

Abstract
We report on the design, synthesis and evaluation of series of furanyl-salicyl-nitroxide derivatives, as effective chemical probes for second site screening against phosphotyrosine phosphatases (PTPs) using NMR-based techniques. The compounds have been tested against a panel of PTPs to asses their ability to inhibit a broad spectrum of these phosphatases. The utility of the derived compounds is illustrated with the phosphatase YopH, a bacterial toxin from Yersinia pestis. Novel chemical fragments were identified during a NMR-based screen for compounds that are capable to bind on the surface of YopH in regions adjacent the catalytic site in presence of the spin-labeled compounds. Our data demonstrate the value of the derived chemical probes for NMR-based second-site screening in PTPs.
Introduction
Protein tyrosine phosphatases (PTPs) have been the focus of considerable drug discovery efforts in recent years owing to evidence of their role in regulating cell-signaling pathways [1, 2, 3]. However, many inhibitors identified so far are peptide-based, and because of the conserved nature of the active site among phosphotyrosine binding pockets, obtaining selectivity is often difficult. Only very recently, several examples point to fragment-based approaches as possible route for the identification of potent and selective protein phosphatase inhibitors [4, 5]. A first example in designing selective PTP1B inhibitors was recently reported by Szczepankiewicz et al. [5]. In this work, the authors screened a library of 10,000 compounds by using heteronuclear [15N,1H] NMR correlations spectra to assess binding. This approach has the advantages that even small fragments that bind fairly weakly can be identified, and that information on the binding mode can also be obtained [6, 7]. This screen resulted in a compound that mimics the natural phosphotyrosine substrate and inhibits PTP1B in the micromolar range. Subsequently this hit was optimized with the aim of fully occupying adjacent sites by modular addition of scaffolds [5]. In fact, an additional compound that bound in a second adjacent site was identified by exploiting another NMR screen. The subsequent chemical linkage of both compounds lead to a bi-dentate molecule that inhibited PTP1B in the nanomolar range [5].
Another example on the use of fragment-based approaches to obtain inhibitors of a PTP is the application of the “click chemistry” [4]. In this case, Srinivasan et al. employed alkyne-containing compounds targeting the PTP active site of PTP1B and azide-containing compounds targeting an already known secondary aryl-phosphate binding site near the catalytic site. With these two types of fragments a library of 66 compounds was synthesized and screened against different PTPs obtaining one compound with an IC50 of 5 μM for PTP1B and a selectivity of 5 to 25 times with respect to the rest of PTPs tested [4].
Recent studies from our laboratories focused on the inhibition of the bacterial protein tyrosine phosphatase YopH [8]. Aided by a combination of chemical library screening, structure-activity relationships analysis and in silico docking of lead compounds, we developed small-molecule inhibitors of YopH [8]. Our inhibitors contain a single salicylate linked to a furanyl moiety as phosphotyrosine mimic and a more variable group that could be exploited to achieve selectivity and higher affinity [8]. In fact, while very small differences can be seen in the phosphotyrosine binding pockets of tyrosine phosphatases, unique sub-pockets can be found in adjacent regions [3, 8]. Therefore, it appears evident that by tailoring a second-site ligand, it should be possible to develop potent and selective inhibitors of therapeutically relevant protein tyrosine phosphatases. An interesting approach to screen for second site binders was recently reported by Jahnke and co-workers [9, 10, 11]. This method utilizes initial binders chemically labeled with organic nitroxide radicals (“spin labels” such as the 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO)) to perform second-site NMR spectroscopic screens of fragment libraries. The binding of a second-site ligand can be simply detected by measuring the relaxation enhancement induced by the spin-labeled first ligand (Figure 1) [9, 10, 11]. Other common approaches for second-site screen via NMR are the well known SAR by NMR method [12], where the second ligand is screened via protein [15N, 1H] correlation spectra, and the SAR by ILOEs method, where the second site ligand is detected via protein-mediated ligand-ligand NOEs [13, 14]. Compared with these methods, a major drawback of the spin-labeling approach consists in the necessity to produce an organic nitroxide radical derivative of a first ligand, which may be different for each target (Figure 1). A more practical approach consists in preparing first-site spin-labeled compounds that are cross-reactive within different members of a protein family, such as protein kinases, as recently reported [15], or phosphatases as we report in this work. Such chemical tools are then useful for the design and synthesis of bi-dentate compounds with increased affinity but also specificity for a given target. In fact, if specificity is a major issue, second site ligands that are specific for a given protein may be selected by performing the NMR screening against counter targets. Based on these premises, we report herein the synthesis and characterization of novel furanyl-salicyl-nitroxide derivatives (Table 1) as versatile probes for NMR-based second-site screening in protein tyrosine phosphatases.
Figure 1.
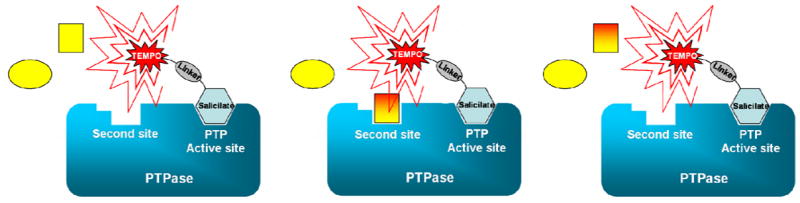
Schematic representation of the second-site screening approach proposed to design potent and selective PTPase inhibitors. The approach is based on the hypothesis that a pharmacophore for potent and selective PTP inhibitors would consist of a salicyl-furanyl moiety, as phosho-Tyrosine mimicking scaffold, connected to a second site binder whose nature would be different for various PTP targets.
Table 1.
Chemical structures and measured IC50 values (μM) for the spin-labeled probes against different PTPs.
| Compound | YopH | PTP1B | HePTP | TCPTP | CD45 | VHR |
|---|---|---|---|---|---|---|
|
1a |
>1,000 | >1,000 | >1,000 | >1,000 | >1,000 | >1,000 |
|
1b |
336 | >1,000 | >1,000 | >1,000 | >1,000 | ~1,000 |
|
2a |
>1,000 | >1,000 | >1,000 | >1,000 | >1,000 | ~850 |
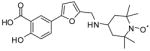
2b |
50 | >1,000 | >1,000 | 685 | >1,000 | 241 |
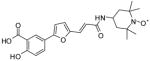
3 |
3.0 | 7.5 | 24 | 14 | 14 | 12 |
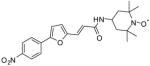
4a |
47 | >1,000 | >1,000 | >1,000 | >1,000 | ~1,000 |
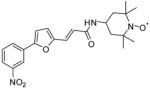
4b |
46 | 211 | ~1,000 | 260 | >1,000 | ~200 |
Results and discussion
Chemical Synthesis
The synthesis of all the nitroxide compounds reported in Table 1 was accomplished employing easy and straightforward synthetic schemes that included as last step the attachment of the 4-amino 2,2,6,6-tetramethylpiperidine 1-oxyl (4-amino TEMPO) through an amide bond to the phosphotyrosine mimic (Figures 2 and 3).
Figure 2.
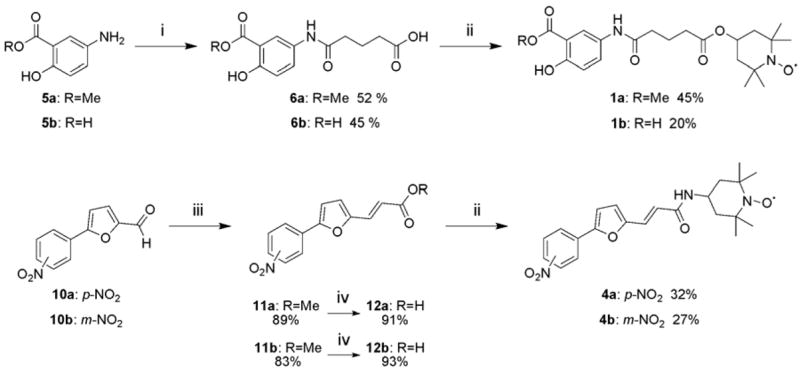
Synthetic scheme for compounds 1a–b and 4a–b: i) Glutaric anhydride (1 eq.), 5a–b (2 eq.) in Pyridine/DCM (1:1) under N2, r.t. 16h; ii) 4-amino-TEMPO (2 eq.), WSC (1.1 eq.) in DCM/DMF (9:1); iii) 10a–b (1 eq.), methyl diethyl phosphonoacetate (1.1 eq.), LiOH (1 eq.) in THF under Ar; iv) MeOH/NaOH 2M (1:1) at 80 °C, 2h.
Figure 3.
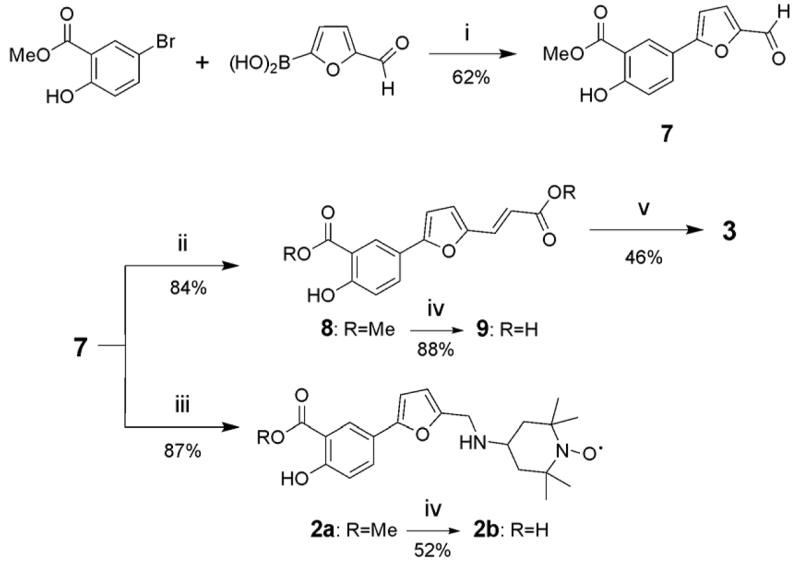
Synthetic scheme for compounds 2a–b and 3: i) 5-Bromo-2-hydroxy-benzoic acid methyl ester (1 eq.), 5-formylfuran-2-ylboronic acid (1.2 eq.), Pd(OAc)2 (3 mol%), K2CO3 (2.5 eq.), Bu4NCl (1 eq.) in H2O/MeOH (1:1); ii) methyl diethyl phosphonoacetate (1.1 eq.), LiOH (1 eq.) in THF under Ar; iii) 4-amino-TEMPO (1 eq.) in toluene with 5% AcOH o.n., then NaBH3CN (1.1 eq.) in MeOH with 0.5% HCl; iv) MeOH/NaOH 2M (1:1) at 80 °C (9) for 2h or r.t. (2a) for 16h; v) 4-amino-TEMPO (2 eq.), WSC (1.1 eq.) in DCM/DMF (9:1).
The compounds 1a–b were obtained through the synthesis of the intermediate carboxylic acids 6a–b by the reaction between the anilines 5a–b and glutaric anhydride. A subsequent amide formation with 4-amino TEMPO, employing water soluble carbodiimide (WSC) as activating agent, led to the stable radical compounds 1a–b (Figure 2).
Compounds 4a–b were obtained from commercial aldehydes 10a–b. A Horner-Wadsworth-Emmons reaction between these aldehydes and methyl diethyl phosphonoacetate afforded the corresponding methyl esters 11a–b in 89% and 83% yields respectively. After saponification and amide formation with 4-amino TEMPO in presence of WSC, compounds 4a–b were isolated (Figure 2).
The first synthetic step towards compounds 2a–b and compound 3 is the same in both cases. A Suzuki coupling between the methyl 5-bromo-2-hydroxybenzoate and the 5-formylfuran-2-ylboronic acid allowed us to obtain aldehyde 7 with a 62% yield. A reductive amination of this aldehyde with 4-amino TEMPO afforded compound 2a with an 87% yield. Then, compound 2b was obtained after the saponification of 2a. In order to obtain compound 3, a Horner-Wadsworth-Emmons reaction was done between aldehyde 7 and the methyl diethyl phosphonoacetate obtaining the methyl diester 8 with 84% yield. This compound was saponificated obtaining the dicarboxylic acid 9 with 88% yield. Knowing by previous experience in the laboratory that the carboxylic acid of the salycilic acid moiety is not particularly reactive, we could obtain compound 3 after an amide bond formation between the compound 9 and 4-amino TEMPO with a 46% yield (Figure 3).
Biochemical Evaluation
In order to characterize affinity of the compounds, we tested their inhibitory activity against a diverse panel of protein tyrosine phosphatases (Table 1). As it can be seen, compound 3 is the most cross-reactive showing inhibition constants in the range from 3.0 μM (YopH) to 24 μM (HePTP), representing an ideal compound candidate for second site screen in the proteins tested, and presumably to many others that have similar phosphotyrosine binding pocket characteristics.
To further characterize the binding of compound 3 to protein tyrosine phosphate binding pockets, we isolated the C-terminal domain (340 residues) of the PTP YopH (468 residues) from Yersinia pestis and monitored the effect of the ligand on resonance intensities in [15N, 1H] correlation spectra (Figure 4). As it can be seen, the binding of compound 3 to the C-terminal domain of YopH cause selective broadening of several resonance lines, presumably those located in the proximity of the binding pocket of the protein due to their closeness to the relaxation enhancer spin-labeled compound. In fact, the main nuclear spin relaxation mechanism in proteins and small molecule binders derives from the dipole-dipole interaction between a given nucleus and surrounding spins. The magnitude of this effect is proportional to the distance between the nucleus of the spins and to the gyromagnetic ratio of the spins. The unpaired electron possesses a gyromagnetic ratio which is 657.4 times that of a hydrogen nucleus, thus producing the most efficient relaxation effect even up to 10–15 Å from a given nucleus. As recently pointed out by Jahnke and co-workers [11], these data also further prove the utility of the chemical probes in the identification of binding site residues in isotopically labeled protein samples, particularly when combined with selective labeling of the target [11]. Finally, we have used compound 3 to screen for fragments that are capable to bind in adjacent pockets on the surface of the C-terminal domain of YopH. This was accomplished by exposing a mixture of potential second-site binders (1 mM) to compound 3 (500 μM) in presence and absence of a substoichiometric amount of YopH (10 μM). 1D 1H-T1ρ NMR experiments are subsequently recorded and the differential loss of signal intensity is used to detect second-site binders. In fact, close proximity of any given second-site binder to the spin-labeled compound will result in rapid nuclear spin relaxation of hydrogen nuclei of the compound with concomitant line broadening in a simple 1D 1H-NMR experiment. In order to amplify this effect, the transverse 1H magnetization of the compounds is “locked” on the xy plane for 50 to 200 ms prior the acquisition of the 1D 1H-NMR experiment. Such technique is usually referred as a T1ρ measurement [16]. Figure 5A shows a typical example of second site binders that can be easily identified with this approach. While binding of a given compound to the protein target would also result in decreased transverse relaxation times and thus reduce signal intensity (Figures 5B,C), the consequence of close proximity with a spin-labeled compound has a much more dramatic effect (Figure 5D). Thus, it is possible to discriminate protein binders in the proximity of the spin-labeled compound among non-binders or even compounds that bind in different locations. Intriguingly, some of the identified compounds, for example the triazole derivative shown in red in Figure 5A, largely resemble scaffolds that were used to obtain potent bi-dentate PTP inhibitors discovered by the SAR by NMR approach [1,5].
Figure 4.
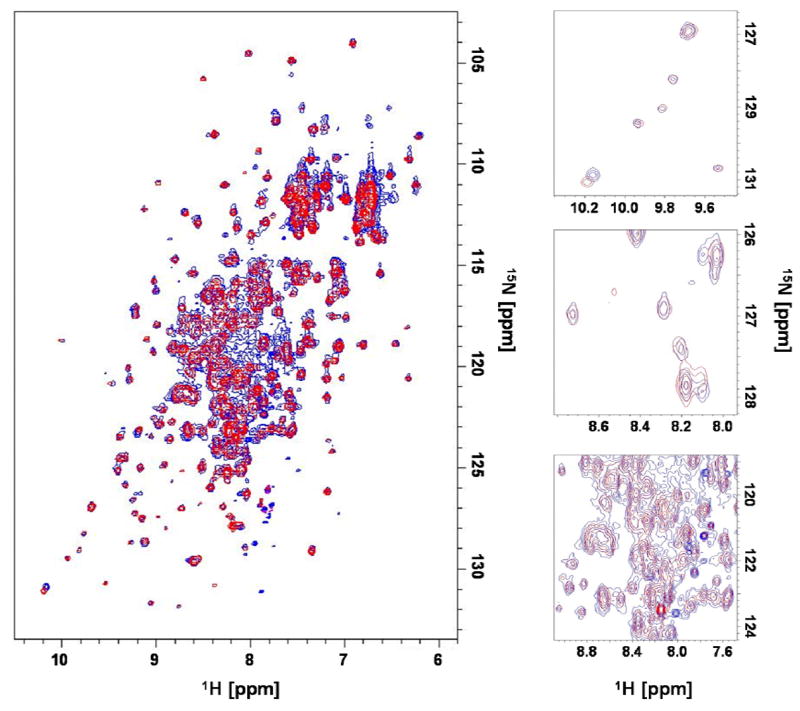
Effect of compound 3 on the [15N, 1H] correlation spectra of the C-terminal domain of YopH. The spectra were recorded in absence (blue) or in presence (red) of such compound. The expanded 2D regions (right) show disappearance or movement of some protein signals as consequence of ligand binding.
Figure 5.
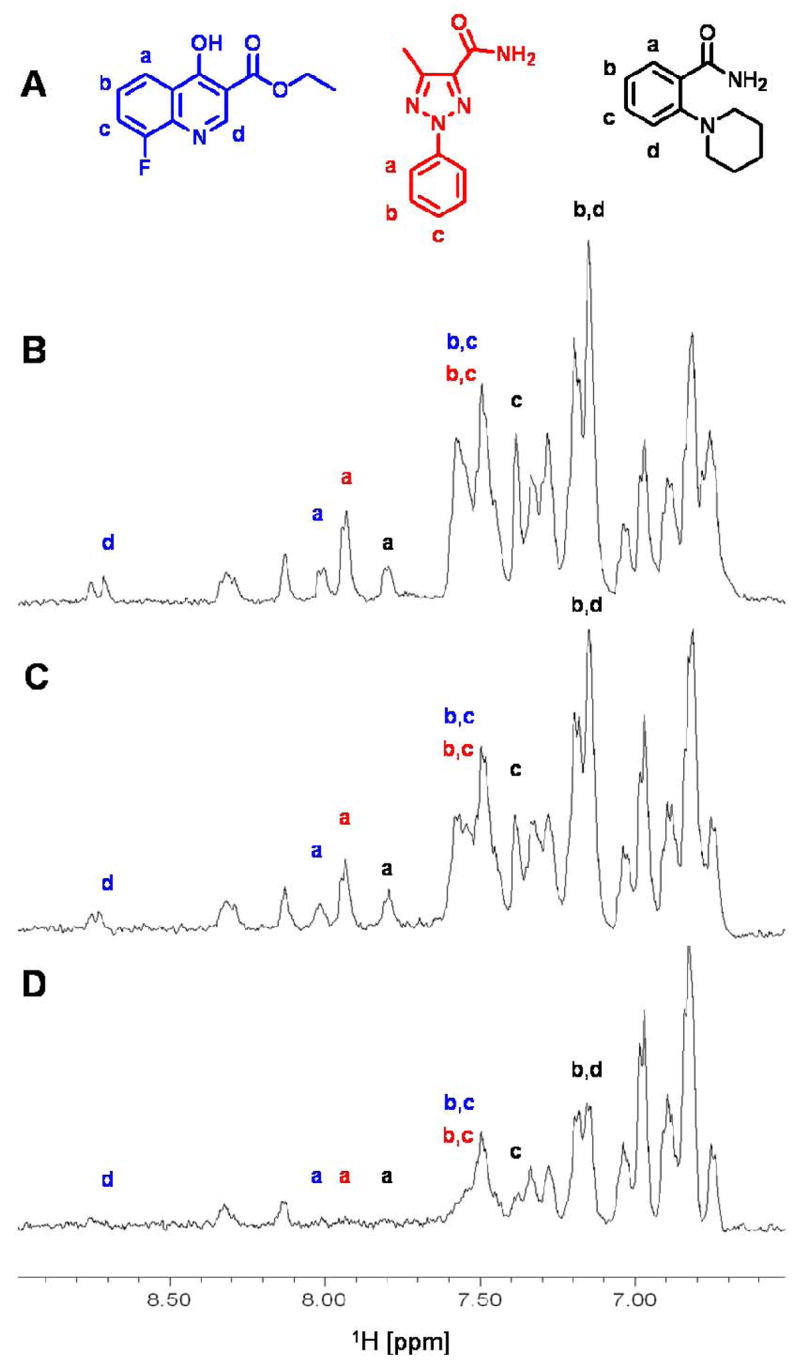
Example of second site binders (A) discovered by using compound 3 as spin-labeled probe. The aromatic region of 1D T1ρ spectra of a mixture of 10 compounds (1 mM each) are reported: B) in the presence of 500 μM of compound 3; C) in the presence of 10 μM of GST-YopH; and, D) in the presence of both 500 μM of compound 3 and 10 μM of GST-YopH. Letters indicate resonances from the compounds shown in panel A in corresponding colors. The additional peak shown around 8.7 ppm comes from an analogue of the quinoline compound (blue in panel A) present in the mixture.
In conclusion, we report here on the synthesis and characterization of a series of furanyl-salicyl-nitroxide derivatives that can be used as probes for NMR-based second-site screening in protein tyrosine phosphatases and to map PTP binding site residues. Although it is clearly emerging that the misregulation of protein tyrosine phosphatase activity is associated with the onset, development and progression of several human malignancies, the discovery of novel, safe and selective inhibitors has not been as successful as for other enzymes, mainly due to the high degree of conservation of the phosphotyrosine binding pocket [17]. We believe that the reported spin-labeled probes and the second-site screening approach proposed here may result very useful in developing novel, potent and selective lead compounds for further target validation and continued drug development.
Experimental Procedures
Chemistry
General methods and materials
Every chemical was used as received from the specified supplier. Anhydrous dimethylformamide (DMF) and anhydrous dichloromethane (DCM) were purchased from AKROS Chemical. Ethyl acetate, hexane and methanol were purchased from Fisher Scientific. Pyridine, toluene, diethylether, tetrahydrofurane (THF), glutaric anhydride, methyl 5-amino salicylate, 5-aminosalicylic acid, 5-bromo-2hydroxybenzoate, 5-formylfuran-2-ylboronic acid, tetrabutylammonium chloride, palladium acetate, potassium carbonate, 4-amino TEMPO, sodium cyanoborohydride, methyl diethyl phosphonate, lithium hydroxide, 5-(4-nitrophenyl)-furfural and 5-(3-nitrophenyl)-furfural were all purchased from SIGMA-ALDRICH. The water soluble carbodiimide (WSC) was purchased from Novabiochem and the silica gel from Silicycle. 1H and 13C NMR data was collected using a 300 MHz Varian apparatus.
5-(4-hydroxy-3-(methoxycarbonyl)phenylamino)-5-oxopentanoic acid 6a
To a suspension of the glutaric anhydride (1.053 g, 9.2 mmol) in 50mL of pyridine-dichloromethane (1:1) under nitrogen it was added the aniline 5a (4.630 g, 27.7 mmol) as powder. The mixture was stirred at r.t. for 16h under N2. After that time, the solvent was removed under reduce pressure and diluted with EtOAc. The organic phase was washed with 2N HCl, water and brine and it was dried over Na2SO4. The solvent was removed under reduce pressure and the resultant oil was treated with diethyl ether to give a light purple solid that it was washed with a small amount of cold ether (1.281 g, 52%).
1H-NMR (300 MHz, DMSO-d6) δ 1.70–1.85 (m, 2H, CH2), 2.15–2.35 (m, 4H, COCH2CH2CH2CO2H), 3.34 (bs, 1H, CONH), 6.89 (d, J = 8.8 Hz, 1H, aromatic), 7.63 (dd, J1 = 8.8 Hz J2 = 2.5 Hz, 1H, aromatic), 8.10 (d, J = 2.5, 1H, aromatic), 9.85 (s, 1H, PhOH), 11.01 (bs, 1H, CO2H); 13C-NMR (75 MHz, DMSO-d6) δ 21.1, 33.6, 35.9, 53.0, 53.4, 113.0, 117.9, 118.4, 120.3, 120.9, 127.8, 128.1, 131.9, 156.5, 171.2; MS(ESI) m/z: 268.1 [M+H] +; Rf (EtOAc) = 0.58.
5-(4-carboxybutanamido)-2-hydroxybenzoic acid 6b
To a suspension of glutaric anhydride (1.053 g, 9.2 mmol) in 50mL of pyridine-dichloromethane (1:1) under nitrogen it was added the aniline 5b (4.245 g, 27.7 mmol) as powder. The mixture was stirred at r.t. for 16h under N2. After that time, the solvent was removed under reduce pressure and diluted with EtOAc. The organic phase was washed with 2N HCl, water and brine and it was dried over Na2SO4. The solvent was removed under reduce pressure and the resultant oil was treated with diethyl ether to give a white solid that it was washed with a small amount of cold ether (1.165 g, 45%).
1H-NMR (300 MHz, DMSO-d6) δ 1.70–1.85 (m, 2H, CH2), 2.15–2.35 (m, 4H, COCH2CH2CH2CO2Me), 3.88 (s, 3H, CO2Me), 6.92 (d, J = 8.8 Hz, 1H, aromatic), 7.61 (dd, J1 = 8.8 Hz J2 = 2.5 Hz, 1H, aromatic), 8.13 (d, J = 2.5, 1H, aromatic), 10.24 (s, 1H, PhOH); 13C-NMR (75 MHz, DMSO-d6) δ 21.1, 33.6, 35.9, 113.0, 117.6, 118.0, 120.7, 121.4, 127.8, 128.3, 131.7, 157.6, 171.1, 172.4; MS(ESI) m/z: 282.1 [M+H] +; Rf (EtOAc) = 0.03.
Compound 1a
To a solution of the carboxylic acid 6a (0.200 g, 0.75 mmol) in a mixture of DCM:DMF (4:1) anhydrous (5 mL) in an ice bath, WSC (0.158 g, 0.82 mmol) was added already dissolved in the same previous solution (5 mL). After 30 min of agitation the 4-amino TEMPO (0.256 g, 1.5 mmol) was added and the reaction was allowed to reach room temperature and to continue for 16h. After this time the solution was diluted with DCM (20 mL) and washed with 0.1 N HCl (3 × 20 mL), H2O (3 × 20 mL) and brine. The organic phase was dried over Na2SO4 and filtered. The solvent was then removed under reduced pressure to afford 0.146 g of an orange solid (45%). HRMS (ESI-TOF) m/z: calcd for [M+H]+ 434.2291, found 434.2285; Anal. (C22H32N3O6) H, calc 7.42, found 7.22; C, calc 60.81, found 59.95.
Compound 1b
The same procedure as before was employed with the carboxylic acid 6b obtaining a yellow solid (20%). HRMS (ESI-TOF) m/z: calcd for [M+H] + 421.2207, found 421.2194; Anal. (C21H30N3O6) H, calc 7.19, found 7.09; C, calc 59.99, found 59.24.
Methyl 5-(5-formylfuran-2-yl)-2-hydroxybenzoate 7
5-bromo-2-hydroxybenzoate (0.344 g, 1.4 mmol), 5-formylfuran-2-ylboronic acid (0.25 g, 1.7 mmol), tetrabutylammonium chloride (0.405 g, 1.4 mmol), palladium acetate (0.006 g, 2 mol %), and potassium carbonate (0.489 g, 3.5 mmol) were added to a 25 mL round-bottom flask. Then ethanol (4 mL) and of deionized water (4 mL) were added in this sequence and the reaction was stirred vigorously for 3h. After this time the ethanol from the mixture was removed under reduced pressure and diluted with water (15 mL). The product was extracted with EtOAc (3 × 50 mL). The organic phase was then stirred over charcoal (~ 1 g) for 30 min and Na2SO4 was added. The solution was filtered and concentrated to give a yellow oil. The residue was purified by silica gel column chromatography eluting with hexanes/EtOAc (4:1) to give 0.216 g of an off white solid (62%).
1H-NMR (300 MHz, CDCl3) δ 4.09 (s, 3H, CO2Me), 6.76 (d, J = 3.7 Hz, 1H, CH furanyl), 7.07 (d, J = 8.8 Hz, 1H, aromatic), 7.33 (d, J = 3.7 Hz, 1H, CH furanyl), 7.89 (dd, J1 = 8.7 Hz J2 = 2.3 Hz, 1H, aromatic), 8.33 (d, J = 2.3, 1H, aromatic), 9.63 (s, 1H, PhOH), 11.02 (s, 1H, CHO); 13C-NMR (75 MHz, DMSO- d6) δ 53.2, 53.5, 108.4, 108.6, 115.1, 119.2, 119.5, 120.9, 126.9, 127.6, 132.5, 133.1, 161.3, 178.0, 178.4; MS(ESI) m/z: 247.1 [M+H] +; Rf (Hexanes/EtOAc (4:1)) = 0.27.
Compound 2a
Compound 7 (0.173 mg, 0.7 mmol) was dissolved in toluene (6 mL) with 5% of acetic acid. Then the 4-amino TEMPO (0.124 g, 0.7 mmol) was added and the reaction was placed at 65 °C under N2 for 16h. After this time the solvent was removed under reduced pressure until dryness. The residue was the dissolved in MeOH (10 mL) and NaBH3CN (0.050 g, 0.8 mmol) was added to the solution at 0 °C. After this another 3ml of 0.4 M HCl in MeOH were added and the reaction was allowed for 30 min at 0 °C. The reaction was warmed up to r.t. for 30 min and the solvent was removed under reduced pressure until almost dryness. The residue was diluted with water (20 mL) and 3M NaOH was added until pH 7. The product was extracted with chloroform (3 × 30 mL). The organic phase was dried over Na2SO4, filtered and the solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography eluting with a gradient from hexanes/EtOAc (4:1) to hexanes/EtOAc (1:4) to give 0.244 g of an orange oil (87%).
HRMS (ESI-TOF) m/z: calcd for [M+H] + 402.2149, found 402.2141; Anal. (C22H29N2O5) H, calc 7.28, found 7.21; C, calc 65.82, found 66.04; Rf (EtOAc/MeOH (4:1)) = 0.48.
Compound 2b
Compound 2a (0.1 g, 0.25 mmol) was dissolved in MeOH (3 mL) and cooled to 0°C. 2N NaOH (3 mL) was added dropwise during 10 min and the reaction was placed at r.t. for 16h. After this time MeOH was removed under reduced pressure and water (15 mL) was added. Extractions with EtOAc (2 × 15mL) were done and the pH of the water solution was acidified to 6. Then, the product was extracted with EtOAc (3 × 15 mL). The organic solution was dried over Na2SO4 and filtered. The solvent was removed under reduced pressure to give a yellow solid (0.051 g) in 52% yield. HRMS (ESI-TOF) m/z: calcd for [M+H] + 388.1993, found 388.2002; Anal. (C21H27N2O5) H, calc 7.02, found 7.35; C, calc 65.10, found 63.85; Rf (EtOAc/MeOH (4:1)) = 0.07.
(E)-methyl 2-hydroxy-5-(5-(3-methoxy-3-oxoprop-1-enyl)furan-2-yl)benzoate 8
A suspension of aldehyde 7 (0.125 g, 0.51 mmol), methyl diethyl phosphonoacetate (0.105 mL, 0.56 mmol) and LiOH (0.014 g, 0.56 mmol) in THF (10 mL) was stirred at r.t under Ar for 4h. After the usual work-up, purification by silica gel column chromatography with hexanes/EtOAc (6:1) as eluent was done to give 0.128 g of a pale yellow solid (84%).
1H-NMR (300 MHz, DMSO-d6) δ 3.72 (s, 3H, CO2Me), 3.93(s, 3H, CO2Me), 6.34 (d, J = 15.7 Hz, 1H, CH=CHCO2Me), 7.05–7.06 (m, 2H, furanyl), 7.08 (d, J = 8.7 Hz, 1H, aromatic), 7.47 (d, J = 15.7 Hz, 1H, CH=CHCO2Me), 7.97 (dd, J1 = 8.7 Hz J2 = 2.3 Hz, 1H, aromatic), 8.13 (d, J = 2.3 Hz, 1H, aromatic), 10.69 (bs, 1H, PhOH); 13C-NMR (75 MHz, DMSO-d6) δ 52.0, 52.3, 53.1, 53.5, 108.3, 108.7, 114.3, 114.6, 114.8, 118.9, 119.3, 119.7, 121.8, 125.9, 126.6, 131.6, 132.4, 150.2, 155.3, 160.5, 167.3, 169.2; MS(ESI) m/z: 303.1 [M+H] +; Rf (Hexanes/EtOAc (4:1)) = 0.49.
(E)-5-(5-(2-carboxyvinyl)furan-2-yl)-2-hydroxybenzoic acid 9
To suspension of compound 8 (0.1 g, 0.33 mmol) in MeOH (5 mL) at 0 °C was added dropwise 2N NaOH (5 mL). After the addition, the solution was warmed up to r.t. and then it was heated to 80 °C for 2h. After this time the reaction was cooled down and the methanol was removed under reduced pressure. The solution was diluted with water (20 mL) and extractions with EtOAc (2 × 15 mL)were done. The water solution was then acidified to pH 5 and the product was extracted with EtOAc (3 × 15 mL). The organic phase was dried over Na2SO4, filtered and the solvent was removed under reduced pressure affording 0.080 g of a yellow solid (88%).
1H-NMR (300 MHz, DMSO-d6) δ 6.27 (d, J = 15.7 Hz, 1H, CH=CHCO2H), 6.95–7.10 (m, 3H), 7.40 (d, J = 15.7 Hz, 1H, CH=CHCO2H), 7.97 (dd, J1 = 8.7 Hz, J2 = 2.3 Hz, 1H, aromatic), 8.16 (d, J = 2.3 Hz, 1H, aromatic); 13C-NMR (75 MHz, DMSO- d6) δ 108.1, 108.5, 114.3, 115.9, 116.2, 118.6, 119.0, 121.7, 126.0, 126.7, 131.1, 150.3, 155.1, 161.8, 168.1, 172.1; MS(ESI) m/z: 275.0 [M+H] +; Rf (EtOAc) = 0.1.
Compound 3
To a solution of the dicarboxylic acid 9 (0.070 g, 0.25 mmol) in a mixture of DCM:DMF (4:1) anhydrous (3 mL) in an ice bath, WSC (0.054 g, 0.28 mmol) was added already dissolved in the same previous solution (2 mL). After 30 min of agitation the 4-amino TEMPO (0.087 g, 0.51 mmol) was added and the reaction was allowed to reach room temperature and to continue for 16h. After this time the solution was diluted with DCM (20 mL) and washed with 0.1 N HCl (3 × 20 mL), H2O (3 × 20 mL) and brine. The organic phase was dried over Na2SO4 and filtered. The solvent was then removed under reduced pressure to afford 0.049 g of a yellow solid (46%). HRMS (ESI-TOF) m/z: calcd for [M+H] + 428.1942, found 428.1938; Anal. (C23H27N2O6) H, calc 6.37, found 6.56; C, calc 64.62, found 64.23.
(E)-methyl 3-(5-(4-nitrophenyl)furan-2-yl)acrylate 11a
A suspension of aldehyde 10a (0.5 g, 2.3 mmol), methyl diethyl phosphonoacetate (0.485 mL, 2.5 mmol) and LiOH (0.062 g, 2.5 mmol) in THF (10 mL) was stirred at r.t under Ar for 4h. After the usual work-up, purification by silica gel column chromatography with hexanes/EtOAc (4:1) as eluent was done to give 0.56 g of a yellow solid (89%).
1H-NMR (300 MHz, DMSO-d6) δ 3.75 (s, 3H, 3H, CH=CHCO2Me), 6.55 (d, J = 15.8 Hz, 1H, CH=CHCO2Me), 7.16 (d, J = 3.6 Hz, 1H, CH furanyl), 7.47 (d, J = 6.3 Hz, 1H, CH furanyl), 7.53 (d, J = 15.8 Hz, 1H, CH=CHCO2Me), 8.11 (d, J = 8.9 Hz, 1H, aromatic), 8.30 (d, J = 8.8 Hz, 1H, aromatic); 13C-NMR (75 MHz, DMSO- 52.2, d6) δ 52.5, 113.5, 116.7, 117.0, 119.2, 119.5, 124.8, 125.3, 125.4, 126.0, 131.2, 131.4, 135.6, 152.3, 167.1; MS(ESI) m/z: 274.1 [M+H] +; Rf (Hexanes/EtOAc (4:1)) =0.83.
(E)-methyl 3-(5-(3-nitrophenyl)furan-2-yl)acrylate 11b
The same procedure as before was employed with the aldehyde 10b obtaining a yellow solid (83%).
1H-NMR (300 MHz, DMSO-d6) δ 3.74 (s, 3H, CH=CHCO2Me), 6.54 (d, J = 15.8 Hz, 1H, CH=CHCO2Me), 7.14 (d, J = 3.5 Hz, 1H, CH furanyl), 7.45 (d, J = 3.6 Hz, 1H, CH=CHCO2Me), 7.52 (d, J = 15.8 Hz, 1H, CH furanyl), 7.76 (t, J = 8.0 Hz, 1H, aromatic), 8.19 (dd, J1= 8.1 Hz J2= 1.5 Hz, 1H, aromatic), 8.30 (dd, J1= 8.1 Hz J2= 1.6 Hz, 1H, aromatic), 8.60 (t, J = 1.6 Hz, 1H, aromatic); 13C-NMR (75 MHz, DMSO-d6) δ 52.1, 52.4, 111.8, 116.2, 119.0, 119.3, 123.2, 123.8, 130.6, 131.1, 131.2, 131.4, 131.6, 149.2, 151.5, 164.6, 167.2; MS(ESI) m/z: 274.2 [M+H] +; Rf (Hexanes/EtOAc (4:1)) =0.83.
(E)-3-(5-(4-nitrophenyl)furan-2-yl)acrylic acid 12a
To solution of compound 11a (0.4 g, 1.5 mmol) in MeOH (10 mL) at 0 °C was added dropwise 2N NaOH (10 mL). After the addition the solution was warmed up to r.t. and then it was heated to 80 °C for 2h. After this time the reaction was cooled down it was acidified to pH 6 after dilution with water (20 mL). The methanol was removed under reduced pressure and the product was extracted with EtOAc (3 × 20 mL). The organic phase was dried over Na2SO4, filtered and the solvent was removed under reduced pressure. Purification by silica gel column chromatography, with hexanes/EtOAc (4:1) as eluent, afforded 0.345 g of a pale orange solid (91%).
1H-NMR (300 MHz, DMSO-d6) δ 6.46 (d, J = 15.7 Hz, 1H, CH=CHCO2H), 7.13 (d, J = 3.6 Hz, 1H, CH furanyl), 7.44 (d, J = 15.6 Hz, 1H, CH=CHCO2H), 7.46 (d, J = 3.6 Hz, 1H, CH furanyl), 8.10 (d, J = 8.9 Hz, 1H, aromatic), 8.30 (d, J = 9.0 Hz, 1H, aromatic); 13C-NMR (75 MHz, DMSO-d6) δ 113.5, 118.3, 118.5, 118.8, 124.8, 125.2, 125.4, 125.5, 130.8, 131.7, 152.5, 168.0; MS(ESI) m/z: 260.1 [M+H] +; Rf (Hexanes/EtOAc (1:1)) =0.46.
(E)-3-(5-(3-nitrophenyl)furan-2-yl)acrylic acid 12b
The same procedure as before was employed with the compound 11b obtaining a yellow solid (93%).
1H-NMR (300 MHz, DMSO-d6) δ 6.45 (d, J = 15.8 Hz, 1H, CH=CHCO2H), 7.10 (d, J = 3.6 Hz, 1H, CH furanyl), 7.43 (d, J = 3.7 Hz, 1H, CH furanyl), 7.45 (d, J = 15.7 Hz, 1H, CH=CHCO2H), 7.76 (t, J = 8.0 Hz, 1H, Arom), 8.19 (dd, J1 = 1.3 Hz, J2 = 8.4 Hz, 1H, Arom), 8.30 (dd, J1 = 1.3 Hz, J2 = 8.3 Hz, 1H, Arom), 8.58 (t, J = 1.4 Hz, 1H, Arom); 13C-NMR (75 MHz, DMSO-d6) δ 111.8, 117.8, 118.1, 118.3, 118.8, 119.3, 123.1, 123.7, 130.5, 131.0, 131.2, 131.5, 151.7, 153.3, 168.0; MS(ESI) m/z: 260.1 [M+H] +; Rf (Hexanes/EtOAc (1:1)) =0.45.
Compound 4a
To a solution of the carboxylic acid 12a (0.133 g, 0.51 mmol) in a mixture of DCM:DMF (4:1) anhydrous (5 mL) in an ice bath, WSC (0.108 g, 0.56 mmol) was added already dissolved in the same previous solution (5 mL). After 30 min of agitation the 4-amino TEMPO (0.205 g, 1.02 mmol) was added and the reaction was allowed to reach room temperature and to continue for 16h. After this time the solution was diluted with DCM (20 mL) and washed with 0.1 N HCl (3 × 20 mL), H2O (3 × 20 mL) and brine. The organic phase was dried over Na2SO4 and filtered. The solvent was then removed under reduced pressure to afford 0.068 g of an orange solid. HRMS (ESI-TOF) m/z: calcd for [M+H] + 413.1945, found 413.1941; Anal. (C22H26N3O5) H, calc 6.35, found 6.61; C, calc 64.06, found 63.67.
Compound 4b
The same procedure as before was employed with the carboxylic acid 12b obtaining a yellow solid (27%). HRMS (ESI-TOF) m/z: calcd for [M+H] + 413.1945, found 413.1943; Anal. (C22H26N3O5) H, calc 6.35, found 6.41; N, calc 10.19, found 9.94.
Expression and purification of recombinant YopH N-and C-terminal domains
Soluble recombinant uniformly 15N-labeled YopH is expressed in E. coli (BL21-DE3) as GST-fusion in minimal-media (with 0.5g of 15NH4Cl per liter as sole source of nitrogen). The GST-YopH was purified from clarified lysates using glutathione-Sepharose beads (GE Healthcare) according to manufacturer’s recommendations. The eluted GST-YopH was dialyzed against 5L of dialysis buffer (30 mM TRIS, 150 mM NaCl) and a second time in dialysis buffer with 1mM DTT. In the case of the C-terminal domain of YopH, after the first dialysis the protein was subject to thrombin cleavage (Thrombin Cleancleave KIT, Sigma-Aldrich, St Louis, MO) for 16h at 4°C following manufacturer’s recommendations. In addition to the thrombin cleavage site between GST and YopH, a second cleavage site is found between the N- and the C-terminal domains (sequence 126GARGHV131). The three components (GST, YopH(1–128) and YopH(129–468)) were separated by a second glutathione-Sepharose purification (to eliminate GST). Pure YopH N- and C-terminal domains were further isolated by passing the elutes through a Sephacryl S-100 gel-filtration column pre-equilibrated with the dialysis buffer. The protein sequences of both domains were further confirmed by tryptic digestion followed by MALDI-TOF analysis.
Enzyme inhibition assay
The phosphatase-catalyzed hydrolysis of p-nitrophenyl phosphate (pNPP; Sigma-Aldrich, St Louis, MO) in the presence of compounds in DMSO (5% final concentration) was assayed at 30°C in 0.15 M Bis-Tris buffer, pH 6.0, having an ionic strength of 150 mM (adjusted with NaCl) and containing 1.0 mM dithiothreitol in a 100-μl 96-well plate format. YopH (expressed and purified as reported before) [8] was at 10 nM, PTP1B (BIOMOL International, L.P., Plymouth Meeting, PA) at 25 nM, HePTP (gift of Dr. Rebecca Page, Brown University) at 75 nM, TCPTP (BIOMOL International, L.P., Plymouth Meeting, PA) at 12 nM, CD45 (BIOMOL International, L.P., Plymouth Meeting, PA) at 5 nM, and VHR (expressed and purified as reported before) [18] at 50 nM. Compound concentrations were 0.0128, 0.064, 0.32, 1.6, 8, 40, 200, and 1000 μM. After preincubation of enzyme and compound for 10 min at room temperature, pNPP was added and the reaction mixture was incubated for 20 min at 30°C. The initial reaction rate at a fixed pNPP concentration, which was equal to the corresponding Km value of 1 mM for YopH, PTP1B, TCPTP, and CD45, 2.5 mM for HePTP, and 1.5 mM for VHR was determined using a ELx808 micro plate reader (Bio-Tek Instruments, Inc., Winooski, VT), measuring absorption of the cleavage product p-nitrophenol at 405 nm after quenching the reaction with 100 μl 1 M NaOH. Nonenzymatic hydrolysis of the substrate was corrected by measuring the control without enzyme. IC50 values were determined by plotting the relative activity versus inhibitor concentration using Prism® (GraphPad Software, San Diego, CA) and fitting to the equation Vi/V0 = IC50/(IC50 + [I]), where Vi was reaction velocity at inhibitor concentration [I], V0 is the reaction velocity without inhibitor and IC50 = Ki + Ki[S]/Km, where Ki is the dissociation constant for binding of inhibitor to enzyme, [S] is the substrate concentration and Km is the Michaelis-Menten constant.
NMR spectroscopy
NMR spectra were acquired on a 500 MHz or 600 MHz Bruker Avance spectrometer equipped with a Bruker TXI probe (500) or a TCI-Cryoprobe (600). All 1D 1H experiments were carried out with samples containing unlabeled GST-YopH at a concentration of 10 μM. 1D T1ρ experiments were measured with a spin-lock time of 100 ms, a recycle delay of 1.5 sec, and water suppression based on the Watergate sequence [19]. A library of 200 compounds (1 mM) was tested in presence of compound 3 (500 μM). Compounds were tested in mixtures of 10 compounds and hits further deconvoluted aided by the 1D NMR spectra of the individual compounds of the library (Maybridge). For the 2D [1H-15N]-HSQC experiments in Figure 4, samples of 280 μM of YopH-C were used to acquire the spectra in the absence and in presence of compound 3 (500 μM).
Acknowledgments
Financial support was obtained thanks to NIH grants AI058123 and AI055789.
Abbreviations
- PTP
phosphotyrosine phosphatase
- SAR
structure-activity relationship
- ILOE
inter-ligand Overhouser effect
- TEMPO
2,2,6,6-tetramethylpiperidine 1-oxyl
- WSC
water soluble carbodiimide
References
- 1.Bialy L, Waldmann H. Inhibitors of protein tyrosine phosphatases: next-generation drugs? Angew Chem Int Ed Engl. 2005;44:3814–3839. doi: 10.1002/anie.200461517. [DOI] [PubMed] [Google Scholar]
- 2.Dewang PM, Hsu NM, Peng SZ, Li WR. Protein tyrosine phosphatases and their inhibitors. Curr Med Chem. 2005;12:1–22. doi: 10.2174/0929867053363504. [DOI] [PubMed] [Google Scholar]
- 3.Tautz L, Pellecchia M, Mustelin T. Targeting the PTPome in human disease. Expert Opin Ther Targets. 2006;10:157–177. doi: 10.1517/14728222.10.1.157. [DOI] [PubMed] [Google Scholar]
- 4.Srinivasan R, Uttamchandani M, Yao SQ. Rapid assembly and in situ screening of bidentate inhibitors of protein tyrosine phosphatases. Org Lett. 2006;8:713–716. doi: 10.1021/ol052895w. [DOI] [PubMed] [Google Scholar]
- 5.Szczepankiewicz BG, Liu G, Hajduk PJ, Abad-Zapatero C, Pei Z, Xin Z, Lubben TH, Trevillyan JM, Stashko MA, Ballaron SJ, Liang H, Huang F, Hutchins CW, Fesik SW, Jirousek MR. Discovery of a potent, selective protein tyrosine phosphatase 1B inhibitor using a linked-fragment strategy. J Am Chem Soc. 2003;125:4087–4096. doi: 10.1021/ja0296733. [DOI] [PubMed] [Google Scholar]
- 6.Pellecchia M. Solution nuclear magnetic resonance spectroscopy techniques for probing intermolecular interactions. Chem Biol. 2005;12:961–971. doi: 10.1016/j.chembiol.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 7.Pellecchia M, Becattini B, Crowell KJ, Fattorusso R, Forino M, Fragai M, Jung D, Mustelin T, Tautz L. NMR-based techniques in the hit identification and optimisation processes. Expert Opin Ther Targets. 2004;8:597–611. doi: 10.1517/14728222.8.6.597. [DOI] [PubMed] [Google Scholar]
- 8.Tautz L, Bruckner S, Sareth S, Alonso A, Bogetz J, Bottini N, Pellecchia M, Mustelin T. Inhibition of Yersinia tyrosine phosphatase by furanyl salicylate compounds. J Biol Chem. 2005;280:9400–9408. doi: 10.1074/jbc.M413122200. [DOI] [PubMed] [Google Scholar]
- 9.Jahnke W. Spin labels as a tool to identify and characterize protein-ligand interactions by NMR spectroscopy. Chembiochem. 2002;3:167–173. doi: 10.1002/1439-7633(20020301)3:2/3<167::AID-CBIC167>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 10.Jahnke W, Floersheim P, Ostermeier C, Zhang X, Hemmig R, Hurth K, Uzunov DP. NMR reporter screening for the detection of high-affinity ligands. Angew Chem Int Ed Engl. 2002;41:3420–3423. doi: 10.1002/1521-3773(20020916)41:18<3420::AID-ANIE3420>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 11.Jahnke W, Blommers MJJ, Fernandez C, Zwingelstein C, Amstutz R. Strategies for the NMR-based identification and optimization of allosteric protein kinase inhibitors. Chembiochem. 2005;6:1607–1610. doi: 10.1002/cbic.200500100. [DOI] [PubMed] [Google Scholar]
- 12.Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Discovering high-affinity ligands for proteins: SAR by NMR. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 13.Becattini B, Sareth S, Zhai D, Crowell KJ, Leone M, Reed JC, Pellecchia M. Targeting apoptosis via chemical design: inhibition of bid-induced cell death by small organic molecules. Chem Biol. 2004;11:1107–1117. doi: 10.1016/j.chembiol.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 14.Becattini B, Pellecchia M. SAR by ILOEs: an NMR-based approach to reverse chemical genetics. Chem Eur J. 2006;12:2658–2662. doi: 10.1002/chem.200500636. [DOI] [PubMed] [Google Scholar]
- 15.Harris TK. Discovering new drug-targeting sites on flexible multidomain protein kinases: Combining segmental isotopic and site-directed spin labeling for nuclear magnetic resonance detection of interfacial clefts. Methods Mol Biol. 2006;316:199–225. doi: 10.1385/1-59259-964-8:199. [DOI] [PubMed] [Google Scholar]
- 16.Hajduk PJ, Olejniczak ET, Fesik SW. One-dimensional relaxation- and diffusion-edited NMR methods for screening compounds that bind to macromolecules. J Am Chem Soc. 1997;119:12257–12261. [Google Scholar]
- 17.Oestman A, Hellberg C, Boehmer FD. Protein-tyrosine phosphatases and cancer. Nat Rev Cancer. 2006;6:307–320. doi: 10.1038/nrc1837. [DOI] [PubMed] [Google Scholar]
- 18.Alonso A, Rahmouni S, Williams S, van Stipdonk M, Jaroszewski L, Godzik A, Abraham RT, Schoenberger SP, Mustelin T. Tyrosine phosphorylation of VHR phosphatase by ZAP-70. Nat Immunol. 2003;4:44–48. doi: 10.1038/ni856. [DOI] [PubMed] [Google Scholar]
- 19.Piotto M, Saudek V, Sklenar V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J Biomol NMR. 1992;2:661–665. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]