

Abstract
This is the first report of papillary thyroid carcinoma combined with multiple endocrine neoplasia type 1 (MEN1) in Korea. MEN1 is a hereditary disease comprising neoplastic disorders such as pituitary, parathyroid and pancreatic neuroendocrine tumor, such as gastrinoma. But papillary thyroid cancer was never regarded as its component before in Korea. Herein we present a 39-year-old woman who manifested typical features of MEN1 with a coincidental papillary thyroid carcinoma. Although the family history of MEN1 was definite, her genetic analysis of DNA had revealed no germline mutation in MEN1 gene locus. Unidentified culprit gene unable us further genetic study to find LOH (loss of heterogeneity) in 11q13, the possible explanation of papillary thyroid carcinoma as a new component of MEN1. As we have first experienced a case of MEN1 combined with papillary thyroid carcinoma in Korea, we report it with the review of literature.
Keywords: Multiple endocrine neoplasia type 1, papillary thyroid carcinoma, prolactinoma, gastrinoma, parathyroid hyperplasia
INTRODUCTION
In 1903, Erdheim first reported concomitant parathyroid hyperplasia with pituitary adenoma and since 1954 when Wermer reported a family with multiple glandular disease, many attentions have been paid on the hereditary characteristics of the disease. The multiple endocrine neoplasia type 1 (MEN1) is a disease that is transmitted in an autosomal dominant way with the incidence of 0.2 - 2 in 100,000 persons, without sexual preference.1 The disease is made by the germline mutation of a tumor suppressor gene on chromosome 11 long arm 13 (11q13), which encodes menin protein, a transcriptional protein.2 MEN1 is characterized by coincidental, multiple endocrine tumors such as parathyroid, anterior pituitary and pancreatic islet cell tumor and less frequently, anterior thymoma, pheochromocytoma, cutaneous lipoma, facial fibroangioma, esophageal leiomyoma, thyroid adenoma may also occur.1-3 Speaking of the thyroid disease, it can be observed in over 25% of MEN1 patients,3,4 and it can be detected incidentally during parathyroid surgery in MEN1 patients. But so far, only two cases of thyroid carcinoma combined with MEN1 were reported, and they confirmed that papillary thyroid cancer did not correlate to MEN1.4,5
Our report which is the first case in Korea, with the following 2 cases above, is anticipated to provide the important information for the characterization of the developmental association of these two diseases.
CASE REPORT
A 39-year-old woman visited the hospital for evaluation of MEN1, as she had her two older brothers diagnosed of the disease one year before. The patient had no past history such as diabetes, hepatitis, hypertension, pulmonary tuberculosis and others. She had two children and normal menstrual cycle, but in recent two years, the cycle became irregular. She presented no particular symptoms except that on physical examination, a 1.5 cm sized nodule in her right thyroid lobe was palpated. Laboratory tests revealed no abnormal results except for increased PTH (201 ng/L, normal value: 13 - 104 ng/L), calcium (2.77 nmol/L), prolactin (149 ug/L, normal value: 3.8 - 31.4 ug/L), and gastrin (555 ng/L, normal value: 0 - 90 ng/L) levels. On cervical ultrasonography, a 1.5 cm sized low density nodule was found on right posterior lobe of the thyroid (Fig. 1) and on sellar MRI, a 0.9 × 0.6 cm sized pituitary mass was seen on the left sellar wing (Fig. 2). Furthermore on abdominal computed tomography (CT) scan, we found a 0.68 cm sized contrast-enhanced mass on pancreatic body portion (Fig. 3). Based on these results, we could conclude that she had hyperparathyroidism, gastrinoma and prolactinoma with thyroid tumor. She underwent parathyroidectomy, thyroidectomy and the pathologic results revealed bilateral parathyroid hyperplasia (Fig. 4) and a typical thyroid papillary cancer (T2N0M0) (Fig. 5). For the treatment of prolactinoma and gastrinoma, she is now taking bromocriptin and proton-pump inhibitor daily. In the analysis of all coding exons of MEN1 of the patient, a silent mutation was detected in the exon 9 and polymorphism in the exon 10, however these results suggest no specific abnormality in MEN1 disease, reports from the Korean Hereditary Tumor Registration Center.
Fig. 1.
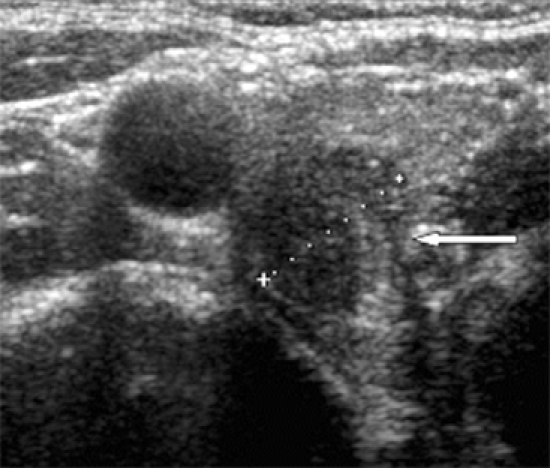
Thyroid US shows about 1.5 cm irregular mass at upper pole of right thyroid gland.
Fig. 2.
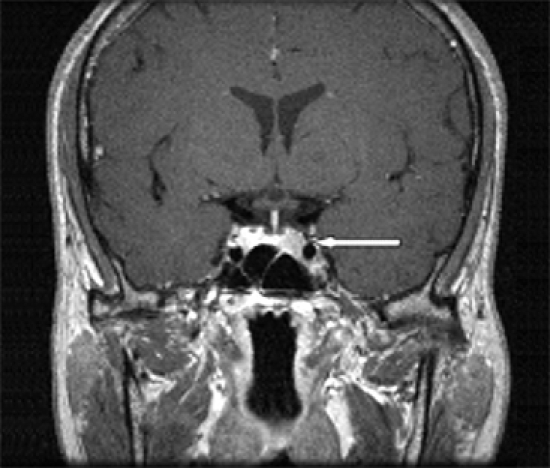
Sellar MRI shows 0.9 × 0.6 mm sized microadenoma in left portion of pituitary gland.
Fig. 3.
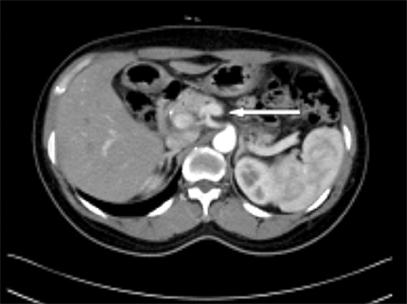
Abdominal dynamic CT shows 0.68 cm sized arterial enhancing mass at pancreatic body.
Fig. 4.
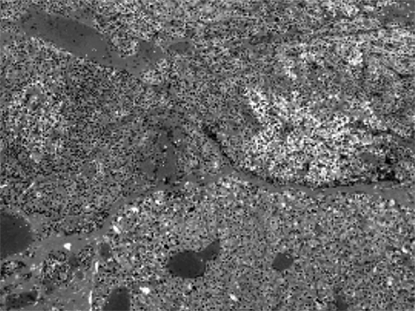
Histologic feature of parathyroid hyperplasia (H&E stain, × 100).
Fig. 5.
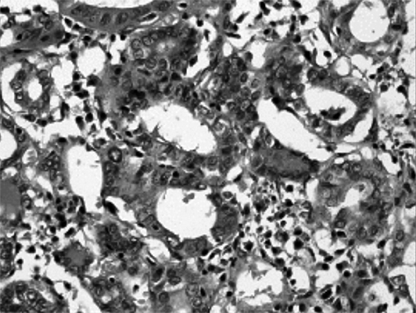
Histologic feature of thyroid papillary carcinoma showing inclusion body (H&E stain, × 100).
DISCUSSION
MEN1 is a familial disease that presents parathyroid, pituitary and pancreatic islet cell tumor or hyperplasia. So far, 16 cases have been reported in Korea, and among them, the family history were confirmed in 5 cases.6 Our case is the first report of thyroid papillary cancer combined with MEN1 in Korea, and the third case report worldwide.4,5
In the year of 1988, using the method of linkage analysis of polymorphic DNA sequencing,7 MEN1 gene was detected in chromosome 11q138 and subsequently loss of heterogeneity in MEN1 gene was found.9 MEN1 gene is a tumor suppressor gene that encodes the nuclear protein, menin, which regulates cell growth and cycle and when this protein deficit occurs, MEN1 develops. Tumors involved endocrine glands in MEN1 patients demonstrate loss of heterogeneity (LOH) at the locus of the MEN1 tumor suppressor gene. Menin, the protein encoded by Menin gene is ubiquitously expressed in endocrine tissues, and less commonly these patients can present with tumors of other endocrine glands including thyroid. To verify whether papillary thyroid cancer is developed as a part of MEN1 disease, one should prove that allelic LOH of 11q13 in thyroid tissue be identical to that of leukocyte DNA.3,4 But as in our case, the lack of obvious LOH of the MEN1 locus in the papillary carcinoma suggests that deletion of the MEN1 tumor suppression might not be etiologically related to the oncogenesis of the papillary thyroid carcinoma. Furthermore papillary thyroid carcinoma was found only in our patient and not in any of the family members involved MEN1. So the possibility that thyroid carcinoma is etiologically associated with MEN1 seems to be low.
In the analysis of MEN1 genetic study, about 20% may have false negative results due to the diversity of the causative mutation and scattered position in the entire open reading frame. Moreover approximately 10% new germline mutations are being detected in the overall MEN1 patients, which is the reason why the association of genotype and phenotype could not be identical in 10 - 30% of patients. Similarly, in our case, we could not find the causative mutation in the patient and family.
In Korea, among five MEN1 families, only four family germline mutations were found which are in concordance with other reports with 80% positive detection rate.6 Among mutations discovered in Korea, three were new ones and the remaining one has already been revealed before.6
In the other hand, thyroid carcinoma has been reported in the range of 1.7% to 6% of patients with primary hyperparathyroidism.10-12 Hypotheses have been presented to address these findings, but no firm conclusions exist at this time regarding the etiology of synchronous thyroid and parathyroid disease.13-15 Only one factor like prior exposure to radiation may play a role in the association. But in our case, the patient had no past history of radiation exposure. So there seems to be little correlation between the two diseases in our case.16
Our case suggests three possibilities of the development of thyroid papillary cancer as a component of MEN1. First possibility is the close association between the two genes. But no data exists supporting this hypothesis yet. Second, the possibility of the incidental occurrence of thyroid papillary cancer for it occurs with a high prevalence. Presently, the opinion that these two diseases' occurrence is separate is prevalent. However, there exit two reports which have evidence for their intimate correlation. Finally, the possibility that it is a new type of familial disease causing two diseases by a genetic abnormality that is not characterized yet.
References
- 1.Lairmore TC, Piersall LD, DeBenedetti MK, Dilley WG, Mutch MG, Whelan AJ, et al. Clinical genetic testing and early surgical intervention in patients with multiple endocrine neoplasia type1 (MEN1) Ann Surg. 2004;239:637–645. doi: 10.1097/01.sla.0000124383.98416.8d. discussion 645-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, et al. Positional cloning of the gene for multiple endocrine neoplasia type1. Science. 1997;276:404–407. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- 3.Dong Q, Debelenko LV, Chandrasekharappa SC, Emmert-Buck MR, Zhuang Z, Guru SC, et al. Loss of heterozygosity at 11q13: analysis of pituitary tumors, lung carcinoids, lipomas, and other uncommon tumors in subjects with familial multiple endocrine neoplasia type1. J Clin Endocrinol Metab. 1997;82:1416–1420. doi: 10.1210/jcem.82.5.3944. [DOI] [PubMed] [Google Scholar]
- 4.Desai D, McPherson LA, Higgins JP, Weigel RJ. Genetic analysis of a papillary thyroid carcinoma in a patient with MEN1. Ann Surg Oncol. 2001;8:342–346. doi: 10.1007/s10434-001-0342-8. [DOI] [PubMed] [Google Scholar]
- 5.Vortmeyer AO, Lubensky IA, Skarulis M, Li G, Moon YW, Park WS, et al. Multiple endocrine neoplasia type 1: atypical presentation, clinical course, and genetic analysis of multiple tumors. Mod Pathol. 1999;12:919–924. [PubMed] [Google Scholar]
- 6.Park JH, Kim IJ, Kang HC, Lee SH, Shin Y, Kim KH, et al. Germline mutations of the MEN1 gene in Korean families with multiple endocrine neoplasia type 1 (MEN1) or MEN1-related disorders. Clin Genet. 2003;64:48–53. doi: 10.1034/j.1399-0004.2003.00091.x. [DOI] [PubMed] [Google Scholar]
- 7.Larsson C, Skogseid B, Oberg K, Nakamura Y, Nordenskjöld M. Multiple endocrine neoplasia type1 gene maps to chromosome 11 and is lost in insulinoma. Nature. 1988;332:85–87. doi: 10.1038/332085a0. [DOI] [PubMed] [Google Scholar]
- 8.Friedman E, Sakaguchi K, Bale AE, Falchetti A, Streeten E, Zimering MB, et al. Clonality of parathyroid tumors in familial multiple endocrine neoplasia type1. N Engl J Med. 1989;321:213–218. doi: 10.1056/NEJM198907273210402. [DOI] [PubMed] [Google Scholar]
- 9.Chandrasekharappa SC, Teh BT. Functional studies of the MEN1 gene. J Intern Med. 2003;253:606–615. doi: 10.1046/j.1365-2796.2003.01165.x. [DOI] [PubMed] [Google Scholar]
- 10.Nishiyama RH, Farhi D, Thompson NW. Radiation exposure and the simultaneous occurrence of primary hyperparathyroidism and thyroid nodules. Surg Clin North Am. 1979;59:65–75. doi: 10.1016/s0039-6109(16)41733-0. [DOI] [PubMed] [Google Scholar]
- 11.Prinz RA, Barbato AL, Braithwaite SS, Brooks MH, Emanuele MA, Gordon DL, et al. Simultaneous primary hyperparathyroidism and nodular thyroid disease. Surgery. 1982;92:454–458. [PubMed] [Google Scholar]
- 12.Attie JN, Vardhan R. Association of hyperparathyroidism with nonmedullary thyroid carcinoma: review of 31 cases. Head Neck. 1993;15:20–23. doi: 10.1002/hed.2880150105. [DOI] [PubMed] [Google Scholar]
- 13.Hedman I, Tisell LE. Associated hyperparathyroidism and nonmedullary thyroid carcinoma: the etiologic role of radiation. Surgery. 1984;95:392–397. [PubMed] [Google Scholar]
- 14.Smith MA, McHenry C, Oslapas R, Hofmann C, Hessel P, Paloyan E. Altered TSH levels associated with increased serum 1,25-dihydroxyvitamin D3: a possible link between thyroid and parathyroid disease. Surgery. 1989;106:987–991. [PubMed] [Google Scholar]
- 15.Arem R, Lim-Abrahan MA, Mallette LE. Concomitant Graves' disease and primary hyperparathyroidism. Influence of hyperthyroidism on serum calcium and parathyroid hormone. Am J Med. 1986;80:693–698. doi: 10.1016/0002-9343(86)90827-2. [DOI] [PubMed] [Google Scholar]
- 16.Regal M, Páramo C, Luna Cano R, Pérez Méndez LF, Sierra JM, Rodríguez I, et al. Coexistence of primary hyperparathyroidism and thyroid disease. J Endocrinol Invest. 1999;22:191–197. doi: 10.1007/BF03343540. [DOI] [PubMed] [Google Scholar]