ile-tRNA synthetase and fragment 585–939, gel filtration chromatography was carried out on a fast protein liquid chromatography Sephacryl S300 column (120 ml bed volume) in 50 mM potassium phosphate (pH 7.5), 0.1 mM NaCl, 50 mM 2-mercaptoethanol. The enzyme and fragment were coincubated in buffer containing guanidine·HCl and refolded as described above. After concentration, the complex was applied to the column, and relevant fractions were analyzed by SDS/PAGE and anti-Ile-tRNA synthetase immunoblotting. Protein standards for the column were: alcohol dehydrogenase (150 kDa; elution volume = 33 ml), Ile-tRNA synthetase (105 kDa; elution volume = 39 ml), and ovalbumin (45 kDa; elution volume = 46 ml).
Detection of in Vivo Complex by Immunoprecipitation.
Plasmid pKS 419 (ColE1 ori, Apr), encoding the 585–939 fragment with a Myc epitope tag (EQKLISEEDL) at its C terminus (21), was constructed by PCR mutagenesis. JM109 cells were cotransformed with plasmids pKS152 (p15A ori, Tcr), encoding full-length Ile-tRNA synthetase, and pKS419. Cell extracts were prepared as described (16). To immunoprecipitate the complex, 1 ng of anti-Myc monoclonal antibody 9E10 (Oncogene Sciences) was added to 10 μl of lysate and incubated at 37°C for 30 min. Pansorbin (10 μl of a 10% suspension from Calbiochem) was added for 10 min at 37°C. Immunoprecipitated samples were washed 4 times with 150 mM NaCl, 1% Triton X-100, 50 mM Tris (pH 7.4), and electrophoresed in a SDS/9% polyacrylamide gel. Proteins in the gel were electroblotted onto polyvinylidene difluoride membranes and detected using anti-Ile-tRNA synthetase antibodies and the Amersham Enhanced Chemiluminescence system. For reprobing experiments, the membranes were stripped by incubation at 50°C for 30 min in 62.5 mM Tris (pH 6.7), 100 mM 2-mercaptoethanol, 2% SDS, washed in Tris-buffered saline-Tween, and reprobed using anti-E. coli Ala-tRNA synthetase antibodies (22).
RESULTS AND DISCUSSION
Growth Inhibition by Fragments of Isoleucyl-tRNA Synthetase.
The dominant negative effect of fragments of isoleucyl-tRNA synthetase was investigated in vivo. Expression of C-terminal fragments of the enzyme leads to growth arrest of E. coli cells. We observed this inhibitory effect for fragments spanning residues 103–939, 211–939, 241–939, 377–939, and 585–939 of isoleucyl-tRNA synthetase. For example, the fragment 585–939 that includes a portion of the N-terminal domain and an intact C-terminal domain (Fig. 1) causes dominant lethality to host cells at 37°C and 42°C (Fig. 1). Smaller C-terminal fragments, such as 795–939 and 887–939, were not inhibitory in this assay (Fig. 1B). Overexpression of fragment 1–583 or other N-terminal fragments of isoleucyl-tRNA synthetase did not have a lethal effect under any growth conditions tested (Fig. 1B and data not shown). Thus, these mapping experiments defined the inhibitory region to be between residues 585–939 of isoleucyl-tRNA synthetase.
Figure 1.
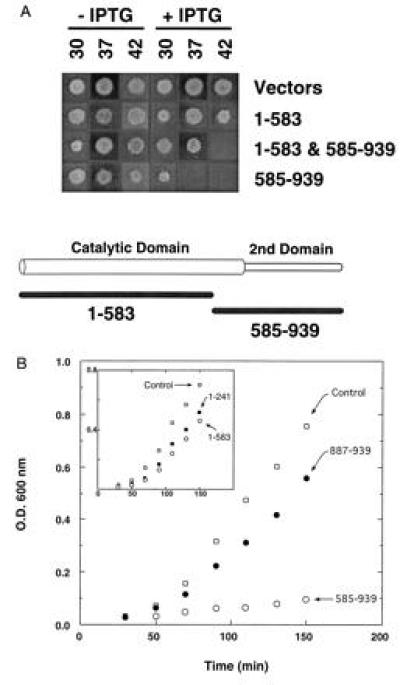
Growth inhibition by fragment 585–939. (A) JM109 cells expressing isoleucyl-tRNA synthetase (IleRS) fragments 1–583 (from plasmid pKS171) or 585–939 (from pKS246) were grown on plates at 30°C, 37°C, and 42°C. (B) Growth inhibition in liquid culture by fragments 887–939 and 585–939, and by 1–241 and 1–583 (Inset).
Because a gel filtration experiment suggested that the C-terminal fragment associated with wild-type isoleucyl-tRNA synthetase in cell lysates (see below), we postulated that the fragment formed a specific complex with intact isoleucyl-tRNA synthetase in vivo, leading to inactivation of the wild-type enzyme. To test this possibility further, we attempted to rescue inhibition by sequestering the 585–939 fragment through coexpression of the “complementary” 1–583 fragment. As expected, this coexpression reduced inhibition of cell growth at 37°C (Fig. 1A).
Association in Vivo Between C-Terminal Fragment and Isoleucyl-tRNA Synthetase.
We confirmed that, in extracts of cells expressing 585–939 alone, isoleucyl-tRNA synthetase activity [but not that of another (proline) tRNA synthetase] was severely reduced. To show an association of 585–939 with full-length protein in vivo, a version of fragment 585–939 with a myc epitope attached to the C terminus was coexpressed with full-length isoleucyl-tRNA synthetase. Lysates of these cells were analyzed in immunoprecipitation reactions using monoclonal anti-myc antibody. In cells expressing both myc-tagged fragment and full-length enzyme, the two proteins coimmunoprecipitate (Fig. 2). The immunoprecipitate did not contain alanyl-tRNA synthetase (Fig. 2), indicating that association with isoleucyl-tRNA synthetase is not due to non-specific interactions. These observations suggested that the observed growth arrest was due to the inactivation of wild-type isoleucyl-tRNA synthetase by association of the C-terminal fragment.
Figure 2.
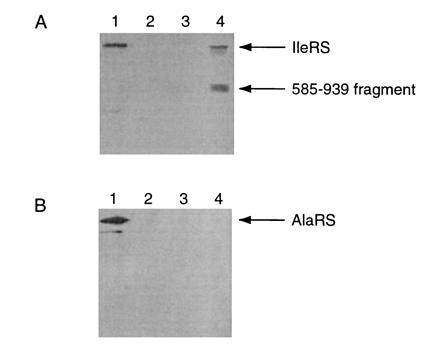
Immunoprecipitation of enzyme complex. (A) Lane 1: lysate from JM109 cells. Lanes 2–4: anti-myc immunoprecipitation reactions (lane 2, JM109; lane 3, JM109 cells expressing IleRS; lane 4, JM109 cells expressing both IleRS and fragment 585–939). (B) The membranes were stripped of IleRS antibody and reprobed using anti-AlaRS antibody.
Inhibition of Isoleucyl-tRNA Synthetase Folding.
Enzyme inhibition by fragment 585–939 was examined in more detail by experiments in vitro with purified proteins. In agreement with the immunoprecipitation experiment described above, the fragment 585–939 was tightly associated with the wild-type enzyme during purification from E. coli cells. We used denaturing gel filtration to separate fragment from full-length enzyme (Fig. 3A). Addition of the purified fragment to the native enzyme had no effect on aminoacylation of tRNAIle in vitro, even at a 10:1 molar ratio of fragment:enzyme (Fig. 3B). The fragment, however, showed a dominant negative effect on wild-type enzyme in a denaturation/refolding experiment, in which proteins were denatured in 6 M guanidine HCl and refolded by rapid dilution into guanidine-free buffer (Fig. 3B). At a 9:1 molar ratio (fragment/enzyme), aminoacylation activity was completely abolished.
Figure 3.
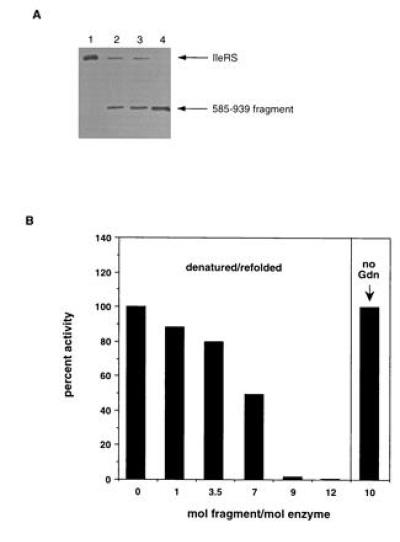
(A) Purification of fragment 585–939. Lysates from JM109/pKS246 cells were fractionated by ammonium sulfate precipitation and analyzed. Peak fractions from the chromatographic runs were analyzed by SDS/PAGE and anti-IleRS Western blotting. Lane 1, purified E. coli IleRS. Lane 2, Sephacryl S-300HR chromatography of the cell lysate. Lane 3, MonoQ chromatography of the cell lysate. Lane 4, MonoQ chromatography followed by Superose-12 gel filtration chromatography under denaturing conditions (6 M guanidine·HCl). Arrows indicate the positions of standards for IleRS and fragment 585–939 of IleRS. (B) Inhibition of IleRS activity by 585–939 fragment. Purified IleRS fragment 585–939 and full-length IleRS were denatured in 6 M guanidinium HCl and renatured. Samples were assayed for aminoacylation activity toward tRNAIle. Percentages of regained activity are expressed relative to a control with no fragment 585–939 (IleRS regains 100% of activity after unfolding and renaturation). In the right panel, a 10-fold excess of fragment 585–939 was incubated with IleRS without unfolding and refolding for 120 min.
The stoichiometry of the inhibited complex was determined by gel filtration of the complex under native conditions. Fractions corresponding to molecular weights from 45–200 kDa were analyzed by SDS/PAGE and Western blotting with anti-isoleucyl-tRNA synthetase antibody (Fig. 4). Fractions corresponding to an apparent molecular weight of 150 kDa contained both full-length isoleucyl-tRNA synthetase and the C-terminal fragment, indicating that the complex formed between the 45-kDa fragment and the wild-type enzyme (105 kDa) has a 1:1 stoichiometry. No complexes of higher apparent molecular weight were evident in this experiment.
Figure 4.
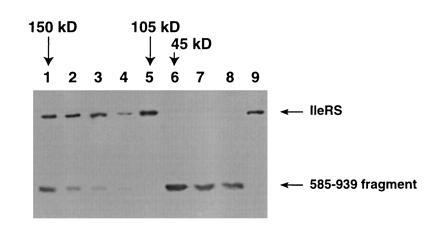
Gel filtration chromatography of inhibited complex. Full-length IleRS was denatured in 6 M guanidinium HCl in the presence of a 12-fold excess of the 585–939 fragment. The sample was renatured, and aminoacylation activity was found to be abolished. The sample was applied to a Sephacryl S300 gel filtration column (120 ml bed volume) in 50 mM potassium phosphate (pH 7.5), 0.1 mM NaCl, 50 mM 2-mercaptoethanol. Peaks of absorbance at 280 nm were analyzed by Western blotting with anti-IleRS antibody. Lanes 1–7 show fractions eluting from the gel filtration column. Lane 8, fragment 585–939 of isoleucyl-tRNA synthetase; lane 9, pure isoleucyl-tRNA synthetase. The retention times of protein standards are indicated by arrows above the figure. Protein standards for the column were: alcohol dehydrogenase (150 kDa; elution volume = 33 ml), IleRS (105 kDa; elution volume = 39 ml), and ovalbumin (45 kDa; elution volume = 46 ml).
Some free isoleucyl-tRNA synthetase (105 kDa) was observed after gel filtration of the inhibited complex (Fig. 4, lane 5). Aminoacylation measurements of the column fractions showed that this 105 kDa fraction had regained activity (data not shown), suggesting that the inhibited complex partially dissociated during the time course of the gel filtration experiment. Future studies will focus on the stability of the enzyme-fragment complex under different experimental conditions.
Concluding Remarks.
Hall and Frieden described differential inhibition of dihydrofolate reductase by polypeptide fragments of the enzyme (11). Our results suggest that this phenomenon may be more general than previously recognized. For dihydrofolate reductase, as well as for isoleucyl-tRNA synthetase, the inhibitory effect is specific, and the phenomenon is only apparent with the refolding enzyme; the folded enzyme is resistant to inhibition. In addition, with isoleucyl-tRNA synthetase, we were able to show that inhibition is enzyme-specific. While the specific mechanism of the inhibitory effect is not known at this time, one possibility is that the inhibitory fragments possess significant secondary structure in solution that serves as an initiation site around which the protein folds incorrectly. For several proteins, including BSA (23), immunoglobulin light chain (24), the β2-subunit of tryptophan synthetase (25), and aspartokinase homoserine dehydrogenase (26), it has been shown that the folding of an isolated domain is as fast or faster than that of the same domain in the context of the entire protein. Work on the kinetic mechanisms of folding for large single-chain proteins suggests that the domains fold independently and rapidly in an initial step, and then pair in a subsequent slower step (26). For enzymes, this slowest step is generally characterized by the reappearance of activity. For dominant negative inhibition of Ile-tRNA synthetase by polypeptide fragments, intermolecular interactions between the folded/unpaired intermediate and the fragments may disrupt the normal folding process and cause inhibition. Because autonomously folding domains have been identified in other systems as well (26–28), our observations suggest that the folding pathways of large monomeric proteins may be exploited to design dominant negative inhibitors.
Acknowledgments
This work was supported by the Human Frontiers Science Program Grant RG-392 (K.S. and W.T.M.) and by Grant GM23562 (P.S.) from the National Institutes of Health.
References
- 1.Herskowitz I. Nature (London) 1987;329:219–222. doi: 10.1038/329219a0. [DOI] [PubMed] [Google Scholar]
- 2.Amaya E, Musci T J, Kirschner M W. Cell. 1991;66:257–270. doi: 10.1016/0092-8674(91)90616-7. [DOI] [PubMed] [Google Scholar]
- 3.Verrall S, Hall Z W. Cell. 1992;68:23–31. doi: 10.1016/0092-8674(92)90203-o. [DOI] [PubMed] [Google Scholar]
- 4.Kintner C. Cell. 1992;69:225–236. doi: 10.1016/0092-8674(92)90404-z. [DOI] [PubMed] [Google Scholar]
- 5.Friedman A D, Triezenberg S J, McKnight S L. Nature (London) 1988;335:452–454. doi: 10.1038/335452a0. [DOI] [PubMed] [Google Scholar]
- 6.Bevec D, Dobrovnik M, Hauber J, Böhnlein E. Proc Natl Acad Sci USA. 1992;89:9870–9874. doi: 10.1073/pnas.89.20.9870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Unger T, Mietz J A, Scheffner M, Lee C L, Howley P M. Mol Cell Biol. 1993;13:5186–5194. doi: 10.1128/mcb.13.9.5186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roman C, Cohn L, Calame K. Science. 1991;254:94–97. doi: 10.1126/science.1840705. [DOI] [PubMed] [Google Scholar]
- 9.Treacy M N, Neilson L I, Turner E E, He X, Rosenfeld M G. Cell. 1992;68:491–505. doi: 10.1016/0092-8674(92)90186-g. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt E, Schimmel P. Proc Natl Acad Sci USA. 1993;90:6919–6923. doi: 10.1073/pnas.90.15.6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hall J G, Frieden C. Proc Natl Acad Sci USA. 1989;86:3060–3064. doi: 10.1073/pnas.86.9.3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Webster T, Tsai H, Kula M, Mackie G A, Schimmel P. Science. 1984;226:1315–1317. doi: 10.1126/science.6390679. [DOI] [PubMed] [Google Scholar]
- 13.Brunie S, Zelwer C, Risler J-L. J Mol Biol. 1990;216:411–424. doi: 10.1016/S0022-2836(05)80331-6. [DOI] [PubMed] [Google Scholar]
- 14.Schimmel P, Shepard A, Shiba K. Protein Sci. 1992;1:1387–1391. doi: 10.1002/pro.5560011018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Starzyk R M, Webster T A, Schimmel P. Science. 1987;237:1614–1618. doi: 10.1126/science.3306924. [DOI] [PubMed] [Google Scholar]
- 16.Shepard A, Shiba K, Schimmel P. Proc Natl Acad Sci USA. 1992;89:9964–9968. doi: 10.1073/pnas.89.20.9964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glasfeld E, Landro J A, Schimmel P. Biochemistry. 1996;35:4139–4145. doi: 10.1021/bi9527810. [DOI] [PubMed] [Google Scholar]
- 18.Shiba K, Schimmel P. Proc Natl Acad Sci USA. 1992;89:1880–1884. doi: 10.1073/pnas.89.5.1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shiba K, Schimmel P. J Biol Chem. 1992;267:22703–22706. [PubMed] [Google Scholar]
- 20.Burbaum J J, Schimmel P. Biochemistry. 1991;30:319–324. doi: 10.1021/bi00216a002. [DOI] [PubMed] [Google Scholar]
- 21.Ellison M J, Hochstrasser M. J Biol Chem. 1991;266:21150–21157. [PubMed] [Google Scholar]
- 22.Jasin M, Regan L, Schimmel P. Cell. 1984;36:1089–1095. doi: 10.1016/0092-8674(84)90059-x. [DOI] [PubMed] [Google Scholar]
- 23.Teale J M, Benjamin D C. J Biol Chem. 1976;251:4603–4608. [PubMed] [Google Scholar]
- 24.Tsunenaga M, Goto Y, Kawata Y, Hamaguchi K. Biochemistry. 1987;26:6044–6051. doi: 10.1021/bi00393a015. [DOI] [PubMed] [Google Scholar]
- 25.Blond S, Goldberg M E. Proc Natl Acad Sci USA. 1987;84:1147–1151. doi: 10.1073/pnas.84.5.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garel J-R. In: Protein Folding. Creighton T E, editor. New York: Freeman; 1992. pp. 405–454. [Google Scholar]
- 27.Chrunyk B A, Matthews C R. Biochemistry. 1990;29:2149–2154. doi: 10.1021/bi00460a027. [DOI] [PubMed] [Google Scholar]
- 28.Wu L C, Grandori R, Carey J. Protein Sci. 1994;3:369–371. doi: 10.1002/pro.5560030301. [DOI] [PMC free article] [PubMed] [Google Scholar]