

SUMMARY
BACKGROUND
FoxA factors are critical regulators of embryonic development and post-embryonic life, but little is know about the upstream pathways that modulate their activity [1]. C. elegans pha-4 encodes a FoxA transcription factor that is required to establish the foregut in embryos, and to control growth and longevity after birth [2–5]. We previously identified the AAA+ ATPase homologue ruvb-1 as a potent suppressor of pha-4 mutations [6].
RESULTS
Here we show that ruvb-1 is a component of the TOR pathway in C. elegans (CeTOR). Both ruvb-1 and let-363/TOR control nucleolar size and promote localization of box C/D snoRNPs to nucleoli, suggesting a role in rRNA maturation. Inactivation of let-363/TOR or ruvb-1 suppresses the lethality associated with reduced pha-4 activity. The CeTOR pathway controls protein homeostasis and also contributes to adult longevity [7, 8]. We find that pha-4 is required to extend adult lifespan in response to reduced CeTOR signaling. Mutations in the predicted CeTOR target rsks-1/S6 kinase or in ife-2/eIF4E also reduce protein biosynthesis and extend lifespan [9–11], but only rsks-1 mutations require pha-4 for adult longevity. In addition, rsks-1, but not ife-2, can suppress the larval lethality associated with pha-4 loss-of-function mutations.
CONCLUSION
The data suggest that pha-4 and the CeTOR pathway antagonize one another to regulate post-embryonic development and adult longevity. We suggest a model in which nutrients promote TOR and S6 kinase signaling, which represses pha-4/FoxA, leading to a shorter lifespan. A similar regulatory hierarchy may function in other animals to modulate metabolism, longevity or disease.
INTRODUCTION
Members of the FoxA family of transcription factors encode critical regulators of development, growth and metabolism. In embryos, FoxA proteins establish the digestive tract and notochord [1, 12], and they contribute to brain development [13, 14]. Post-embryonically, FoxA factors control metabolism, developmental progression and lifespan in response to dietary restriction induced in liquid media or by mutation (eat-2) [1, 3, 4, 15]. These functions depend on the appropriate dosage of FoxA activity, and reduced FoxA is associated with developmental abnormalities and disease. For example, animals with a lower dose of FoxA2 in mice or pha-4 in worms often arrest at birth [5, 16–18]. FoxA2 heterozygotes lose dopaminergic neurons and develop symptoms that resemble Parkinson’s Disease [13, 14]. The dosage sensitivity of FoxA factors may reflect the contribution of DNA binding site affinity for FoxA target gene selection [5, 19]. Suboptimal DNA binding sites that associate weakly with FoxA may lose occupancy when FoxA levels are reduced.
Given the involvement of FoxA proteins in metabolism and growth, it is appealing to consider that nutrient signaling pathways might regulate FoxA. Wolfrum and colleagues suggested that insulin induces FoxA2 nuclear exclusion via Akt phosphorylation [1]. However, others have found that FoxA2 associates with target genes in liver nuclei regardless of the status of insulin signaling [1]. Thus, the relationship between FoxA and the insulin pathway is unclear. A second nutrient sensing pathway is the TOR pathway, which couples growth factors and nutrients to protein homeostasis [20]. Regulation of protein synthesis depends on substrates involved in translation including the eIF4E binding protein 4E-BP and ribosomal S6 kinase (S6K) [20]. TOR also modulates ribosome biogenesis, autophagy and transcription [20]. C. elegans possesses homologues of TOR complex 1 (TORC1) components, including TOR kinase (let-363 [21]), Raptor (daf-15; [15]) and LST8 (http://www.wormbase.org/). Both let-363/TOR and daf-15/Raptor influence larval growth, protein synthesis, adult aging and autophagy [8, 9, 11, 15, 22]. let-363 has been implicated in dietary restriction induced by eat-2 or pep-2 mutations [9, 23], making it an attractive candidate to function with FoxA. However, little is known about how CeTOR controls growth and aging, or its involvement, if any, with FoxA.
To identify regulators of FoxA factors, we previously undertook a genetic screen for mutations that could suppress the lethality associated with pha-4, which encodes the sole C. elegans FoxA protein [6]. This screen identified the AAA+ ATPase homologue ruvb-1 as a potent suppressor of pha-4 mutations [6]. Here we show that ruvb-1 is a component of the CeTOR pathway, which established a genetic connection between pha-4/FoxA and CeTOR. In larvae, both ruvb-1 and let-363/TOR promote Box C/D snoRNP localization to the nucleolus, which is required for robust protein synthesis. In adults, inactivation of CeTOR or rsks-1/S6 kinase prolongs lifespan, and this effect requires pha-4 activity. Another regulator of protein translation, ife-2/eIF4E, also modulates lifespan but is pha-4-independent. The data suggest that CeTOR and rsks-1 antagonize pha-4/FoxA to control post-embryonic development and adult longevity. Other animals may rely on an analogous regulatory relationship to control metabolism, longevity or disease states.
RESULTS
Similarity in phenotypes between ruvb-1, a pha-4 suppressor, and CeTOR mutants
We initiated our study by analysis of the loss of function phenotype associated with ruvb-1. Heterozygous ruvb-1/+ animals appear wildtype and suppress loss of pha-4 function [6]. The most striking phenotype associated with ruvb-1 homozygous mutants is an arrest during the third larval stage (L3), as deduced by body size, perturbed vulval development and blocked gonadogenesis (Figure 1, Table S1). Previous studies had found that mutations that disrupt insulin signaling (e.g. daf-2/insR), and the CeTOR kinase pathway (e.g. let-363/TOR, daf- 15/raptor), led to a Dauer or L3 arrest [21, 24], suggesting ruvb-1 might belong to one of these pathways. Like daf-2, let-363 and daf-15, ruvb-1 mutants had increased intestinal lipids and an abundance of granules in the epidermis (Figure 1, Table S1). All four mutants also had small nucleoli, suggesting reduced or defective ribosome biogenesis (Figure 1, Table S1). Further inspection revealed that the ruvb-1 phenotypes were distinct from those of daf-2. For example, daf-2 mutants have cuticular ridges called alae, the buccal cavity is sealed against the external environment, the intestinal lumen constricts, and the pharynx shrinks radially and ceases to pump [24]. Mutants for ruvb-1 lacked all of these features: they did not have alae, their digestive tract remained open and their pharynx continued to pump despite the larval arrest (Figure 1, Table S1). Thus, ruvb-1 larvae lacked phenotypes typically associated with daf-2/insR and resembled let-363/TOR mutants.
Figure 1. Similarity of phenotypes associated with ruvb-1 and CeTOR.

The phenotypes of wild-type (WT), pha-4(RNAi), ruvb-1(px34), let-363(h111)/TOR, daf-15(m81)/Raptor and daf-2(e1370)/insR mutants were compared at the third larval stage (L3). daf-2 mutants were examined as Dauer larvae except for lipid accumulation, which was examined in arrested larvae [60]. pha-4(RNAi) was initiated at the L1 stage and analyzed at the L3 stage. Lipid accumulation was visualized by Nile Red staining [57]. Mouth opening (arrowhead, scale bar=5µm), epidermal granules (Epi, scale bar=5µm), cuticular alae (arrowhead), arrested gonad development (red, scale bar=50µm) and epidermal nucleoli (scale bar=5.12µm) were examined by light microscopy. Larvae were monitored at 20°C except for daf-2, which was temperature-sensitive and therefore examined at 25°.
As a second means to probe the relationship between ruvb-1, insulin and CeTOR, we examined interactions with daf-16/FoxO. Mutations in daf-16 are epistatic to all components of the insulin pathway, but not to those of CeTOR [8, 24]. We found that reduction of ruvb-1 by RNAi led to an L3 arrest, high fat and epidermal granules even in combination with a null allele of daf-16 (Figure 2A). This result indicates that ruvb-1 functions independently of daf-16.
Figure 2. Genetic interactions between Fox factors and ruvb-1 or CeTOR.
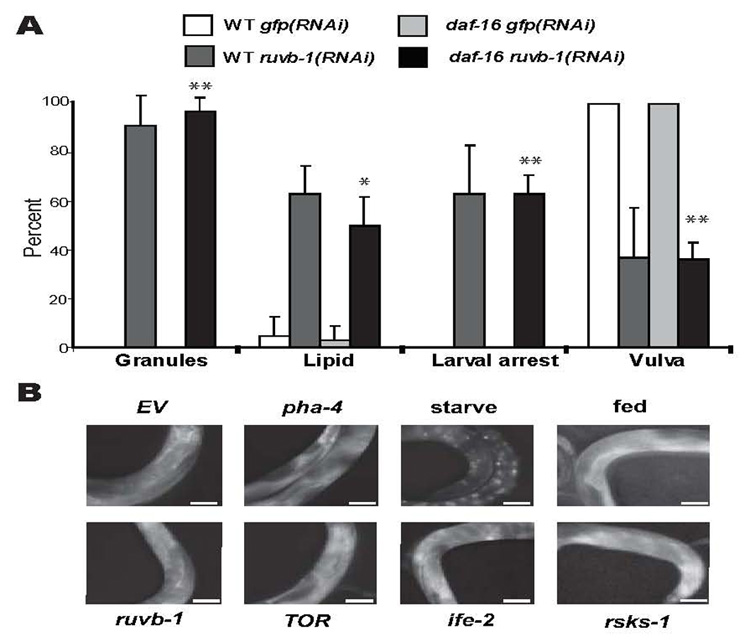
A) ruvb-1 phenotypes do not depend on daf-16/FoxO. RNAi against GFP or ruvb-1 was induced in either wild-type (WT) or daf-16(mu86) hermaphrodites, and their progeny scored by light microscopy for larval arrest, epidermal granules (Granules) or a mature vulva. Lipid accumulation of L3 progeny was determined by Nile Red staining [57] (20°, 3 experiments, n≥24 animals for each condition, error bars denote standard deviation, **p<0.0002, *p=0.0038). B) DAF-16 localization. DAF-16 localization was monitored with daf-16p::daf- 16::GFP [25]. Worms subjected to OP50 (fed), empty vector (EV), ruvb-1(RNAi), pha- 4(RNAi), ife-2(RNAi), rsks-1(RNAi) or let-363(RNAi); daf-15(RNAi) (TOR) show cytoplasmic localization compared to worms starved for 24 hrs, which display nuclear localization (n≥9 worms for each condition, scale bar=50µm).
Next we examined the subcellular localization of DAF-16. Normally, insulin signaling promotes cytoplasmic retention of a DAF-16::GFP reporter, whereas reduced insulin signaling leads to nuclear accumulation [25–28]. DAF-16::GFP remained cytoplasmic in ruvb-1(RNAi), pha-4 (RNAi) or let-363(RNAi); daf-15(RNAi) animals (Figure 2B), suggesting that ruvb-1 functions in parallel or downstream of the insulin pathway. As a control, we observed nuclear DAF-16::GFP when animals were starved, in agreement with previous studies [25, 28] (Figure 2B). In sum, the constellation of phenotypes and genetic interactions we observed for ruvb-1 was identical to those associated with let-363/TOR and its interacting partner daf-15/Raptor [15, 21, 29]; no other known pathway has a matching set of attributes in C. elegans. We conclude that ruvb-1 is a likely component of the CeTOR pathway.
ruvb-1 and CeTOR are critical for box C/D snoRNP localization
How might ruvb-1 contribute to the CeTOR pathway? Biochemical studies in other organisms have identified RUVB orthologues as members of several multiprotein complexes [30–32]. To assess which of these complexes could account for the role of ruvb-1 in the CeTOR pathway, we screened members of these complexes by RNAi to determine if any were associated with let-363/TOR-like phenotypes (Table S2, Figure S3). Inactivation of nol- 5, K07C5.4 or fib-1 each resulted in a larval arrest with excess epidermal granules and small nucleoli (Table S2). We did not observe a high-fat phenotype for nol-5, K07C5.4 or fib-1, indicating that either a separate RUVB-1-containing complex is responsible for lipid accumulation, or that inactivation of these four genes affects a common target to different extents. The similarity of phenotypes suggested that nol-5, K07C5.4 and fib-1 could account for at least part of the CeTOR phenotype of ruvb-1.
nol-5, K07C5.4 and fib-1 encode proteins predicted to be members of the Box C/D snoRNP [33]. Box C/D snoRNPs function in the nucleolus and methylate pre-rRNAs during ribosome maturation [33]. In other organisms, the Box C/D snoRNP complex is assembled and stabilized in the nucleus by association with multiple proteins, including RUVB [31, 34]. Once stabilized, the mature box C/D snoRNP is transported into the nucleolus where it methylates rRNAs [32]. We used an antibody to the predicted box C/D snoRNP component FIB-1 to localize box C/D snoRNPs in wild-type and mutant worms. We observed robust FIB-1 in 100% of the nucleoli of wild-type animals, and this signal was lost after fib-1(RNAi), indicating the stain was specific (Figure 3). FIB-1 levels were reduced, and FIB-1 failed to localize to the nucleolus in the majority of ruvb-1 mutant larvae (Figure 3). In affected animals, we observed a faint ring of FIB-1 at the nucleolar periphery and, to a lesser degree, within the nucleoplasm. Thus, C. elegans ruvb-1 is required for snoRNP localization and accumulation, similar to its human and yeast counterparts. Inactivation of let-363/TOR produced similar defects, with low levels of FIB-1, which were localized to the nucleoplasm and nucleolar periphery (Figure 3). This result is consistent with ruvb-1 and CeTOR functioning in the same pathway. We also tested if snoRNP localization required pha-4 activity. FIB-1 localization was unperturbed by pha-4(RNAi) (Figure 3). Moreover, pha- 4(RNAi) did not restore FIB-1 localization to ruvb-1 or let-363 mutants (Figure S1). The data indicate that PHA-4 is not involved in Box C/D snoRNP localization.
Figure 3. Localization of the Box C/D snoRNP complex requires food, ruvb-1 and let 363/TOR.
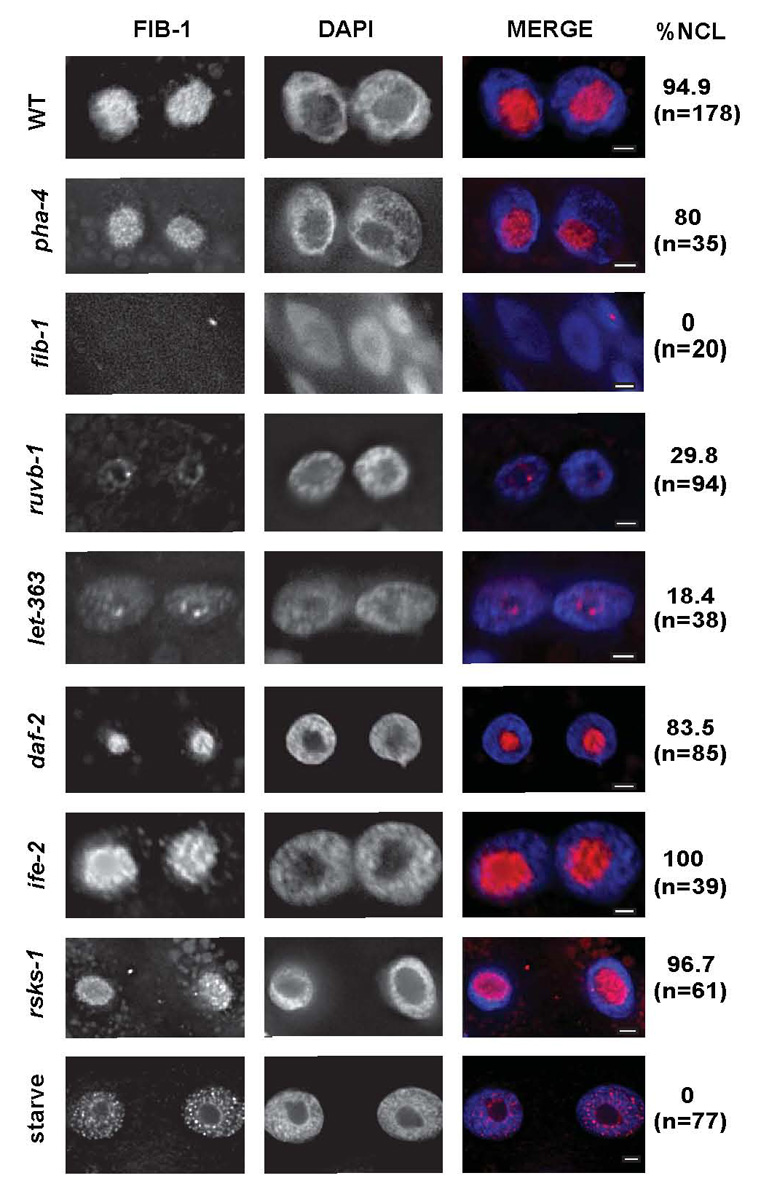
Fed (WT) or starved (8 hours, starve) wild-type, pha-4(RNAi), ruvb-1(px34), let- 363(h111)/TOR, daf-2(e1368ts)/InsR, rsks-1(ok1255) or ife-2(ok306)/eIF4E larvae were stained for FIB-1 (pink) and DNA (DAPI, blue) at the L3 stage (scale bar=2 µm). pha-4(RNAi) was initiated at the L1 stage and analyzed at the L3 stage. fib-1(RNAi) was initiated at the L4 stage and L1 progeny were analyzed. Data were quantified for percent nucleolar (% NCL) and number of nuclei (n).
TOR is responsive to nutrient status [20], and CeTOR may be part of the dietary restriction (DR) pathway for lifespan extension [9, 23], suggesting that FIB-1 localization might be regulated by nutritional status. We analyzed worms that had undergone eight hours starvation and observed that FIB-1 levels were reduced and restricted to the nucleoplasm (Figure 3). Regulation of FIB-1 was specific, since nucleolar FIB-1 was observed in daf-2/insR, rsks-1/S6 kinase or ife-2/eIF4E mutants (Figure 3). These data reveal that food and CeTOR signaling promote accumulation of FIB-1 in the nucleolus, and by extension rRNA maturation.
Given the effect of let-363/TOR and ruvb-1 on snoRNP localization, we predicted that protein biosynthesis should require these factors, as well as Box C/D snoRNP components. fib-1 or TOR pathway members were inactivated in adult worms, and 35S incorporation monitored over a five-hour period. Reduction of let-363 led to a 50% decrease in 35S incorporation, in agreement with previous studies (Figure 4)[9, 11]. We observed a 20% decrease in 35S incorporation when we inactivated pha-4, genes required for the Box C/D snoRNP (ruvb-1 or fib-1), or ife-2 for translation initiation (Figure 4). An empty vector controlled that produced a small, nonspecific double-stranded RNA (dsRNA), served as a negative control (Figure 4). These data suggest that one way CeTOR controls protein biosynthesis is by modulating the accumulation and localization of Box C/D snoRNPs. In addition, pha-4 is required for protein synthesis independent of the Box C/D snoRNP complex.
Figure 4. Inhibition of CeTOR or Box C/D snoRNPs decreases the rate of newly synthesized proteins.
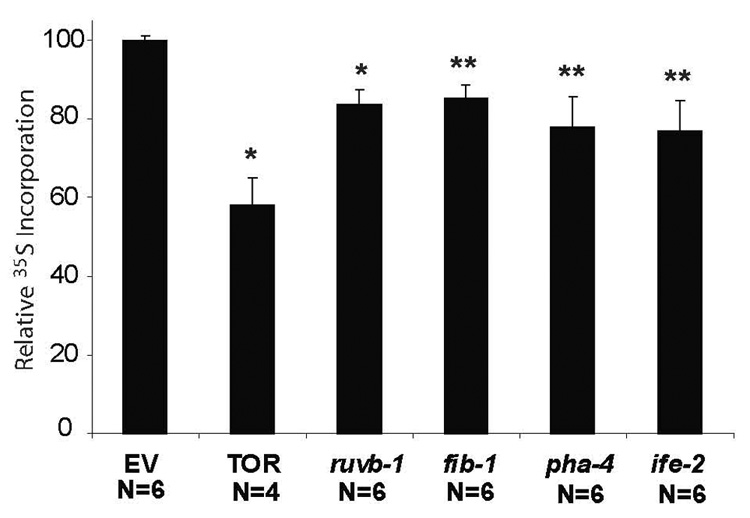
Relative levels of 35S-methionine incorporation in 2-day-old fog-1(q253ts) adult animals treated with either empty vector (EV), let-363(RNAi); daf-15(RNAi) (TOR), ruvb-1(RNAi), fib-1(RNAi), pha-4(RNAi) or ife-2(RNAi). RNAi was initiated at the L4 stage (“day 0” of adulthood) at 25°C. Bar graphs represent average 35S-methionine incorporation normalized to total protein levels for different RNAi treatments compared to EV(RNAi) (**p<0.006, *p<0.0002, one-sided paired t-test. error bars represent SEM, N, number of measurements).
let-363/TOR and daf-15/Raptor antagonize pha-4
ruvb-1 was originally discovered as a suppressor of the lethality associated with partial inactivation of pha-4 [6], prompting us to test whether let-363/TOR and daf-15/Raptor could also suppress pha-4. We examined suppression in two ways. First, we used RNAi to inactivate let-363/TOR and daf-15/Raptor, and we examined the effect of reduced CeTOR signaling on pha-4 mutants. We engineered pha-4 to be cold sensitive (pha-4(ts); Experimental Procedures) and chose an intermediate temperature when worms die due to intermediate levels of PHA-4 protein (20°; [16]). The intermediate temperature provided a sensitive means to uncover genetic interactions between CeTOR pathway components and pha-4. We inactivated let-363 and daf-15 together, to ensure the strongest possible reduction in CeTOR signaling, and scored the proportion of pha-4(ts) progeny that progressed past the first larval stage (L1). We found that the fraction of survivors for pha-4(ts); let-363(RNAi); daf-15(RNAi) was similar to that observed for pha-4(ts); ruvb-1(RNAi) and about two-fold higher than pha-4(ts) alone (Figure S2). This result suggests that inactivation of canonical CeTOR pathway components suppresses pha-4 mutations.
Second, we examined survival of let-363/+ or daf-15/+ heterozygotes treated with pha-4 dsRNA. To sensitize our ability to detect genetic interactions, pha-4 dsRNA was diluted with GFP dsRNA to generate a partial inactivation of pha-4 (Experimental Procedures). Alone, let-363/+ and daf-15/+ animals appeared superficially wild-type, which allowed us to score the number of animals that lived when subjected to pha-4(RNAi) [6](data not shown). This experiment revealed that approximately twice as many let-363/+; pha- 4(RNAi) larvae lived compared to pha-4(RNAi) alone (Figure S2). daf-15/+ heterozygotes failed to rescue pha-4(RNAi) to a significant extent, which may reflect distinct roles for ruvb- 1, let-363 and daf-15, or dissimilar genetics such as maternal effects or genetic dominance [6]. Together, the data show that reduced CeTOR activity (i.e. let-363/TOR or ruvb-1) can suppress the lethality associated with reduced pha-4, and that therefore the CeTOR pathway antagonizes pha-4 during development.
pha-4 is required for lifespan extension due to decreased CeTOR
Reduced CeTOR signaling leads to prolonged lifespan whereas reduced pha-4 shortens life, and both genes are implicated in dietary restriction [3, 8, 9, 11, 15]. To explore the relationship between these genes, we inactivated both pha-4 and CeTOR components conditionally, beginning at the fourth larval stage and continuing through adulthood. Alone, pha-4(ts) animals had a slightly shortened lifespan, as had been observed previously [3]. Reduction of let-363 and daf-15 together led to a statistically-significant increase in lifespan in 3/4 experiments (p<0.05; Figure 5A, Table S4). To examine the effect of pha-4, we analyzed our datasets by multivariate Cox regression modelling, which allowed four experimental conditions to be compared simultaneously [35]. This analysis revealed that pha-4 was required for CeTOR-induced lifespan extension since we observed a statistically significant decrease in longevity for pha-4 with reduced CeTOR compared to pha-4 alone (p<0.0001; Figure 5A, Table S4, Figure S5). Strikingly, pha-4(ts); let-363(RNAi); daf-15(RNAi) animals had shorter life spans than pha-4(ts) alone. This result suggests that pha-4 is crucial for survival when the TOR pathway is inactivated. The effect of pha-4 on longevity was specific since pha-4 was not required for the pronounced lifespan extension induced by daf-2/insR mutations (Figure 5B, Table S4), in agreement with previous studies [3]. Moreover, inactivation of pha-4 had only minor effects on the number of eggs laid, indicating that these worms were generally healthy and fecund (115±37 eggs for pha-4(RNAi) mothers vs. 127±33 for wild-type (n=18)). We conclude that pha-4/FoxA is selectively required for lifespan extension by reduced CeTOR.
Figure 5. Longevity due to reduced CeTOR or rsks-1 requires pha-4.
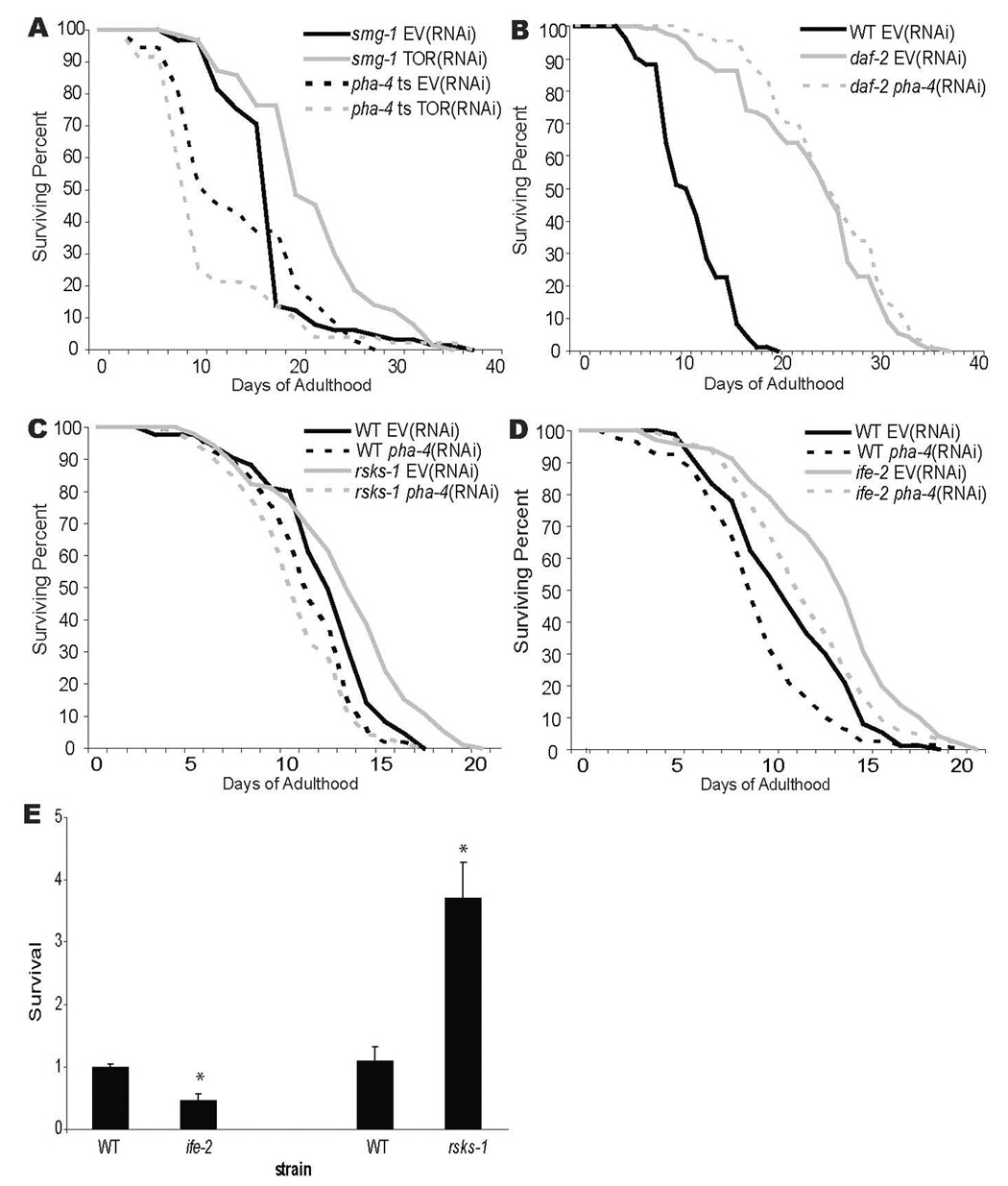
A–D) Survival curves for A) pha-4(zu225);smg-1(cc546ts) (pha-4(ts)) or smg-1(cc546ts) grown at 24°C and shifted to 15°C beginning at the L4 stage. Worms were subjected to let-363(RNAi); daf-15(RNAi) (TOR(RNAi)) to inactivate TOR signaling compared to empty vector control (EV). Mean lifespan was 18.1 days for smg-1; EV(RNAi) (control), 21.3 days for smg-1; TOR(RNAi) (p=0.0077 vs. control), 13.8 days for pha-4(ts); EV(RNAi) (p=0.0017 vs. control), and 12.2 days for pha-4(ts); TOR RNAi (p=<0.0001 vs. control) B) daf-2(e1368ts) worms were grown at 15°C and shifted to 25°C beginning at the L4 stage to inactivate insulin signaling. Worms were subjected to pha-4(RNAi) or an empty vector control (EV). Wild-type (WT) worms were grown at 25°C. Mean lifespan was 11.6 days for WT; EV(RNAi) (control), 23.6 days for daf-2; EV(RNAi) (p<0.0001 vs. control) and 25.2 days for daf-2; pha-4(RNAi) (p<0.0001 vs. control). C) Wild-type (WT) and rsks-1(ok1255) worms were grown at 25°C. Worms were subjected to pha-4(RNAi) or an empty vector control (EV). Mean lifespan was 11.96 days for WT; EV(RNAi) (control), 11.1 days for WT; pha-4(RNAi) (p=0.0075 vs. control), 12.98 days for rsks-1; EV(RNAi) (p=0.0011 vs. control), 10.4 days for rsks-1; pha-4(RNAi) (p=0.0001 vs. control). D) Wild-type (WT) and ife-2(ok306) worms were grown at 25°C. Worms were subjected to pha-4(RNAi) or an empty vector control (EV). Mean lifespan was 11.16 days for WT; EV(RNAi) (control), 9.5 days for WT; pha-4(RNAi) (p=0.0032 vs. control), 13.6 days for ife-2; EV(RNAi) (p=<0.0001 vs. control), 12.15 days for ife-2; pha-4(RNAi) (p=0.0664 vs. control). In all experiments, RNAi was initiated at the L4 stage [61]. E) Suppression of the lethality associated with reduced pha-4 by rsks-1, not ife-2. Wildtype animals and animals mutant for ife-2(ok306) or rsks-1(ok1255) were subjected to weak pha-4(RNAi) (Experimental Procedures). The proportion of mutant animals that survived beyond the L1 stage was counted and normalized against wildtype worms also subjected to pha-4(RNAi) (25°, 2 experiments, n≥800 animals for each strain, error bars denote standard error, *p=0.001).
To investigate how CeTOR signaling negatively regulates pha-4, we examined whether PHA-4 levels or localization changed in response to CeTOR inactivation, starvation or aging. By monitoring a translational PHA-4::mCherry reporter in adults, we observed strong expression in the pharynx and lower levels in the intestine under all experimental conditions (Figure S4). We also observed constitutively nuclear expression in agreement with Panowski et al. and Zhang et al., but distinct from Wolfrum et al. [1, 3]. Our data suggest that the levels of nuclear PHA-4 protein do not change in response to aging or starvation. Instead, the transcriptional activity may be altered by CeTOR signaling.
rsks-1 antagonizes pha-4
The TOR pathway controls protein homeostasis at many levels, including ribosome biogenesis, translation and autophagy [20]. We wondered which of these downstream pathways relied on pha-4 for lifespan extension. One appealing candidate was rsks-1, which is homologous to the TOR target S6 kinase [20] and which leads to lifespan extension when inactivated in C. elegans [9–11]. We observed a ~10% extension in lifespan for rsks-1(ok1255) in 4/4 experiments, similar to previous studies (Figure 5C, Table S5)[10]. This effect was dependent on pha-4 since pha-4(RNAi) caused worse survival in rsks-1 compared to wild-type (p=0.007 by multivariate Cox modeling [35]; Figure 5C, Table S5, Figure S5).
Next we tested ife-2, which encodes one of five eIF4E isoforms (WS180 www.wormbase.org). In other animals, TOR promotes eIF4E activity by inactivating the eIF4E repressor 4E-BP [20]. In 3/3 experiments, we observed extended longevity for ife-2 mutants (Figure 5D, Table S5), similar to previous work [9, 11]. Lifespan decreased when pha-4 was inactivated by RNAi (4/5 experiments, Figure 5D, Table S5). However, inactivation of pha-4 reduced longevity to a similar extent as inactivation of pha-4 in wild-type animals, and never returned ife-2 lifespan to baseline (p=0.158 by multivariate Cox modeling [35], Table S5, Figure S5). These data implicate alternative processes for lifespan extension by ife-2. We suggest that pha-4/FoxA plays a critical role for lifespan extension due to decreased let-363 and rsks-1, but not ife-2.
We were surprised that rsks-1 and ife-2 had different genetic interactions with pha-4 for adult aging. To extend this finding, we tested whether rsks-1 or ife-2 mutations could suppress the larval lethality associated with loss of pha-4 function. pha-4 was partially inactivated by RNAi in wild-type, rsks-1 or ife-2 mutants. We observed a ≥2x suppression of pha-4 by rsks-1 but no suppression by ife-2 (Figure 5E, Table S6). These results bolster the conclusion that rsks-1 is a negative regulator of pha-4, whereas ife-2 is not.
DISCUSSION
We have identified the pha-4 suppressor ruvb-1 as a new component of the C. elegans TOR pathway, which lead us to probe the genetic interactions between CeTOR and pha-4. Our findings reveal that CeTOR antagonizes pha-4 during larval development and adult aging. let-363/TOR and ruvb-1 are both needed to accumulate Box C/D snoRNPs in the nucleolus, and loss of these proteins leads to decreased protein synthesis. Downstream of CeTOR, reduced rsks-1/S6 kinase, but not ife-2/eIF4E, relies on pha-4 activity to prolong life. Moreover, rsks-1/S6 kinase mutations, but not ife-2/eIF4E, can suppress pha-4-associated larval lethality. These data suggest that nutrients activate the CeTOR pathway and rsks-1/S6 kinase to repress pha-4 during larval development and adult aging (Figure 6). Reduced nutrients, for example during dietary restriction, leads to enhanced pha-4/FoxA activity and prolonged lifespan, via reduced TOR signaling.
Figure 6. Two models for lifespan extension due to decreased protein synthesis.
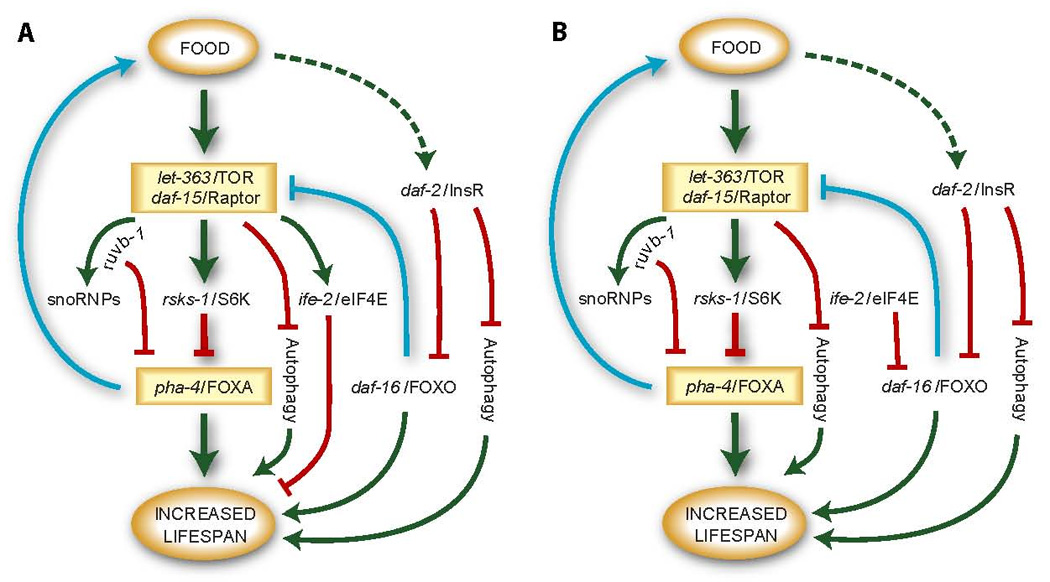
See the Discussion for an explanation of these models.
CeTOR and ruvb-1 share common phenotypes
We have shown that ruvb-1 and let-363/TOR control the accumulation and localization of Box C/D snoRNPs within nucleoli, providing an explanation for ruvb-1 function in the CeTOR pathway. Box C/D snoRNPs methylate rRNAs during maturation, and loss of Box C/D snoRNPs is predicted to reduce ribosome biogenesis. This function likely explains, at least in part, why inactivation of CeTOR [9, 15, 21] or ruvb-1 (this study) leads to decreased protein biosynthesis and arrested larval development. In our hands, CeTOR had the most pronounced effect on protein synthesis rates compared to other genes, which may reflect multiple levels of regulation of protein homeostasis by CeTOR, similar to other organisms [20]. By contrast, rsks-1/S6 kinase and ife-2/eIF4E each promoted protein biosynthesis, but neither was necessary to complete larval development or to localize FIB-1 ([9–11], this study). C. elegans possesses five predicted eIF4E factors [36], which may explain why ife-2/eIF4E was associated with weaker phenotypes than ruvb-1 or CeTOR.
CeTOR and one of the core components of the Box C/D snoRNP complex, W01B11.3/Nop58, have been implicated in lifespan extension [8, 37], raising the question of whether ruvb-1 or other snoRNP components contribute to longevity as well. Inactivation of ruvb-1 beginning at the L4 stage did not cause a reproducible extension of lifespan (Table S3). One possibility is that ruvb-1 and the Box C/D snoRNP may be important for lifespan extension, but pleiotropic phenotypes associated with other RUVB-1-containing complexes may mask a role in aging. Different experimental conditions may reveal an aging role.
Models for lifespan extension by reduced protein biosynthesis
Large-scale screens in C. elegans have identified a multitude of genes that can modulate adult lifespan (reviewed in [38]). An ongoing challenge is to organize these genes into defined pathways. Analysis has been complicated by the large number of genes that can impinge on aging, the complex genetic interactions between these genes, the different approaches to induce lifespan extension by DR and the need for conditional or partial inactivation of essential genes [3, 39–44]. Previous studies have shown that reduction of CeTOR or the translation machinery can extend lifespan [8–11, 45, 46]. Given that CeTOR controls protein homeostasis and responds to nutrients [9, 20, 23], one might predict that inhibition of translation would extend longevity by the same pathway as reduction of CeTOR signaling. However, genetic studies have lead to seemingly contradictory conclusions regarding the relationship between TOR and protein translation, or between these factors and DR [9–11, 25, 45]. pha-4 epistasis offers another approach to group genes within common pathways. Based on our analysis with pha-4/FoxA and results from other studies, we suggest two models for coupling nutrients to protein translation and aging (Figure 6). These models both place pha-4 downstream of S6 kinase and CeTOR, but differ with regard to positioning ife-2 and the translation initiation factors within an aging network. We recognize that the two models are not mutually exclusive, and the translation initiation factors may function in multiple contexts.
For both models, we place CeTOR and rsks-1/S6 kinase downstream of nutrients, based on genetic epistasis experiments in C. elegans [9, 10, 23], common effects on snoRNP localization and protein biosynthesis (this study, [9, 10]), and studies with other species [20]. Inactivation of rsks-1 and CeTOR together resembles inactivation of CeTOR alone for lifespan extension [9], consistent with these genes functioning in a common pathway. Moreover, neither gene is dependent on daf-16/FoxO [9, 10], suggesting this pathway is parallel or downstream of insulin signaling. The genetic interactions between pha-4/FoxA and either CeTOR or rsks-1, tentatively place pha-4 downstream of these kinases. We position ruvb-1 and snoRNPs downstream of CeTOR but separate from rsks-1 or ife-2, based on a lack of aging phenotypes for ruvb-1 and common snoRNP phenotypes for ruvb-1 and let-363, but not ife-2 or rsks-1. Finally, we draw a dotted arrow between food and the insulin pathway, to reflect examples of daf-16 regulation or function during starvation or dietary restriction induced by some means but not others [25, 28, 40].
In the first model, we position ife-2/eIF4E and presumably additional translation initiation factors downstream of CeTOR (Figure 6A). In other organisms, 4E-BP is a negative regulator of translation initiation that binds and sequesters eIF4E [20]. TOR phosphorylates 4E-BP, leading to eIF4E release. If a similar regulatory hierarchy exists in worms, CeTOR may activate translation initiation machinery. An advantage of this model is that all genes that affect protein biosynthesis (CeTOR, rsks-1, translation initiation factors, pha-4 as well as food deprivation; this study, [9–11]) are consigned to one branch of the aging network.
A critical feature of the first model is a pair of feedback loops. In the first loop, pha-4 promotes food uptake (Figure 6A, blue; [2]), which may explain why inactivation of pha-4 leads to reduced protein synthesis. In the second feedback loop, daf-16 negatively regulates daf-15/Raptor [15], to modify CeTOR activity (Figure 6A, blue). This feedback loop may explain two perplexing genetic interactions. First, there have been differing claims regarding the dependence on daf-16/FoxO for lifespan extension after inactivation of translation initiation factors [9–11, 37, 45]. We suggest that inactivation of daf-16 may increase CeTOR activity, which is predicted to boost translation and thereby suppress ife-2/eIF4E mutations. This scenario can explain genetic interactions between daf-16 and ife-2 (or other translation factors)[9, 11], despite the absence of nuclear-enriched DAF-16 (Figure 2). Second, although DR and CeTOR appear largely independent of insulin signaling for aging [8, 41, 43], let-363/TOR and daf-2/InsR fail to synergize for lifespan extension when they are inactivated together [8, 9]. One possibility is that the negative feedback loop decreases CeTOR activity in daf-2 mutants, such that daf-2 single mutants resemble daf-2; let-363 double mutants for aging (Figure 6A, blue). We note, however, that daf-2 mutants do not alter protein synthesis rates [9] or suppress pha-4 mutations (data not shown), suggesting that daf-16 does not repress TOR completely or in all cells.
A second model separates ife-2 and the translation initiation factors from CeTOR, where they impinge on daf-16 more directly (Figure 6B). For example, the absence of food or inactivation of translation factors may induce a stress response that activates DAF-16. In yeast, one of the two isoforms of eIF4E is up-regulated by stress and required for the stress response [47]. C. elegans also possesses multiple isoforms of eIF4E [36], and future studies will determine if any of these isoforms are involved in stress. A link to stress may explain why inactivation of some translation factors impacts the nuclear localization of DAF-16 to some extent [45]. A stress response may also explain why food deprivation induces nuclear localization of DAF-16 [25, 28], even though dietary restriction does not [40].
Both DR and TOR are important regulators of autophagy, which cells use to survive periods of starvation by recycling macromolecules and nutrient transporters [48]. Autophagy is necessary for lifespan extension due to mutations that inactivate feeding (eat-2), CeTOR or insulin signaling throughout the life of the animal [22, 49–52]. However, inactivation of let-363/TOR in adults, rather than throughout life, can prolong lifespan in the absence of an obvious autophagic response [22]. Neither ife-2/eIF4E nor rsks-1/S6K mutants have increased autophagy, yet they extend longevity [22]. Induction of autophagy in daf-2/InsR; daf-16/FoxO mutants is not sufficient for increased lifespan [22]. These three observations suggest that there must be additional processes beyond autophagy involved in lifespan extension, and that these processes depend on pha-4/FoxA. pha-4 can also impinge on autophagy. Long-term reduction (>1 generation) of pha-4 blocks the induction of autophagy in response to long-term reduction in feeding (eat-2) or daf-15/Raptor heterozygotes [22]. These effects could reflect a developmental role for these proteins, particularly pha-4, which is required for embryonic and larval development [4, 5]. Alternatively, pha-4 may be an acute regulator of the autophagic response. Comparison of long-term vs. short-term inactivation of these proteins will clarify their roles.
In summary, our analysis of ruvb-1 has revealed that the activity of pha-4 is modulated by the CeTOR pathway. This interaction could be relatively direct, for example, by PHA-4 modification. Alternatively, it could be indirect, if both pha-4 and CeTOR impinge on common processes. An intriguing avenue for future studies will be to determine if the developmental or metabolic roles of FoxA proteins in other animals are modified by TOR signaling.
EXPERIMENTAL PROCEDURES
See Supplemental Data for additional experimental procedures.
RNA Interference
RNAi by bacterial feeding was performed essentially as described in [6]. HT115 bacteria [53] expressing dsRNA for GFP, ruvb-1, pha-4, let-363, daf-15, dpy-1, nhr-23, fib-1, rsks-1 or ife-2 were grown in overnight cultures and seeded onto plates containing 5mM IPTG (Sigma) and 60ug/ml Carbenicillin (Sigma) or 1mM IPTG only for lifespan analysis. All RNAi clones were derived from the Ahringer library [54] except for pha-4 (bSEM 865) [55], GFP [53], let-363 (bSEM 911), daf-15 (bSEM 912) and rsks-1 obtained from the C. elegans ORF-RNAi library v1.1 (Geneservice Ltd.). Clones were confirmed by restriction enzyme digest.
daf-16 epistasis
Wild-type or daf-16(mu86) [56] hermaphrodites were subjected to ruvb-1(RNAi) beginning at the L4 stage. Progeny were scored for phenotypes after 3–4 days incubation at 20°C. L3 larval arrest was determined by body size, gonad extension and presence/absence of a mature vulva with either wild-type or protruding morphology. Fat was detected by Nile Red staining [57] and scored if increased from average wild-type staining by visual inspection. Epidermal granules are refractile storage vesicles in the epidermis and were scored for increased abundance compared to the wild type.
Immunostaining
Immunostaining was performed as described previously [58] with the following changes. Microscope slides were treated with a 0.1% poly-L-lysine solution overnight (Sigma-Aldrich Product #P8920). In situ antibody staining for FIB-1 was performed using a 1:200 dilution of α-FIB-1 mouse monoclonal antibody (EnCor BioTechnology®, Catalog #MCA-38F3) and detected using a 1:200 Cy3 conjugated α-IgG secondary antibody (Jackson ImmunoResearch Inc). Mounting medium consisted of 50% glycerol in PBS with DAPI and p-phenylenediamine.
Worms were subjected to pha-4(RNAi), Empty Vector(RNAi) or OP50 bacteria at the L4 or L1 stage. L1 progeny or L3 stage worms were picked off of plates, washed with water and placed in 2% paraformaldehyde and only L3 worms were cut to release gonad and intestine. Worms were fixed in 2% paraformaldehyde and permeabilized by freeze-crack method for 30 min, then submersed in ice-cold methanol for 3 min. Following methanol treatment slides were rinsed twice, 5 min each, in 2X TBST (Tris-Buffered Saline Tween). Slides were later blocked for 30 min in TNB (0.1 M Tris–HCl, 0.15 M NaCl, 0.05% Tween 20, pH 7.5 containing blocking reagent (NEN)) and 10% NGS (Normal Goat Serum) followed by overnight incubation with the primary antibody at 15°C. Following overnight incubation slides were washed 3 times in 2X TBST and secondary antibodies were added. Slides incubated with the secondary antibody at room temperature for 2 hrs. Images were captured using DeltaVision RT Deconvolution system and SoftWoRx software (Applied Precision).
Lifespan Analysis
Lifespan analysis was performed as described previously [9] with the following changes. Worms were grown for at least 2 generations at 25°C or 20°C, as indicated, before the experiment was initiated. Hermaphrodites were allowed to lay eggs for 4–8 hours on OP50, and progeny were grown to the L4 stage. In all experiments, L4 larvae were transferred to new plates with 1mM IPTG and appropriate bacteria to initiate pha-4(RNAi) or ruvb-1(RNAi) vs. a vector control. The first day of adulthood was counted as day one of the experiment. Worms were moved daily until reproduction ceased. Worms were moved every 3–4 days for the rest of the lifespan assay. Lifespan analysis was conducted with wild-type, daf-2(e1368) [59], ife-2(ok306) [36], and rsks-1(ok1255) [10].
For experiments with reduced CeTOR, smg-1(cc546ts); pha-4(zu225) [16] and control smg-1(cc546ts) (www.addgene.org/labs/Fire/Andrew/Vec97.pdf) worms were used. Worms were grown at the permissive temperature of 24°C for at least two generations prior to beginning the experiment. Hermaphrodites were allowed to lay eggs for 4–6 hours and removed. L4 progeny were moved to the non-permissive temperature of 15°C and RNAi was initiated. We used a 1:1 mixture of bacteria for let-363(RNAi); daf-15(RNAi). The first day of adulthood was counted as day one of the experiment. Worms were moved every other day until reproduction ceased. Worms were moved every 5–7 days for the rest of the lifespan assay. Censoring within each experiment included animals that ruptured, crawled off the plate or exhibited progeny hatching internally. We have reported the number of ruptured animals.
Statistical Methods for Lifespan Analysis
Log rank tests were used in pair-wise comparisons of wildtype, mutant, RNAi, and control groups. To determine if pha-4 had an effect beyond its effect on wild-type worms, we combined data from multiple experiments using the same conditions and applied multivariate Cox proportional modelling (Cox regression) using Stata Software [35]. This statistical approach enabled us to compare four experimental conditions at once, and determine if pha-4 had a greater effect on our experimental strain relative to the wild-type control. It also included information from the censored subjects.
Suppression of pha-4
pha-4(RNAi) suppression was performed as reported previously [6] using bacteria expressing pha-4 and GFP dsRNA at a ratio of 1:4 or 1:8, to give an intermediate inactivation of pha-4. 5–10 L4 stage wild-type, unc-42 ruvb-1(px34)/evl-1, let-363(h111)/ dpy- 5 and daf-15(m81)/ unc-24 worms were picked to 2–4 RNAi plates per experiment and incubated at 25°C. P0 were allowed to lay eggs for 1 day and then removed. Progeny were counted 2 days later for the percentage of animals older than L1 (n≥100 animals/plate). P-values were determined by t-test.
pha-4(ts) suppression was performed as reported previously [6] using bacteria expressing ruvb-1(RNAi), a mix of let-363(RNAi); daf-15(RNAi) or bacteria containing empty vector as a control. Ten L4 pha-4(ts) worms were picked to 2–5 RNAi plates per experiment and incubated at 20°C. P0 were allowed to lay eggs for 1 day and then removed. Progeny were counted 2 days later for the percentage of animals older then L1 (n≥20 animals/plate). Strength of RNAi was monitored by observation of progeny for larval arrest (ruvb-1(RNAi)) or slow growth and sterility (TOR(RNAi)). P-values were determined by t-test.
Supplementary Material
ACKNOWLEGMENTS
We thank K. Ashrafi, H. Gabel, J. Kim, D. Riddle, G. Ruvkun, and the modENCODE TF group for reagents and discussion, A. Brunet, C. Thummel, J. Rutter and A. Schier for comments on the manuscript, L. Pappas for help with statistics and D. Lim for Figure 6. This work was funded by T32 HD007491 to D.U. and K.S. S.E.M. was supported by NIH R01DK070184, NIH R01GM056264, the Huntsman Cancer Institute/Foundation and the Dept. of Oncological Sciences. Some strains were obtained from the Caenorhabditis Genetics Center and OMRF Knockout Group. We acknowledge the Mitani Lab and National Bioresource Project for providing uri-1(tm939). University of Utah Core facilities were supported by NIH P30 CA42014.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Friedman JR, Kaestner KH. The Foxa family of transcription factors in development and metabolism. Cell Mol Life Sci. 2006;63:2317–2328. doi: 10.1007/s00018-006-6095-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mango SE. The C. elegans pharynx: a model for organogenesis. WormBook; 2007. oi/10.1895/wormbook.1.7.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Panowski SH, Wolff S, Aguilaniu H, Durieux J, Dillin A. PHA-4/Foxa mediates diet-restriction-induced longevity of C. elegans. Nature. 2007;447:550–555. doi: 10.1038/nature05837. [DOI] [PubMed] [Google Scholar]
- 4.Ao W, Gaudet J, Kent WJ, Muttumu S, Mango SE. Environmentally induced foregut remodeling by PHA-4/FoxA and DAF-12/NHR. Science. 2004;305:1743–1746. doi: 10.1126/science.1102216. [DOI] [PubMed] [Google Scholar]
- 5.Gaudet J, Mango SE. Regulation of organogenesis by the Caenorhabditis elegans FoxA protein PHA-4. Science. 2002;295:821–825. doi: 10.1126/science.1065175. [DOI] [PubMed] [Google Scholar]
- 6.Updike DL, Mango SE. Genetic suppressors of C. elegans pha-4/FoxA identify the predicted AAA helicase ruvb-1/RuvB. Genetics. 2007 doi: 10.1534/genetics.107.076653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaeberlein M, Kennedy BK. Protein translation, 2007. Aging cell. 2007;6:731–734. doi: 10.1111/j.1474-9726.2007.00341.x. [DOI] [PubMed] [Google Scholar]
- 8.Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Muller F. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature. 2003;426:620. doi: 10.1038/426620a. [DOI] [PubMed] [Google Scholar]
- 9.Hansen M, Taubert S, Crawford D, Libina N, Lee SJ, Kenyon C. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging cell. 2007;6:95–110. doi: 10.1111/j.1474-9726.2006.00267.x. [DOI] [PubMed] [Google Scholar]
- 10.Pan KZ, Palter JE, Rogers AN, Olsen A, Chen D, Lithgow GJ, Kapahi P. Inhibition of mRNA translation extends lifespan in Caenorhabditis elegans. Aging cell. 2007;6:111–119. doi: 10.1111/j.1474-9726.2006.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Syntichaki P, Troulinaki K, Tavernarakis N. eIF4E function in somatic cells modulates ageing in Caenorhabditis elegans. Nature. 2007;445:922–926. doi: 10.1038/nature05603. [DOI] [PubMed] [Google Scholar]
- 12.Mango SE, Lambie EJ, Kimble J. The pha-4 gene is required to generate the pharyngeal primordium of Caenorhabditis elegans. Development. 1994;120:3019–3031. doi: 10.1242/dev.120.10.3019. [DOI] [PubMed] [Google Scholar]
- 13.Kittappa R, Chang WW, Awatramani RB, McKay RD. The foxa2 gene controls the birth and spontaneous degeneration of dopamine neurons in old age. PLoS Biol. 2007;5:e325. doi: 10.1371/journal.pbio.0050325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferri AL, Lin W, Mavromatakis YE, Wang JC, Sasaki H, Whitsett JA, Ang SL. Foxa1 and Foxa2 regulate multiple phases of midbrain dopaminergic neuron development in a dosage-dependent manner. Development. 2007;134:2761–2769. doi: 10.1242/dev.000141. [DOI] [PubMed] [Google Scholar]
- 15.Jia K, Chen D, Riddle DL. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development. 2004;131:3897–3906. doi: 10.1242/dev.01255. [DOI] [PubMed] [Google Scholar]
- 16.Kaltenbach LS, Updike DL, Mango SE. Contribution of the amino and carboxyl termini for PHA-4/FoxA function in Caenorhabditis elegans. Dev Dyn. 2005;234:346–354. doi: 10.1002/dvdy.20550. [DOI] [PubMed] [Google Scholar]
- 17.Ang S-L, Rossant J. HNF-3beta is essential for node and notochord formation in mouse development. Cell. 1994;78:561–574. doi: 10.1016/0092-8674(94)90522-3. [DOI] [PubMed] [Google Scholar]
- 18.Weinstein DC, Ruiz i Altaba A, Chen WS, Hoodless P, Prezioso VR, Jessell TM, Darnell JE. The winged-helix transcription factor HNF-3beta is required for notochord development in the mouse embryo. 1994;78:575–588. doi: 10.1016/0092-8674(94)90523-1. [DOI] [PubMed] [Google Scholar]
- 19.Gaudet J, Muttumu S, Horner M, Mango SE. Whole-genome analysis of temporal gene expression during foregut development. PLoS Biol. 2004;2:e352. doi: 10.1371/journal.pbio.0020352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 21.Long X, Spycher C, Han ZS, Rose AM, Muller F, Avruch J. TOR deficiency in C. elegans causes developmental arrest and intestinal atrophy by inhibition of mRNA translation. Curr Biol. 2002;12:1448–1461. doi: 10.1016/s0960-9822(02)01091-6. [DOI] [PubMed] [Google Scholar]
- 22.Hansen M, Chandra A, Mitic LL, Onken B, Driscoll M, Kenyon C. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 2008;4:e24. doi: 10.1371/journal.pgen.0040024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meissner B, Boll M, Daniel H, Baumeister R. Deletion of the intestinal peptide transporter affects insulin and TOR signaling in Caenorhabditis elegans. J Biol Chem. 2004;279:36739–36745. doi: 10.1074/jbc.M403415200. [DOI] [PubMed] [Google Scholar]
- 24.Hu PJ. Dauer. WormBook; 2007. pp. 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Henderson ST, Johnson TE. daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr Biol. 2001;11:1975–1980. doi: 10.1016/s0960-9822(01)00594-2. [DOI] [PubMed] [Google Scholar]
- 26.Lee RY, Hench J, Ruvkun G. Regulation of C. elegans DAF-16 and its human ortholog FKHRL1 by the daf-2 insulin-like signaling pathway. Curr Biol. 2001;11:1950–1957. doi: 10.1016/s0960-9822(01)00595-4. [DOI] [PubMed] [Google Scholar]
- 27.Liang B, Moussaif M, Kuan CJ, Gargus JJ, Sze JY. Serotonin targets the DAF-16/FOXO signaling pathway to modulate stress responses. Cell Metab. 2006;4:429–440. doi: 10.1016/j.cmet.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 28.Weinkove D, Halstead JR, Gems D, Divecha N. Long-term starvation and ageing induce AGE-1/PI 3-kinase-dependent translocation of DAF-16/FOXO to the cytoplasm. BMC Biol. 2006;4:1. doi: 10.1186/1741-7007-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110:177–189. doi: 10.1016/s0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- 30.Gallant P. Control of transcription by Pontin and Reptin. Trends Cell Biol. 2007;17:187–192. doi: 10.1016/j.tcb.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 31.McKeegan KS, Debieux CM, Boulon S, Bertrand E, Watkins NJ. A Dynamic Scaffold of Pre-snoRNP Factors Facilitates Human Box C/D snoRNP Assembly. Mol Cell Biol. 2007;27:6782–6793. doi: 10.1128/MCB.01097-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watkins NJ, Lemm I, Ingelfinger D, Schneider C, Hossbach M, Urlaub H, Luhrmann R. Assembly and maturation of the U3 snoRNP in the nucleoplasm in a large dynamic multiprotein complex. Mol Cell. 2004;16:789–798. doi: 10.1016/j.molcel.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 33.Reichow SL, Hamma T, Ferre-D'Amare AR, Varani G. The structure and function of small nucleolar ribonucleoproteins. Nucleic Acids Res. 2007;35:1452–1464. doi: 10.1093/nar/gkl1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.King TH, Decatur WA, Bertrand E, Maxwell ES, Fournier MJ. A well-connected and conserved nucleoplasmic helicase is required for production of box C/D and H/ACA snoRNAs and localization of snoRNP proteins. Mol Cell Biol. 2001;21:7731–7746. doi: 10.1128/MCB.21.22.7731-7746.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bewick V, Cheek L, Ball J. Statistics review 12: survival analysis. Critical care (London, England) 2004;8:389–394. doi: 10.1186/cc2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keiper BD, Lamphear BJ, Deshpande AM, Jankowska-Anyszka M, Aamodt EJ, Blumenthal T, Rhoads RE. Functional characterization of five eIF4E isoforms in Caenorhabditis elegans. J Biol Chem. 2000;275:10590–10596. doi: 10.1074/jbc.275.14.10590. [DOI] [PubMed] [Google Scholar]
- 37.Chen D, Pan KZ, Palter JE, Kapahi P. Longevity determined by developmental arrest genes in Caenorhabditis elegans. Aging cell. 2007;6:525–533. doi: 10.1111/j.1474-9726.2007.00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kennedy BK. The genetics of ageing: insight from genome-wide approaches in invertebrate model organisms. Journal of internal medicine. 2008;263:142–152. doi: 10.1111/j.1365-2796.2007.01903.x. [DOI] [PubMed] [Google Scholar]
- 39.Bishop NA, Guarente L. Two neurons mediate diet-restriction-induced longevity in C. elegans. Nature. 2007;447:545–549. doi: 10.1038/nature05904. [DOI] [PubMed] [Google Scholar]
- 40.Greer EL, Dowlatshahi D, Banko MR, Villen J, Hoang K, Blanchard D, Gygi SP, Brunet A. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr Biol. 2007;17:1646–1656. doi: 10.1016/j.cub.2007.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Houthoofd K, Braeckman BP, Johnson TE, Vanfleteren JR. Life extension via dietary restriction is independent of the Ins/IGF-1 signalling pathway in Caenorhabditis elegans. Exp Gerontol. 2003;38:947–954. doi: 10.1016/s0531-5565(03)00161-x. [DOI] [PubMed] [Google Scholar]
- 42.Kaeberlein TL, Smith ED, Tsuchiya M, Welton KL, Thomas JH, Fields S, Kennedy BK, Kaeberlein M. Lifespan extension in Caenorhabditis elegans by complete removal of food. Aging cell. 2006;5:487–494. doi: 10.1111/j.1474-9726.2006.00238.x. [DOI] [PubMed] [Google Scholar]
- 43.Lakowski B, Hekimi S. The genetics of caloric restriction in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1998;95:13091–13096. doi: 10.1073/pnas.95.22.13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee GD, Wilson MA, Zhu M, Wolkow CA, de Cabo R, Ingram DK, Zou S. Dietary deprivation extends lifespan in Caenorhabditis elegans. Aging cell. 2006;5:515–524. doi: 10.1111/j.1474-9726.2006.00241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Curran SP, Ruvkun G. Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet. 2007;3:e56. doi: 10.1371/journal.pgen.0030056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hansen M, Hsu AL, Dillin A, Kenyon C. New genes tied to endocrine, metabolic, and dietary regulation of lifespan from a Caenorhabditis elegans genomic RNAi screen. PLoS Genet. 2005;1:119–128. doi: 10.1371/journal.pgen.0010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ptushkina M, Malys N, McCarthy JE. eIF4E isoform 2 in Schizosaccharomyces pombe is a novel stress-response factor. EMBO Rep. 2004;5:311–316. doi: 10.1038/sj.embor.7400088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 49.Toth ML, Sigmond T, Borsos E, Barna J, Erdelyi P, Takacs-Vellai K, Orosz L, Kovacs AL, Csikos G, Sass M, et al. Longevity pathways converge on autophagy genes to regulate life span in caenorhabditis elegans. Autophagy. 2008;4:330–338. doi: 10.4161/auto.5618. [DOI] [PubMed] [Google Scholar]
- 50.Jia K, Levine B. Autophagy is required for dietary restriction-mediated life span extension in C. elegans. Autophagy. 2007;3:597–599. doi: 10.4161/auto.4989. [DOI] [PubMed] [Google Scholar]
- 51.Melendez A, Talloczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science. 2003;301:1387–1391. doi: 10.1126/science.1087782. [DOI] [PubMed] [Google Scholar]
- 52.Hars ES, Qi H, Ryazanov AG, Jin S, Cai L, Hu C, Liu LF. Autophagy regulates ageing in C. elegans. Autophagy. 2007;3:93–95. doi: 10.4161/auto.3636. [DOI] [PubMed] [Google Scholar]
- 53.Timmons L, Court DL, Fire A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene. 2001;263:103–112. doi: 10.1016/s0378-1119(00)00579-5. [DOI] [PubMed] [Google Scholar]
- 54.Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421:231–237. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- 55.Kiefer JC, Smith PA, Mango SE. PHA-4/FoxA cooperates with TAM-1/TRIM to regulate cell fate restriction in the C. elegans foregut. Dev Biol. 2007;303:611–624. doi: 10.1016/j.ydbio.2006.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin K, Dorman JB, Rodan A, Kenyon C. daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science. 1997;278:1319–1322. doi: 10.1126/science.278.5341.1319. [DOI] [PubMed] [Google Scholar]
- 57.Ashrafi K, Chang FY, Watts JL, Fraser AG, Kamath RS, Ahringer J, Ruvkun G. Genome-wide RNAi analysis of Caenorhabditis elegans fat regulatory genes. Nature. 2003;421:268–272. doi: 10.1038/nature01279. [DOI] [PubMed] [Google Scholar]
- 58.Horner MA, Quintin S, Domeier ME, Kimble J, Labouesse M, Mango SE. pha-4, an HNF-3 homologue, specifies pharyngeal organ identity in Caenorhabditis elegans. Genes Dev. 1998;12:1947–1952. doi: 10.1101/gad.12.13.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gems D, Sutton AJ, Sundermeyer ML, Albert PS, King KV, Edgley ML, Larsen PL, Riddle DL. Two pleiotropic classes of daf-2 mutation affect larval arrest, adult behavior, reproduction and longevity in Caenorhabditis elegans. Genetics. 1998;150:129–155. doi: 10.1093/genetics/150.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277:942–946. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- 61.Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.