

Abstract
We have developed a coupled helicase–polymerase DNA unwinding assay and have used it to monitor the rate of double-stranded DNA unwinding catalyzed by the phage T4 DNA replication helicase (gp41). This procedure can be used to follow helicase activity in subpopulations in systems in which the unwinding-synthesis reaction is not synchronized on all the substrate-template molecules. We show that T4 replication helicase (gp41) and polymerase (gp43) can be assembled onto a loading site located near the end of a long double-stranded DNA template in the presence of a macromolecular crowding agent, and that this coupled “two-protein” system can carry out ATP-dependent strand displacement DNA synthesis at physiological rates (400 to 500 bp per sec) and with high processivity in the absence of other T4 DNA replication proteins. These results suggest that a direct helicase–polymerase interaction may be central to fast and processive double-stranded DNA replication, and lead us to reconsider the roles of the other replication proteins in processivity control.
Keywords: helicase–polymerase, DNA unwinding, ATPase, kinetics, macromolecular crowding
The bacteriophage T4 DNA replication (elongation) complex is composed of seven phage-coded proteins (1, 2). The DNA polymerase (gene 43 product, gp43) plays the central role in replication, in that it catalyzes the template-directed incorporation of nucleotide triphosphates (dNTPs) into the nascent DNA in both leading- and lagging-strand synthesis. Under physiological conditions, the polymerase together with the three T4 polymerase accessory proteins (gp44/62 and gp45) and the single-stranded (ss) DNA binding protein (gp32) interact to form a holoenzyme complex that is capable of rapid and processive DNA chain elongation on ssDNA templates. The addition of the T4 replication helicase (gp41) and primase (gp61) results in the formation of a seven-protein complex that can catalyze rapid, processive, and coupled leading- and lagging-strand DNA replication on double-stranded templates. The helicase and the primase work together to form a primosome subassembly within the replication complex, which is responsible for unwinding the double-stranded (ds) DNA template downstream of the replication fork and for producing and positioning pentameric oligoribonucleotides to prime the synthesis of Okazaki fragments on the lagging strand.
Determinations of the unwinding properties of replication helicases have generally used an assay in which a prebound oligodeoxyribonucleotide that is complementary to part of a tailed DNA template is released from the template on the addition of ATP (e.g., see refs. 3–5). Such analyses can reveal helicase directionality (depending on where on the template the complementary oligomer is located) and how many DNA oligomers have been completely removed from their complementary templates. However, these assays cannot be used to determine how fast the helicase moves through the dsDNA in carrying out the unwinding reaction. Furthermore the “product” of such assays is, of course, simply two separated DNA strands that will spontaneously rehybridize unless some means is found to “trap” the products without interfering with the reaction. Due to difficulties in achieving efficient and synchronized loading of the T4 DNA helicase (which has a fairly low affinity for DNA; see ref. 6), recently developed methods that are based on observing overall populations (7–9) have not been useful in studying the unwinding of dsDNA by the gp41 helicase.
In this study we present an assay that can monitor the unwinding of dsDNA by individual helicase molecules. The method, which we have called the “polymerase-spying assay,” is based on coupling the activity of the helicase with that of a following polymerase so that the polymerase “reports” the movement of the helicase through a dsDNA sequence by synthesizing a radioactively labeled DNA strand, which serves to define the template sequence exposed by the helicase. We show that this method can be used to measure the rate and processivity of DNA unwinding, even if only a small fraction of the available helicase substrates are used at any given time. Thus the assay is especially useful to monitor systems in which synchronized initiation of the helicase reaction is difficult to achieve. It is clear that the polymerase is an active partner in this helicase assay, and thus the reaction that is really being studied is the coupled activity of this two-protein complex. We show here that once properly assembled onto an appropriate DNA substrate-template, the two-protein T4 helicase–polymerase complex can carry out rapid and processive strand-displacement DNA synthesis at physiological rates.
EXPERIMENTAL PROCEDURES
Preparation of the Helicase–Polymerase Substrate-Template Construct.
The DNA constructs used as the helicase substrate and polymerase template in this study were prepared using a modified form of the procedure developed by Cha and Alberts (10). By optimizing the concentrations of DNA template and T4 DNA polymerase holoenzyme, we have obtained singly-nicked and completely double-stranded 7.25-kb M13 DNA circles with a short ssDNA tail (see Fig. 1) in sufficient quantities for physical biochemical study. Our most successful preparative conditions use 100 nM ssM13 mp18 DNA, 2 μM tailed primer, 500 nM gp43, 1 μM gp45 (trimers), and 270 nM gp44/62 complexes in synthesis buffer [25 mM Hepes/60 mM KOAc/6 mM Mg(OAc)2/1 mM DTT/0.1 mM EDTA, pH 7.6], as well as 1 mM each of rATP and rGTP and 250 μM each of dATP, dGTP, dCTP, and dTTP.
Figure 1.
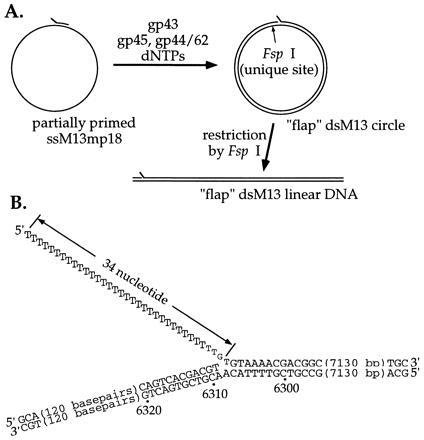
The DNA substrate-template construct used for the helicase assay. (A) Outline of the preparative procedure. The flap-containing dsM13 mp18 circle was formed by primer extension under conditions that exclude strand-displacement synthesis, starting with a tailed primer (hybridized to ssM13 mp18 DNA at positions 6288 to 6308) that carries a short single-stranded overhanging DNA sequence [oligo(dT)]. The product was then digested with FspI restriction nuclease, which results in blunt-end cleavage of the DNA at position 6427, to give the 7.25-kb dsDNA molecule carrying a helicase and polymerase assembly site located 120 bp from the end of the fragment. (B) Schematic drawing of the DNA helicase–polymerase substrate-template construct showing details of the sequence at the helicase and polymerase assembly site. Positions are labeled according to the numbering of the M13 mp18 DNA sequence.
Preparative reactions (in 400 μl aliquots) were initiated by annealing the primer to the ssM13 DNA by heating to 90°C for 2 min in synthesis buffer (without Mg2+), followed by slow cooling to 37°C. The above concentrations of nucleotides and enzymes were added, and dsDNA synthesis was initiated by making the solution 6 mM in Mg(OAc)2. After 60 min of synthesis at 37°C, the resulting flap-containing circular dsDNA was purified by electrophoresis through 1% low-melting agarose gels and cleaved by FspI restriction endonuclease, producing the final (flap-containing) linear helicase–polymerase substrate-template (see Fig. 1). This material was purified by phenol-chloroform extraction and ethanol precipitation and redissolved in TE buffer (10 mM Tris·Cl/1 mM EDTA, pH 7.8).
Preparation of the T4 DNA Replication Helicase and Polymerase.
T4 helicase (gp41) and DNA polymerase (gp43) were purified as described (11, 12). Protein concentrations were determined by UV absorbance at 280 nm, using calculated (13) extinction coefficients (ɛM,280) of 7.6 × 104 M−1·cm−1 for the helicase and 1.3 × 105 M−1·cm−1 for the polymerase.
The Coupled Helicase–Polymerase Assay.
Coupled dsDNA unwinding and template-dependent primer extension assays were performed at 37°C in synthesis buffer containing 7.5% polyethylene glycol of average molecular weight 12,000 Da (PEG 12000), which served as a macromolecular crowding agent (14). Typically, reaction solutions contained 10 nM linear (flap-containing) nicked DNA molecules, 10 nM polymerase, 50 nM gp41 helicase hexamers, 75 μM dATP, 150 μM each of dGTP, dCTP, and dTTP, 1 mM rATP, and a trace amount (<0.1 μM) of [α-32P]dATP. To avoid activation of the 3′ → 5′ exonuclease activity of the polymerase, which would have degraded the DNA substrate-template in advance, samples were prepared by mixing the two enzymes and the DNA template in synthesis buffer (containing 7.5% PEG) without Mg2+. The reaction was then started by adding a premixed solution containing all of the required nucleotide triphosphates and Mg(OAc)2, also in synthesis buffer. Aliquots of the reaction were quenched at various times by mixing with equal volumes of 500 mM EDTA, passed through BioSpin-6 columns (Bio-Rad) to remove unincorporated nucleotides, and analyzed by 1% alkaline agarose gel electrophoresis. The radioactively-labeled products of this strand-displacement helicase–polymerase catalyzed reaction were visualized and quantitated by radiometric scanning using the AMBIS (AMBIS, San Diego) and (in some cases) the PhosphorImager (Molecular Dynamics) systems.
RESULTS
Characterization of the Helicase–Polymerase Substrate-Template Construct.
A DNA construct to monitor dsDNA unwinding by the T4 replication helicase–polymerase assay must include several structural components. These are: (i) a small fork with a 20–32 nucleotide residue single-stranded 5′-overhanging sequence to serve as a loading site for the helicase (3, 4, 15); (ii) a primed template so that the polymerase can be loaded “behind” the helicase and commence primer extension synthesis as soon as dsDNA unwinding by the helicase begins (replication polymerases are generally incapable of de novo DNA synthesis); and (iii) a long dsDNA template extending downstream of the loading site at the initial fork, so that the unwinding process (which is typically several hundred bp per sec under physiological conditions at the replication fork) will last long enough to be followed kinetically by (in this case) manual methods.
The long flap-containing linear dsM13 mp18 DNA construct, prepared as shown in Fig. 1, meets all of these criteria and, in addition, carries a “built-in” primer strand resulting from the linearization process (see Fig. 1A). This feature assures that the construct has a 1:1 primer/template ratio and avoids the necessity of annealing a primer onto the construct, which can be difficult. Using optimized reaction conditions (see Experimental Procedures), we have prepared the flap-containing circular dsM13 intermediate and subsequently the linear flap-containing helicase–polymerase substrate-template in near milligram quantities. Analysis using agarose gel electrophoresis shows that the yields in both steps in our preparation were very high (>80%) (not shown). The purified helicase–polymerase substrate-template runs as a single discrete band in agarose gels.
Characteristics of the Helicase–Polymerase Reaction.
The reaction steps involved in the helicase–polymerase assay are summarized in Fig. 2A. The polymerase and helicase are preincubated with DNA template-substrate in the absence of nucleotides and Mg2+ (see Experimental Procedures), so that the polymerase associates with the DNA and reaches binding equilibrium (Fig. 2A, step 1), while the helicase remains inactive until the reaction is started by the addition of nucleoside triphosphates and Mg2+ (Fig. 2A, step 2).
Figure 2.
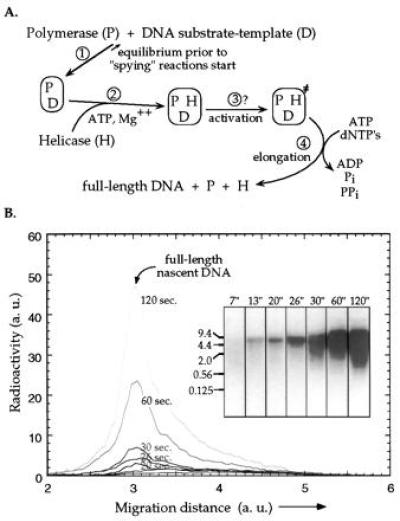
Strand-displacement DNA synthesis on the long dsDNA helicase–polymerase substrate-template construct catalyzed by gp41 and gp43. (A) Reaction pathway for the coupled helicase–polymerase assay. The binding of DNA polymerase to the DNA construct (step 1) is an equilibrium process in which association is greatly favored. The helicase and polymerase reactions are then triggered by the loading of the ATP-activated helicase (step 2). The fully assembled ternary complex unwinds the dsDNA template and synthesizes a new DNA strand by elongating the existing primer. An activation step (step 3) may or may not be involved after the assembly of the ternary complex and before the two-protein replication system enters the elongation phase (see Discussion). The lag-time in full-length DNA production corresponds to the total time required for the activation and elongation reactions (steps 3 and 4). (B) Analysis of the products of the coupled assay by DNA strand-separating alkaline agarose gel electrophoresis. Reactions were performed in the presence of 7.5% PEG and a trace amount of [α-32P]ATP. The resulting radioactively-labeled DNA strands were analyzed by alkaline agarose gel (1%) electrophoresis (Inset) and detected using the AMBIS radioactivity scanning system. The numbers at the top of the lanes denote reaction time (in seconds) while those at the left side of the gel indicate the positions to which various DNA fragments have migrated. The scans represent the distribution of radioactivity along the lanes of the gel. Both the amount of radioactivity and the migration distance are in arbitrary units. Note that even though the images in the gel show significant broadening of the DNA bands at longer times, the scans (which have a much higher dynamic range) indicate little change in the relative distributions of fragments within the lanes.
It is known that the polymerase binds primed DNA templates stably with a dissociation constant in the nanomolar range (16). [The Kd for the binding of the gp43 polymerase to a miniature DNA fork construct has been measured to be ≈20 nM in our laboratory (G. J. Latham, unpublished data).] Thus, a significant amount of polymerase-DNA complex was present (together with the free components) in equilibrium at the polymerase and DNA concentrations used in our experiments. To better control the experiments, we wished to start the reaction with polymerase bound to all of the DNA template-substrate molecules. To this end, we have used macromolecular crowding conditions [7.5% (vol/vol) PEG 12,000] to drive the reaction between the polymerase and DNA primer-template to completion, as demonstrated by single-round primer extension reactions using a 30/50-mer primer-template DNA construct (data not shown). Therefore, under these conditions all of the DNA template-substrate molecules are initially bound with gp43, and the strand-displacement DNA synthesis reaction begins when an active helicase hexamer [formed by interacting gp41 with ATP (11)] is loaded onto a polymerase-bound DNA template-substrate molecule.
If the DNA unwinding and synthesis reactions proceed in synchrony in the assay (i.e., reactions are initiated on all template-substrate constructs simultaneously and then elongated in phase), we would expect to see a homogeneous population of nascent DNA strands growing in length together, with all becoming full-length when the synthesis reaction reaches the end of the template. However, our results show clearly that this is not the case.
A typical experiment that illustrates the progress of the assay is shown in Fig. 2B. As the gel shows, and as is more clearly seen in the radiometric scans of the gel lanes, only small amounts of short DNA products are made in the early phases of the reaction. These short products then grow rapidly and become (mostly) full-length. This initial phase of the reaction is followed by a much longer phase in which full-length strand-displacement DNA synthesis products accumulate; Fig. 2B shows that this accumulation of full-length product is characterized by an initial lag of 10–20 sec and then the amount increases approximately linearly with time. This finding suggests that, despite the use of macromolecular crowding conditions, the helicase “loading” reaction (which represents the initiation event in the coupled helicase–polymerase assay; Fig. 2A, step 2) is rate-limiting. As a consequence, only a very small fraction of the DNA template-substrate constructs are being unwound and extended at any given time, i.e., the reaction is very asynchronous. However, after successful loading of the helicase, the rate of conversion of each substrate molecule to full-length DNA (Fig. 2A, steps 3 and 4) is very rapid.
Because the helicase is present in excess and the efficiency of DNA template-substrate utilization by the helicase in the assay is very low, the concentration of the free helicase remains approximately constant during the course of the reaction. As a result, the assembly of helicase onto the polymerase-bound DNA constructs (to form active complexes that can initiate strand-displacement DNA synthesis) should follow first-order reaction kinetics with respect to the concentration of the DNA template-substrate construct. The rate of full-length DNA formation should then follow the same pseudo-first-order kinetics as the assembly reaction, but be delayed by the time interval (tsyn) that is required for the assembled helicase–polymerase substrate-template complex to be converted into full-length synthesis product. This delay (tsyn) is equal to the lag time observed in the formation of full-length DNA after the helicase reaction is initiated (tlag), which corresponds to the time required for the initially assembled complexes to elongate fully. Thus, the kinetics of helicase loading and of full-length DNA production can be described by Eqs. 1 and 2, respectively:
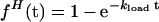 |
1 |
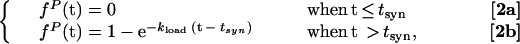 |
where fH(t) is the fraction of the DNA template-substrate construct that has been activated by helicase binding at time t, fP(t) is the fraction of full-length product formed at time t, and kload is the rate constant for the helicase loading reaction.
The Helicase and Polymerase Reactions Are Coupled and Highly Processive.
As shown in Fig. 2, this two-protein system can readily perform strand-displacement leading-strand DNA synthesis under our assay conditions. Control experiments using gp43 polymerase alone on this construct and under these conditions yielded no detectable primer-extension product (data not shown). Thus, the appearance of full-length DNA product within a short time (20 sec) in the assay indicates that the helicase and the polymerase must be tightly coupled, i.e., the polymerase and helicase remain engaged throughout the reaction so that primer-extension is completed before reanealing of the double-stranded template can occur. Since helicase loading is very slow, the early stage of the helicase–polymerase reaction proceeds as a single-hit process, with the majority of the DNA substrate-template molecules remaining unhit. Therefore, the initial efficient production of full-length DNA within only a small fraction of the total template-substrate construct pool indicates that the coupled reaction is also highly processive.
Rate of the Helicase–Polymerase Catalyzed Strand-Displacement DNA Synthesis Reaction.
Based on the above considerations, we know that the initiation of primer-extension DNA synthesis as a consequence of helicase loading follows pseudo-first-order (in DNA construct concentration) reaction kinetics and that the production of full-length DNA must follow the same kinetics offset by a lag corresponding to tsyn.
Both the rate constant for the helicase loading reaction (kload) and the conversion time for full-length product (tsyn) can be determined by analyzing data such as that presented in Fig. 2. In Fig. 3, the amounts of full-length DNA produced at various times in the assay are fitted to a first-order reaction with a lag, as described in Eq. 2; the hypothetical curve representing the helicase loading process (Eq. 1) is shown as the dashed curve in Fig. 3. From these results, and additional experiments of the same kind carried out at varying substrate concentrations, the conversion time for full-length DNA production (tsyn) can be shown to be 16 ± 2 sec, while the first-order rate constant for the helicase loading reaction (kload) is measured as 0.007 ± 0.002 sec−1. The ≈16 sec tsyn measured for the formation of full-length product is also consistent with the results of many individual assays in which the initial lag time of full-length product formation (tlag) is measured by stopping the polymerase–helicase reaction after 10–30 sec of DNA unwinding and synthesis, followed by gel analysis to determine whether full-length products exist. In addition, results from many experiments performed over a hundred-fold range of DNA template-substrate concentration have also confirmed that the reaction does indeed follow first-order kinetics in DNA concentration, since these experiments all yielded the same value for kload. We note that the rate constant for the helicase loading reaction (kload) and the lag-time for synthesis (tsyn) characterize two different and distinct processes in this multistep reaction (see Fig. 2A).
Figure 3.
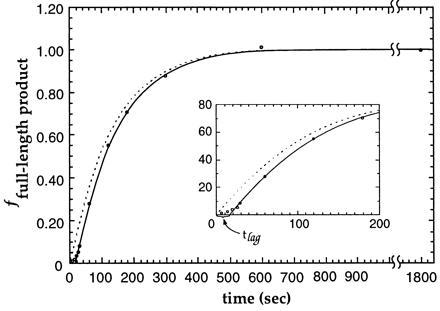
Quantitative analysis of the full-length DNA product of the coupled assay. The amount of radioactivity that corresponds to the full-length DNA product was plotted as a function of reaction time. Data were fitted to a first-order reaction with a lag for product formation using Eq. 2. The longer time data plotted here are not shown in Fig. 2. All data points were used in the fitting, while those corresponding to 5–95% product formation range (more accurately reflecting the reaction kinetics) were given more weight in fitting. The dotted curve represents the kinetics of the helicase loading reaction, which, of course, is not directly observed. The inset is an expanded view of the kinetics during the initial 200 sec of the reaction.
If we divide the length of the helicase–polymerase template (7130 bp) by the observed tsyn, we see that the rate of the coupled helicase–polymerase elongation reaction is ≈450 bp per sec. Since the conversion time for the formation of full-length product (tsyn) corresponds to the total time required for the assembled helicase–polymerase–DNA complex to be activated (tact) and to complete the coupled DNA unwinding-primer extension reaction (telong), the above calculation assumes that the activation time (Fig. 2A, step 3) is negligible compared with the time required for elongation of the DNA primer to the end of the ≈7 kb template. If the activation time (tact) were longer, the elongation time (telong) would be shorter. Thus, the estimated helicase rate of 450 bp per sec represents a lower limit for the elongation rate for the coupled helicase–polymerase reaction. The fact that a polymerase elongation rate of ≈450 nucleotide residues per sec is close to that measured (per polymerase) for T4 DNA replication in vivo is consistent with our assumption that tact is likely to be small compared with telong for this two-protein system under the assay conditions used.
DISCUSSION
Processive and Efficient Leading-Strand DNA Replication Is an Intrinsic Property of the Coupled T4 Helicase–Polymerase System.
Our results have shown that the T4 DNA replication helicase and polymerase can carry out coupled dsDNA template unwinding and template-dependent DNA synthesis reactions with high rates and processivity. We note that this rapid and processive leading-strand replication process catalyzed by these two enzymes must reflect intrinsic properties of the T4 replication helicase–polymerase complex, and not the presence of polyethylene glycol as a macromolecular crowding agent. This follows because macromolecular crowding serves primarily to increase assembly rates, and generally perturbs neither the rate of a putative intracomplex activation step nor the dissociation rate of the complex (17).
The Coupled Helicase–Polymerase Assay Can Monitor Helicase-Catalyzed Unwinding of dsDNA in Replication Systems for which Helicase Loading Is Inefficient.
An additional advantage of the coupled assay is its ability to follow the unsynchronized unwinding of dsDNA. This method selectively detects only newly synthesized DNA, and thus can monitor the rate of DNA unwinding either by following the extension rate of the first DNA fraction to be labeled or (as illustrated in Fig. 3) by determining the time required to complete the primer-extension synthesis (tsyn) of a known length of DNA.
In such a system, the fraction of DNA template-substrate construct [fUt(t)] that is actively utilized at any time (t) can be calculated as:
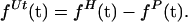 |
3 |
Thus, we can calculate that the first-order rate constant of 0.007 sec−1 measured for the helicase loading reaction in this system corresponds to a fractional helicase occupancy of ≈0.1 of the total DNA template-substrate molecules present during the initial 16 sec of the reaction, and that the fractional occupancy decreases during the course of the reaction. This assay method should, in principle, also be useable for replication systems with much lower efficiencies of helicase loading, as long as the synthesis time (tsyn) or the initial lag time (tlag) for full-length product formation can be measured. Thus, this approach should be useful for monitoring unwinding reactions driven by other replication helicases that may also be “difficult to load.”
Helicase and Polymerase Are Directly Coupled in Leading-Strand DNA Replication.
The observed coupling between the DNA template unwinding process catalyzed by the T4 helicase, and the template-dependent primer extension reaction carried out by the T4 polymerase, suggests that there must be a direct and continuing molecular interaction between these enzymes throughout the replication process. Previous studies of the T4 system have suggested the presence of an interaction between the gp41 helicase and either the polymerase or the accessory proteins, or both (18–21), and have implicated a 20 amino acid residue sequence at the C terminus of gp41 as being critical in the interaction (19). It has also recently been shown that the helicase and polymerase of the Escherichia coli DNA replication complex are also coupled; in this system, the coupling is mediated by the τ subunit of polmerase III (22).
However, the interactions observed in these earlier studies were determined in the presence of other replication proteins. Here we show that effective coupling can occur in the much simpler two-protein system, indicating direct helicase–polymerase interactions. Furthermore, this two-protein interaction is geometrically defined, since we know that the gp43 polymerase is located at the primer-template junction on the leading strand, while the gp41 helicase (with a 5′ → 3′ directionality) assembles on the lagging strand (and interacts with both strands; see ref. 4) of the replication fork. Thus, our findings here are consistent with earlier results and also demonstrate that the helicase–polymerase interaction is at least partially direct, regardless of any additional interactions that may exist between the helicase and either the lagging-strand polymerase or the accessory proteins (associated with either polymerase) within the full replication complex.
Coupling of Helicase and Polymerase in Other DNA Replication Systems.
Most replication helicases function inefficiently (as well as nonprocessively and slowly) in the absence of other components of the replication system. This may reflect the inability of the isolated helicases to load efficiently and correctly onto the replication fork models that have been used. Evidence that the helicases can function efficiently in the right context is manifested by the fact that various “complete” replication elongation systems can replicate both leading and lagging-strand DNA at physiological rates and processivities. Clearly this process requires a fully functional helicase because strand-displacement DNA synthesis in the absence of a helicase—even under favorable conditions—is orders of magnitude slower and less processive (e.g. see ref. 22).
Recently, several elegant experiments have been performed to ask how the helicase component might be integrated into the full replication complex. Thus, the experiments of Kim et al. (22) have shown that the τ subunit of polymerase III is central in coupling the dnaB helicase into the replication complex of E. coli. In addition, Schrock and Alberts (21) have shown for the complete T4 replication complex that gp41 is sufficiently stabilized by the rest of the T4 system to permit unwinding of the entire T4 genome in a fully processive manner. Though these experiments were carried out with complete replication complexes, they demonstrate conclusively that helicase coupling exists and is required in all complete DNA replication systems. Schrock and Alberts (21) also speculate that the leading- and lagging-strand polymerases in the T4 complex must somehow be complexed and coupled by the gp41 helicase.
The Polymerase May Not be a Passive “Reporter” of Helicase Activity in Coupled Helicase–Polymerase Assays.
The fact that the helicase and polymerase reactions are coupled in our assay also raises the question of whether the polymerase serves as a passive reporter of helicase unwinding or whether it participates actively in the unwinding process. On one hand, the ≈450 bp per sec per polymerase rate corresponds both to the in vivo replication rate for T4 and to the fastest rate that has been measured for the full replication system in vitro. Thus, one could argue that the helicase alone is capable of unwinding dsDNA still more rapidly, and that the apparent upper limit observed in replication experiments is set by other components of the complex. On the other hand, it is also possible that the helicase alone cannot unwind dsDNA at such a high rate, but that it is helped to do so by the coupled polymerase. An argument can be made that the helicase unwinding rate observed in the coupled assay represents the intrinsic rate of the helicase itself by noting that Liu and Alberts (23), in a very different kind of experiment, have previously estimated that gp41 alone can translocate along a ssDNA template (i.e., with no unwinding involved) at about 500 residues per sec.
From an energetics point of view, the optimal ATPase activity of gp41 is ≈20 ATP per sec per gp41 monomer (6), corresponding to ≈120 ATP per sec per active helicase hexamer. The unwinding of 450 bp per sec of dsDNA per helicase at an ATP hydrolysis rate of 120 sec−1 suggests that nearly 4 bp are unwound by gp41 for every ATP molecule hydrolyzed. This suggests either that the gp41 helicase must be very efficient in converting the free energy of ATP hydrolysis to the mechanical energy required to separate the base pairs (much more efficient than any ATP-driven helicase characterized to date, e.g., the E. coli Rep helicase and the RecBCD helicase; see ref. 24 and references therein) or that it may be assisted by the coupled (and homologous) polymerase moving along behind. The elongation reaction catalyzed by the polymerase is an energetically favored process,† and thus the polymerase could assist the helicase if the favorable free energy change associated with leading-strand DNA synthesis on a single-stranded template is somehow transferred to the unwinding reaction. While it remains an open question for the T4 helicase and polymerase, the existence of such a polymerase “pushing” effect on a homologous helicase is consistent with the results of a recent experiment with the E. coli polymerase III-dnaB helicase system (22), in which uncoupling the system (by omitting the τ subunit of polymerase III) results in a slowdown of the replication fork movement.
A Model for the T4 DNA Replication (Elongation) Complex.
The direct interactions between the gp41 helicase and the gp43 DNA polymerase revealed in this study may be centrally involved in the structural and functional integration of the complete DNA replication complex. A schematic illustration of the functional replication complex as we presently understand it is shown in Fig. 4. The complex contains three subassemblies (two holoenzymes and one primosome) that must work together (and presumably remain associated) to achieve coordinated DNA replication on both leading- and lagging-strand templates. Each of the two holoenzymes shown in Fig. 4 consists of a single gp43 polymerase subunit and a trimeric gp45 processivity ring, whereas the primosome consists of a hexameric gp41 helicase and a single gp61 primase subunit (11, 26). In addition to the five proteins shown explicitly in Fig. 4, the gp44/62 accessory proteins complex must also associate (at least transiently) with the polymerase holoenzymes to drive ATP-dependent loading (and reloading, after Okazaki fragment formation in lagging-strand synthesis) of the sliding processivity clamps onto the polymerase and the DNA.
Figure 4.
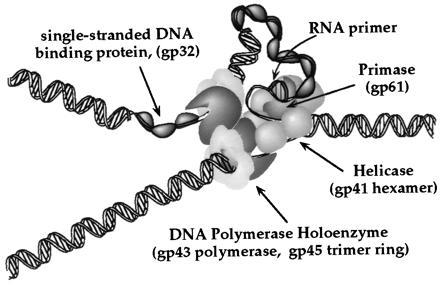
A functional model of the phage T4 DNA replication (elongation) complex (see text).
The primosome subassembly and the leading-strand polymerase holoenzyme may be coupled through the same set of helicase–polymerase interactions that are responsible for the coupling of these enzymes in our helicase assay. A direct interaction between the polymerase and the helicase may not only increase the processivity and the rate of the coupled reaction, but may also serve to insure that the primosome remains in contact with the polymerase holoenzymes during editing and repair or when the replication complex is slowed by other “traffic” along the template. The molecular nature of these coupling reactions remains to be elucidated. It has been suggested by others that functional coupling between the leading- and lagging-strand syntheses may require the two holoenzyme subassemblies to communicate with one another, either by allosteric interactions or perhaps by a “geared spiraling” interaction (e.g., see refs. 27 and 28). Such ideas make it tantalizing to consider that a coupled (“geared”) movement of the helicase (a ring-shaped hexamer with a probable D3 or C3 symmetry) in the primosome with either the processivity clamp (a ring-shaped gp45 trimer with a real C3 and a pseudo 6-fold symmetry) or the polymerase in the lagging-strand holoenzyme might also be involved as part of a coordinating mechanical linkage to allow coordinated movement of all three subassemblies (i.e., the entire replication fork).
The Role of the Other Subunits in Stability and Processivity Control.
In this paper, we have shown that rapid and processive strand displacement leading-strand DNA synthesis can be achieved with a properly loaded and activated two-protein helicase–polymerase system. Yet it is also clear that under physiological salt conditions even minimal processive synthesis by polymerase alone on a single-stranded template requires participation of the gp44/62 and gp45 accessory proteins (or their equivalents in other replication systems) to form and to load the sliding processivity clamp and thus to tether the polymerase (and the nascent DNA strand) onto the template (28–31). More recently it has also been shown (26) that an essential role of the gp61 primase subunit is to equivalently tether and stabilize the hexameric gp41 helicase onto the DNA to form a primosome complex that is sufficiently stable to complete the replication of an entire T4 genome.
Our present results lead us to consider whether these tethering and stabilizing activities of the other T4 proteins may be less central in DNA replication at the replication fork than is currently assumed. We suggest that perhaps the most important role of these proteins might be in permitting facile control of the processivity of the DNA replication complex and its constituent subassemblies by other interacting DNA regulatory systems involved in transcription, repair, and recombination, which share the same DNA templates in carrying out their roles in the expression and maintenance of the genome.
Acknowledgments
We are grateful to Drs. W. H. Konigsberg and N. G. Nossal for providing the pTL43W and pDH518 clones, and to Ms. Hongwei Jing of our laboratory for her help with preparing the gp41 helicase. We also thank Drs. B. M. Alberts, T. M. Lohman, and N. G. Nossal for useful advice and discussions during the course of this work. This research was supported by U. S. Public Health Service Research Grants GM-15792 and GM-29158 (to P.H.v.H.), by Damon Runyon–Walter Winchell Cancer Research Fellowship DRG-1152 (to F.D.), and by a grant to the Institute of Molecular Biology at the University of Oregon from the Lucille P. Markey Charitable Trust. P.H.v.H. is an American Cancer Society Research Professor of Chemistry.
Footnotes
Abbreviations: ss, single-stranded; ds, double-stranded.
The single-step nucleotide addition reaction catalyzed by various polymerases (reviewed in ref. 25) is characterized by a forward equilibrium constant of approximately 100, corresponding to a favorable standard free energy change (ΔGo) of ≈−2.7 kcal/mol and a significantly larger (more favorable) ΔG value under physiological reactant and product concentrations. Some of this free energy might be available to help “drive” the coupled helicase–polymerase reaction.
References
- 1.Alberts B M. Philos Trans R Soc London Ser B. 1987;317:395–420. doi: 10.1098/rstb.1987.0068. [DOI] [PubMed] [Google Scholar]
- 2.Nossal N G. In: Molecular Biology of Bacteriophage T4. Karam J D, editor. Washington, DC: Am. Soc. Microbiol.; 1994. pp. 43–53. [Google Scholar]
- 3.Venkatesan M, Silver L L, Nossal N G. J Biol Chem. 1982;257:12426–12434. [PubMed] [Google Scholar]
- 4.Richardson R W, Nossal N G. J Biol Chem. 1989;264:4725–4731. [PubMed] [Google Scholar]
- 5.Raney K D, Carver T E, Benkovic S J. J Biol Chem. 1996;271:14074–14081. doi: 10.1074/jbc.271.24.14074. [DOI] [PubMed] [Google Scholar]
- 6.Young M C, Schultz D E, Ring D, von Hippel P H. J Mol Biol. 1994;235:1447–1458. doi: 10.1006/jmbi.1994.1100. [DOI] [PubMed] [Google Scholar]
- 7.Eggleston A K, Rahim N A, Kowalczykowski S C. Nucleic Acids Res. 1996;24:1179–1186. doi: 10.1093/nar/24.7.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raney K D, Sowers L C, Millar D P, Benkovic S J. Proc Natl Acad Sci USA. 1994;91:6644–6648. doi: 10.1073/pnas.91.14.6644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roman L J, Eggleston A K, Kowalczykowski S C. J Biol Chem. 1992;267:4207–4214. [PubMed] [Google Scholar]
- 10.Cha T A, Alberts B M. Cancer Cells. 1988;6:1–10. [Google Scholar]
- 11.Dong F, Gogol E P, von Hippel P H. J Biol Chem. 1995;270:7462–7473. doi: 10.1074/jbc.270.13.7462. [DOI] [PubMed] [Google Scholar]
- 12.Reddy M K, Weitzel S E, von Hippel P H. J Biol Chem. 1992;267:14157–14166. [PubMed] [Google Scholar]
- 13.Pace C N, Vajdos F, Fee L, Grimsley G, Gray T. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jarvis T C, Ring D M, Daube S S, von Hippel P H. J Biol Chem. 1990;265:15160–15167. [PubMed] [Google Scholar]
- 15.Hinton D M, Richardson R W, Nossal N G. In: DNA Replication and Recombination: UCLA Symposium on Molecular Cellular Biology. McMacken R, Kelly T J, editors. Vol. 47. New York: Liss; 1987. pp. 173–182. [Google Scholar]
- 16.Capson T L, Peliska J A, Kaboord B F, Frey M W, Lively C, Dahlberg M, Benkovic S J. Biochemistry. 1992;31:10984–10994. doi: 10.1021/bi00160a007. [DOI] [PubMed] [Google Scholar]
- 17.Minton A P. Mol Cell Biochem. 1983;55:119–140. doi: 10.1007/BF00673707. [DOI] [PubMed] [Google Scholar]
- 18.Cha T A, Alberts B M. J Biol Chem. 1989;264:12220–12225. [PubMed] [Google Scholar]
- 19.Richardson R W, Nossal N G. J Biol Chem. 1989;264:4732–4739. [PubMed] [Google Scholar]
- 20.Spacciapoli P, Nossal N G. J Biol Chem. 1994;269:447–455. [PubMed] [Google Scholar]
- 21.Schrock R D, Alberts B. J Biol Chem. 1996;271:16678–16682. doi: 10.1074/jbc.271.28.16678. [DOI] [PubMed] [Google Scholar]
- 22.Kim S, Dallmann H G, McHenry C S, Marians K J. Cell. 1996;84:643–650. doi: 10.1016/s0092-8674(00)81039-9. [DOI] [PubMed] [Google Scholar]
- 23.Liu C C, Alberts B M. J Biol Chem. 1981;256:2813–2820. [PubMed] [Google Scholar]
- 24.Lohman T M, Bjornson K P. Annu Rev Biochem. 1996;65:169–214. doi: 10.1146/annurev.bi.65.070196.001125. [DOI] [PubMed] [Google Scholar]
- 25.Erie D A, Yager T D, von Hippel P H. Annu Rev Biophys Biomol Struct. 1992;21:379–415. doi: 10.1146/annurev.bb.21.060192.002115. [DOI] [PubMed] [Google Scholar]
- 26.Dong F, von Hippel P H. J Biol Chem. 1996;271:19625–19631. doi: 10.1074/jbc.271.32.19625. [DOI] [PubMed] [Google Scholar]
- 27.McHenry C S. Annu Rev Biochem. 1988;57:519–550. doi: 10.1146/annurev.bi.57.070188.002511. [DOI] [PubMed] [Google Scholar]
- 28.Kelman Z, O’Donnell M. Annu Rev Biochem. 1995;64:171–200. doi: 10.1146/annurev.bi.64.070195.001131. [DOI] [PubMed] [Google Scholar]
- 29.Fairfield F R, Newport J W, Dolejsi M K, von Hippel P H. J Biomol Struct Dyn. 1983;1:715–727. doi: 10.1080/07391102.1983.10507477. [DOI] [PubMed] [Google Scholar]
- 30.Jarvis T C, Newport J W, von Hippel P H. J Biol Chem. 1991;266:1830–1840. [PubMed] [Google Scholar]
- 31.Kaboord B F, Benkovic S J. Curr Biol. 1995;5:149–157. doi: 10.1016/s0960-9822(95)00036-4. [DOI] [PubMed] [Google Scholar]