

Abstract
Cortical spreading depression (CSD) leads to dramatic changes in cerebral hemodynamics. However, mechanisms involved in promoting and counteracting cerebral vasodilator responses are unclear. Here we review the development and current status of this important field of research especially with respect to the role of perivascular nerves and nitric oxide (NO). It appears that neurotransmitters released from the sensory and the parasympathetic nerves associated with cerebral arteries, and NO released from perivascular nerves and/or parenchyma, promote cerebral hyperemia during CSD. However, the relative contributions of each of these factors vary according to species studied. Related to CSD, axonal and reflex responses involving trigeminal afferents on the pial surface lead to increased blood flow and inflammation of the overlying dura mater. Counteracting the cerebral vascular dilation is the production and release of constrictor prostaglandins, at least in some species, and other possibly yet unknown agents from the vascular wall. The cerebral blood flow response in healthy human cortex has not been determined, and thus it is unclear whether the cerebral oligemia associated with migraines represents the normal physiological response to a CSD-like event or represents a pathological response. In addition to promoting cerebral hyperemia, NO produced during CSD appears to initiate signaling events which lead to protection of the brain against subsequent ischemic insults. In summary, the cerebrovascular response to CSD involves multiple dilator and constrictor factors produced and released by diverse cells within the neurovascular unit, with the contribution of each of these factors varying according to the species examined.
Keywords: cerebral circulation, cerebral arteries, nitric oxide, calcitonin-gene related peptide, prostaglandins, neurons, ischemia, NMDA receptors, rats, rabbits, piglets, cats, migraine
1. Introduction
As first described by Leão (1944), cortical spreading depression (CSD) represents a wave of neuronal depolarization followed by repolarization, which once initiated by a variety of chemical, electrical, and mechanical stimuli (Ayata and Moskowitz, 2006; Grafstein, 1956; Shibata et al., 1990; Somjen, 2001; Van Harreveld, 1959; Verhaegen et al., 1992; Verhaegen et al., 1992) progresses outward across the surface of the cerebral cortex of both lissencephalic and gyrencephalic brains at a speed of approximately 2-5 mm/minutes (Leão, 1944; Van Harreveld et al., 1956; Goadsby, 1996; Grafstein, 1956; Lauritzen, 1987; Shimizu et al., 2000a; Shibata et al., 1990; Smith et al., 2006; Wahl et al., 1996). Although leading to a transient depression of cortical activity, repeated CSDs in the healthy brain do not lead to pronounced neural damage (Nedergaard and Hansen, 1988) and may even be beneficial by promoting the development of protection against ischemic stress (Horiguchi et al., 2005b; Kawahara et al., 1995; Kiss et al., 2004; Kobayashi et al., 1995; Matsushima et al., 1996; Otori et al., 2003). However, CSD in physiologically impaired neural tissue can be detrimental (Somjen, 2006) and the induction of inflammatory genes even in healthy brain has unknown consequences (Thompson and Hakim, 2005). Spreading depression-like events occur in other brain locations such as cerebellum (Case et al., 2002) and hippocampus (Herreras and Somjen, 1993; Kunkler and Kraig, 1998), and can also be induced in retina (de Oliveira Castro and Martins-Ferreira, 1970; Martins-Ferreira and de Oliveira Castro, 1966; 1971; Farkas et al., 2008). Cortical spreading depression has been shown to occur in mature brains of all mammalian species studied including man (Van Harreveld et al., 1956; Somjen, 2001; Lauritzen, 1987a; Goadsby, 1996; Yokota et al., 2002; Shibata et al., 1990; Fabricius et al., 2006; Lauritzen, 1987 & 2001; Goadsby, 2005; Dalkara et al., 2006), and is associated with major changes in extracellular levels of ions (Hansen and Zeuthen, 1981; Hansen et al., 1980), neurotransmitters (Davies et al., 1995), metabolic rate (Mayevsky and Weiss, 1991), and cerebral blood flow (CBF) (Table 1). For example, CSD is associated with a dramatic but short-lived rise in K+ concentration from 3 to more than 50 mM (Hansen and Zeuthen, 1981). Similarly, glutamate levels in extracellular fluid also increases transiently during CSD (Fabricius et al., 1993; Iijima et al., 1998; Scheller et al., 2000). In contrast, it appears that neonatal brains of many, but not all, species are relatively resistant to initiation of CSD (Domoki et al., 1999a; Richter et al., 1998; Somjen, 2001) although the underlying basis is not clear.
Table 1.
Vasoactive stimuli and cerebral vascular tone during CSD
| Year | Investigators | Preparation | Species | Observation | |
|---|---|---|---|---|---|
| A. Basic Characteristics | |||||
| 1944 | Leão | Cranial Window | Rabbit | Dilation Only | |
| 1952 | Van Harreveld & Stamm | Cortical photocell | Rabbit | Vasoconstriction | |
| 1956 | Van Harreveld & Ochs | Frozen cortical sections | Rabbit, Cat | Vasoconstriction followed by dilation in rabbit, dilation only in cat | |
| 1980 | Hansen et al. | 14C iodoantipyrine | Rat | Hyperemia following increased extracellular potassium | |
| 1981 | Mies et al. | 131I iodoantipyrine | Rat | Coupled CBF and metabolism | |
| 14C deoxyglucose | |||||
| 1981 | Olesen et al. | 133Xenon | Human | Hyperemia followed by oligemia with migraine | |
| 1982 | Lauritzen et al. | 14C iodoantipyrine | Rat | Hyperemia followed by oligemia | |
| 1983a | Lauritzen et al. | 133Xenon | Human | Reduced CBF responsiveness to hypercapnia during induced migraines | |
| 1983b | Lauritzen et al. | 133Xenon | Human | Oligemia following migraine | |
| 1984 | Mies and Paschen | 14C iodoantipyrine | Rat | Dilation with reduced ATP levels | |
| 1984 | Lauritzen | 14C iodoantipyrine | Rat | Oligemia with reduced CO2 responsiveness but preserved autoregulation | |
| 1986 | Lauritzen & Diemer | 14C iodoantipyrine | Rat | Reduced CBF with intact metabolic rate (14C-deoxyglucose) | |
| 1987a | Lauritzen | 14C iodoantipyrine | Rat | Increased CBF which is resting CBF dependent | |
| 1987 | Wahl et al. | Cranial window | Rat, Cat | Post CSD vasoconstriction in rat but not cat, reduced responsiveness to constrictor and dilator stimuli | |
| 1990 | Shibata et al. | Cranial Window | Rabbit | Dilation of pial arteries and arterioles | |
| 1991 | Piper et al. | Laser Doppler | Cat | Hyperemia followed by oligemia, reduced CO2 responsiveness | |
| 1991 | Mayevsky & Weiss | 14C Iodoantipyrine | Rat | Hyperemia, oxygen delivery matched to metabolic need | |
| 1991 | Duckrow | 14C Iodoantipyrine | Rat | Different rCBF patterns in awake or anesthetized animals | |
| 1992 | Shibata et al. | Cranial Window | Rabbit | Hyperemia, oligemia in some but not all animals | |
| 1992 | Lacombe et al. | Mass spectrometry | Rat | Hyperemia, oligemia, and reduction in post-CSD CO2 responsiveness | |
| 1992 | Mraovitch et al | 14C iodoantipyrine | Rat | Long lasting cerebrovascular and metabolic changes in brainstem | |
| 14C deoxyglucose | |||||
| 1993 | Fabricius & Lauritzen | 14C iodoantipyrine | Rat | Post-CSD oligemia | |
| 1993 | Busija & Meng | Cranial window | Rabbit | Post-CSD responsiveness intact | |
| 1994 | Florence et al. | Laser Doppler | Rabbit | Post-CSD response to hypotension and hypercapnia reduced | |
| 1994 | Lambert & Michalicek | Laser Doppler | Cat | Reduced meningeal blood flow | |
| 1995 | Fabricius et al. | Laser Doppler | Rat | Brief hypoperfusion, hyperemia, and prolonged oligemia L-arginine restored normal response to hypercapnia | |
| 1995 | Lauritzen & Fabricius | LDPI | Rat | Increased blood flow is segment-dependent | |
| 1996 | Shimazawa & Hara | Laser Doppler | Rat | Vasoactive responses are similar in awake animals | |
| 1997 | Wolf et al. | Laser Doppler | Rat | Dilation unaffected by hyperglycemia | |
| 2000b | Shimizu | Laser Doppler | Rat | CBF increases restricted by cortical but not subcortical infarcts | |
| 2001 | Dunn et al. | Laser Speckle | Rat | 2-3 mm area of increased CBF | |
| 2002 | Shimizu et al. | Laser Dopppler | Rat | Cerebral hyperemia independent of intact endothelium | |
| 2002 | Zhang et al. | Laser Doppler | Rat | Post-CSD vasoactivity is restored by NO donor | |
| 2002 | Otsuka et al. | Laser Doppler | Rat | Reduced CBF response after venous occlusion, increased infact area | |
| 2002 | Yokota et al | PET | Monkey | Hyperemia without preceding hypoperfusion or oligemia | |
| 2002 | Dreier et al | Laser Doppler | Rat | CSD initiated by endothelin-1 receptor agonist | |
| 2003 | Otori et al. | Laser Doppler/14C Iodoantipyrine | Rat | CBF depression for 3 days, preserves CBF during subsequent MCAO | |
| 2004 | Sietz et al. | Isolated MCA | Rat | Reduced ex vivo vasoactivity of MCA by previous CSD | |
| 2004 | Gursoy-Ozdemir et al. | BBB | Rat | Increased MMP-9 activity increases BBB permeability | |
| 2004 | Ayata et al. | Laser Speckle | Mouse | Initial pronounced hypoperfusion and return to baseline CBF | |
| Rat | Initial mild hypoperfusion followed by hyperemia | ||||
| 2005 | Tomita et al. | Capillary Flow | Rat | CSD-induced astoglial swelling stops capillary flow and triggers increased flow | |
| 2006 | Sonn and Mayevsky | Laser Doppler | Rat | CBF increases in awake and anesthetized animals | |
| 2006 | Scheckenbach et al. | Laser Doppler | Rat | Restoration of post-CSD reactivity by exogenous NO or cGMP | |
| 2006 | Akin & Bilensoy | fNIRS | Human | Reduced CBF to hypercapnia associated with migraines | |
| 2007 | Shin et al. | Cranial Window | Mouse | CSD-hypoperfusion unaffected by cerebral amyloid deposition | |
| 2007 | Brennan et al. | Cranial Window | Mouse | Dilation, constriction, followed by persistent dilation | |
| Rat | Dilation followed by persistent constriction | ||||
| 2007 | Takano et al. | Two photon imaging | Mouse | Prolonged dilation of pial and parenchymal arterioles followed by oligemia, tissue hypoxia and neuronal swelling | |
| 2007 | Chuquet et al. | Two photon imaging | P15 Rat | Constriction of pial and parenchymal arterioles followed by differential responses | |
| 2008 | Sukhotinsky et al. | Laser Doppler | Rat | Hypoxia and hypotension convert hyperemia to reduction in CBF | |
| 2008 | von Baumgarten et al. | Contusion volume | Mice | Unaffected by post trauma CSDs | |
| B. Mediators of Vascular Responses | |||||
| Year | Investigators | Preparation | Species | Approach | Effect |
|
| |||||
| 1987 | Lauritzen | 14C iodoantipyrine | Rat | Indomethacin | Increased CBF from reduced baseline |
| 1990 | Shibata et al. | Cranial Window | Rabbit | Indomethacin | Enhanced dilation from same baseline |
| 1991 | Duckrow | 14C iodoantipyrine | Awake rat | Indomethacin | Increased CBF from reduced baseline |
| 1991a | Shibata et al. | Cranial Window | Rabbit | Superfusion with aCSF | Fails to reduce arterial dilation |
| 1991b | Shibata et al. | Cranial Window/Laser Doppler | Rabbit | Indomethacin | Enhanced dilation/increased CBF |
| 1992 | Shibata et al. | Cranial Window | Rabbit | Indomethacin | Enhanced dilation and prevention of post-CSD oligemia |
| Superfusion with aCSF | Prevented post-CSD oligemia | ||||
| 1992 | Goadsby et al. | Laser Doppler CBF | Cat | NOS inhibition | Complete blockade of CBF increase |
| 1992 | Goadsby | Laser Doppler | Cat | Tirilazad | Lack of ROS contribution |
| 1992 | Duckrow & Beard | 14C iodoantipyrine | Rat | Tirilazad | Lack of ROS contribution |
| 1993 | Duckrow | 14C iodoantipyrine | Awake Rat | NOS inhibition | Uncovered brief hypoperfusion |
| 1993 | Piper et al. | Exernal Jugular Vein | Cat | Sampling for CGRP | No detectable CGRP in venous blood |
| 1994a | Colonna et al. | Cranial Window | Rabbit | CGRP receptor blockade | Decreased dilation by 39% |
| 1994b | Colonna et al. | Cranial Window | Rabbit | NOS inhibition | Reduced dilation by 50% |
| 1994 | Wahl et al. | Cranial Window | Cat | CGRP Receptor blockade | Decreased dilation 50% |
| NOS inhibition | Decreased dilation by 50% | ||||
| Combined inhibition/blockade | Decreased dilation by 75% | ||||
| 1994 | Zhang et al. | CBF | Rat | NOS inhibition | No effect |
| 1994 | Fabricius et al | Laser Doppler | Rat | Topical/intravenous NOS inhibition | Enhanced hyperemia |
| 1995 | Meng et al. | Cranial Window | Rabbit | Indomethacin | Blocks effects of NOS inhibition |
| 1995 | Fabricius et al. | Laser Doppler | Rat | NOS inhibition | Potentiated hypoperfusion and oligemia but not hyperemia |
| L-Arginine administration | Accentuated hypoperfusion but no effect on hyperperfusion; accelerated return of responsiveness | ||||
| Tetrodotoxin | Accentuated hypoperfusion but no effect on hyperperfusion | ||||
| 1996a | Goadsby et al. | Laser Doppler | Cat | Endothelin receptor antagonist | No effect on oligemia |
| 1996 | Meng & Busija | Cranial Window | Rabbit | ROS scavenger | No effect |
| 1996 | Wolf et al. | Laser Doppler | Rat | NOS inhibition | No effect |
| 1997 | Colonna et al. | Microspheres | Rabbit | Specific nNOS inhibition | Decreased CBF by 36% |
| 1997 | Reid et al | NO electrode | Cat | NOS inhibition | Reduced NO production |
| Laser Doppler | NOS inhibition | Reduced hyperemia | |||
| 1998 | Reuter et al. | Laser Doppler | Rat | Acute nasociliary nerve section | No effect |
| Chronic nasociliary nerve section | Reduced CBF by 23% | ||||
| Chronic nasociliary and parasympathetic nerve section | Reduced CBF by 55% | ||||
| Muscarinic receptor blockade | Reduced CBF by 41% | ||||
| CGRP receptor blockade | Reduced CBF by 49% | ||||
| Combined muscarinic and CGRP receptor blockade | Reduces CBF by 69% | ||||
| 1998 | Read & Parsons | NO electrode | Rat | NOS inhibition | Increased NO blocked |
| Laser Doppler | NOS inhibition | No reduction in CBF | |||
| 1998 | Gold et al. | Laser Doppler | Rat | Serotonin receptor blockers | Reduced cerebral hyperemia |
| 1998 | Dreier et al. | Laser Doppler | Rat | NOS inhibition | Reduction in CBF |
| 1999 | Kaube et al. | Laser Doppler | Cat | Amyl nitrite and isoproterenol application | No effect |
| 2000 | Bari et al. | Laser Doppler | Rat | Topical or intravenous capsaicin | Reduced CBF |
| 2000a | Shimizu et al. | Laser Doppler | Rat | Glibenclamide | Enhanced increase in CBF |
| Indomethacin | No effect | ||||
| 2000b | Shimizu et al. | Laser Doppler | Rat | NOS inhibition | No effect on CBF |
| 2000 | Read & Parsons | NO electrode | Rat/Cat | Sumatriptan | Reduced NO levels |
| O2 electrode | Rat/Cat | Sumatriptan | Low O2 levels during CSD | ||
| 2002 | Shimizu et al. | Laser Doppler CBF | Rat | Indomethacin, miconazole, | No effect |
| NOS inhibition | No effect | ||||
| Endothelial “stunning” | No effect | ||||
| Dietary insulin resistance | No effect | ||||
| 2002 | Obrenovitch et al. | NO microdialysis | Rat | NOS inhibition | Prolonged NO production occurs during CSD |
| 2004 | Ayata et al. | Laser Speckle | Mouse | NOS inhibition | Reduced normalization of CBF |
| Specific nNOS inhibition | No effect | ||||
| 2005 | Horiguchi et al. | Laser Doppler | Rat | Adenosine-1 receptor blocker | Enhanced CBF increase |
| 2007 | Shin et al. | Cranial Window | Mouse | Cerebral amyloid deposition | CSD-hypoperfusion unaffected |
| 2008 | Petzold et al. | Brain slices | Mouse, Rat Human | NOS inhibition | CSD threshold maintained by endothelial NO |
CSD, cortical spreading depression; fNIRS, near-infrared spectroscopy; NO, nitric oxide; NOS, nitric oxide synthase; nNOS, neuronal nitric oxide synthase; ROS, reactive oxygen species; CGRP, calcitonin gene-related peptide; CBF, cerebral blood flow; MMP-9, metalloproteinase-9; MCAO, middle cerebral artery occlusion; BBB, blood brain barrier; LPDI, laser-Doppler perfusion imaging; aCSF, artificial cerebral spinal fluid.
Cortical spreading depression has been induced experimentally in animals to study diverse topics such as memory (Case et al., 2002), learning (Koroleva and Bures, 1993), preconditioning (Shen and Gundlach, 1999; Horiguchi et al. 2005a; 2006), and CBF regulation (Table 1). In the presence of a constant initiating presence, such as a surgical sponge saturated with KCl on the intact surface of the dura mater, there is an innate frequency of spontaneous CSDs which is affected by level and/or type of anesthetic (Figure 1) (Horiguchi et al., 2005a) or other pathophysiological conditions (Back et al., 1994). In addition, CSD is thought to be closely associated with some types of migraines in people (Lauritzen, 1987 & 2001; Goadsby, 2005; Dalkara et al., 2006). It also occurs following head trauma and strokes, and exacerbates brain injury in experimental animals and people (Back et al., 1994; Mayevsky et al., 1996; Fabricius et al. 2006; Dreier et al., 2006). The documentation of CSD-like episodes in man has been possible only recently because of the current availability of sensitive imaging technologies and the combined efforts of clinical investigators at multiple centers (Dreier et al., 2006; Fabricius et al., 2006; Hadjikhani et al., 2001; Strong et al., 2007).
Figure 1.
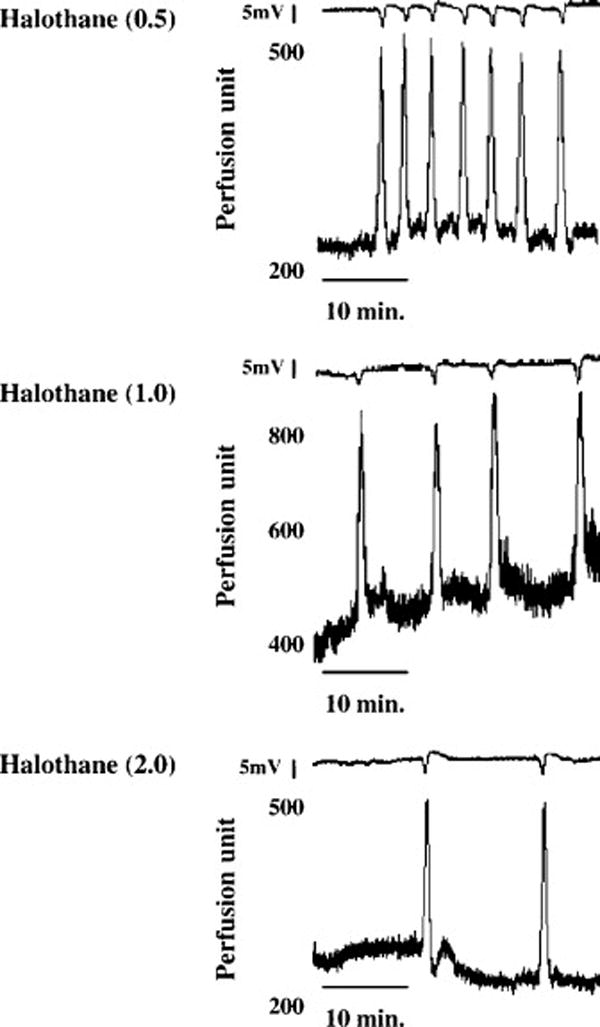
Representative tracings of direct current deflections in mV (smaller tracings) and cerebral blood flow changes (CBF; larger tracings) associated with repeated, spontaneous CSDs. CSDs were elicited by the presence of a surgical sponge soaked with a KCl solution on the dura mater in rats exposed to 0.5% (0.5; top panel), 1.0% (1.0; middle panel), or 2.0% (2.0; bottom panel) halothane. While numbers of CSDs varied from 27 ± 3, 12 ± 2, and 6 ± 1 per minute with 0.5%, 1.0% and 2.0 % halothane exposure, respectively, the duration and amplitude of the DC deflections and the peak changes in CBF were not different among groups (n=8 for each group). From Horiguchi et al. (2005a), with permission.
Of particular interest to many investigators are the dramatic changes in CBF which occur during and following CSD. However, we are unaware of a recent, comprehensive review of the studies which has examined the mechanisms underlying hemodynamic responses caused by CSD. Therefore, the purpose of this review is to critically examine the literature concerning the regulation of cerebral hemodynamics during CSD. The focus of this review will be on the hyperemia that occurs during CSD since this is the most characteristic and reproducible aspect of the cerebral arterial response. In particular, we will address three relevant issues. First, we will interpret the complex, underlying mechanisms of cerebral vascular responses during CSD in light of new information concerning the participation and interactions of diverse cell types composing the neurovascular unit (Xu and Pelligrino, 2007; Iadecola and Nedergaard, 2007; Jackovcevic and Harder, 2007). Second, we will evaluate whether information acquired in experimental animals concerning cerebrovascular control mechanisms during CSD can be extrapolated to humans during normal and pathological conditions such as migraines (Lauritzen, 1987 & 2001; Goadsby, 2005; Dalkara et al., 2006). Third, we will explore the enigmatic role and possible importance of nitric oxide (NO) production during CSD, where this substance is produced by the neurovascular unit (Horiguchi et al., 2005b; Read et al., 1997; Obrenovitch et al., 2002) but apparently does not mediate vasodilation at least in rodents (Shimizu et al., 2002).
2. Defining the “Playing Field” for integrated blood flow responses to CSD
To fully understand the mechanisms involved in regulation of the cerebral circulation during CSD, we must consider the cerebral blood vessels, astroglia, neurons, and perivascular nerves as functionally inter-related components of the neurovascular unit (Hawkins and Davis, 2005). The tight coupling of brain metabolism and blood flow is a well established concept (Roy and Sherrington, 1890; Busija and Leffler, 1987; Busija, et al., 1988) and provides the functional basis for the neurovascular unit. However, the precise inter-relationships among the components of the neurovascular unit are only now being defined for various stimuli such as CSD. While primary cortical neurons can exert a direct effect on cerebral vascular tone, a prominent role of astroglia (Fellin et al., 2004; Filosa et al., 2004; 2006; Mulligan and MacVicar, 2004; Zonta, et al., 2003; Koehler et al., 2006; Lalo et al., 2006) and local circuit neurons (interneurons) (Cauli et al., 2004; Vaucher et al., 2000) in conveying signals between primary neurons and blood vessels has been described recently with the use of several novel imaging approaches. Neurotransmitter released from perivascular nerves can also directly affect cerebral resistance vessels. Neuronal- and astroglial-derived factors act directly on vascular smooth muscle (Domoki et al., 2002; Filosa et al., 2006) or secondarily following production and release of endothelium-dependent dilator or constrictor agents (Murphy et al., 1994).
Neurons affect cerebral vascular tone either directly or indirectly following activation of interneurons and/or astroglia. For example, we and others have shown that application of N-methyl-D-aspartate (NMDA) is able to activate receptors on cortical neurons and produce NO, which apparently diffuses to vascular smooth muscle and dilates cerebral arteries and arterioles without substantial involvement of endothelium or astroglia (reviewed by Busija et al., 2007). In addition, increased neuronal activity leads to production of other metabolites, such as adenosine, which following release into the cerebrospinal fluid (CSF), are able to directly dilate resistance vessels in the cerebral circulation (Phillis, 2004). Synaptically connected interneurons can also adapt local blood flow responses to corresponding metabolic demand following increases in afferent signals from subcortical pathways (Hamel, 2006).
Neuronal interactions with astroglia also participate in CBF regulation. Virtually the entire parenchymal segment of the cerebral vasculature is surrounded by astroglial endfeet, and this anatomical arrangement, with neurons also in contact with the astroglia, is thought to be an important means for adjusting CBF to local metabolic activity (Xu and Pelligrino, 2007; Iadecola and Nedergaard, 2007; Jackovcevic and Harder, 2007). In particular, astroglial swelling has been described as an early event in the progression of CSD, at least in mice, and this event may be involved in the subsequent cerebral vascular responses in this species (Takano et al., 2007; Tomita et al., 2005). While the surface of pial cerebral arteries and arterioles are not usually thought to be invested with astroglia (Mercier and Hatton, 2002), they are positioned over the glia limitans (Niermann et al., 2001) and thus can be influenced by glial-derived factors released into the CSF which is in contact with the blood vessels. Flushing of the cortical surface with appropriately gassed artificial CSF (aCSF) does not normally affect basal pial arteriolar tone, thus suggesting a minimal contribution of vasoactive influences from CSF under quiescent conditions in anesthetized animals. On the other hand, the ubiquitous positioning of the astroglia among cerebral blood vessels and neurons and the well known interactions involving these cells appear optimal for rapid and integrated control of local vascular tone in order to meet metabolic demands. Bulk CSF could also act as a “sink” and thus dilute the local vasodilator effects of metabolites released from the cortical surface during CSD.
Cortically-derived astroglial cells are avid producers of prostanoids (prostaglandins and thromboxane) (Nam et al., 1996; Thore et al., 1994; 1996) and cerebral arteries normally located close to cortical astroglia are responsive to even small concentrations of dilator and constrictor prostanoids which are present in CSF (Busija and Leffler, 2002; Leffler and Busija, 1985; Wagerle and Busija, 1990). In particular, the profile of prostanoids produced by cerebral arteries varies considerably from that produced by cortical astrogia of the same species (Busija, 1997; 2002) and thus the CSF bathing the cerebral arteries contains substantial amounts of vasoactive prostanoids arising from the brain parenchyma. Other studies have shown that similar to neurons, astroglia can produce and release a wide range of vasodilator substances such as NO, carbon monoxide (CO), adenosine, hydrogen ion, potassium ion, lipoxygenase products, and P-450 monooxygenase products (Bhardwaj et al., 2002; Zonta et al., 2003; Filosa et al., 2006; Koehler et al., 2006; Leffler et al., 2005).
Further complicating the situation is the presence of many types of perivascular innervation which may mediate or modulate cortical influences on cerebral resistance vessels. The cerebral arteries are richly innervated by sympathetic (Busija, 1996), parasympathetic (Goadsby and Hoskin, 1994; Dauphin and MacKenzie, 1995; El-Assouad and Tayebati, 2002) and sensory (Edvinsson et al., 1987; 1998; 2001) nerve fibers which release a large array of different neurotransmitters which can affect cerebrovascular tone via direct actions on smooth muscle (Edvinsson et al., 1987; 1998) or indirectly via endothelium (Rosenblum et al., 1993). In general, activation of sympathetic nerves causes cerebrovascular constriction while activation of parasympathetic and sensory nerves promotes dilator responses (Busija, 1996). Perivascular nerves can be activated centrally in a traditional manner involving pre-ganglionic pathways (Goadsby and Hoskin, 1994; Edvinsson et al., 1998), or via direct activation of post-ganglionic fibers by local factors such as potassium ions (Reuter et al., 1998). The presence of functional perivascular innervation provides the underlying basis of dramatic deviation from the concept of tight coupling of blood flow and metabolism, such that changes in cerebrovascular resistance can take place prior to and/or be independent of changes in metabolic demand. It is also possible that sustained activation of perivascular nerves could affect the slope of the blood flow/metabolism relationship.
3. Studies of cerebral hemodynamics during CSD
3.1. Responses during normal conditions
Leão (1944) was the first investigator to demonstrate that spreading depression is associated with dramatic changes in the tone of the cortical resistance vasculature. In anesthetized rabbits, he was able to show pronounced but transient dilation of pial arterioles to CSD. The veracity of these early observations by Leão (1944) on pial arteries and arterioles is such that they are very similar to more recent findings made in the same species over 40 years later (Shibata et al., 1990). The pioneering research of Leão (1944) was soon followed by cerebral vascular studies in the rabbit and cat by Van Harreveld and Stamm (1952) and Van Harreveld and Ochs (1956), respectively. During this period, the characteristics and mechanisms of CSD were examined in detail (Grafstein, 1956; Van Harreveld, 1959).
Following the development of new approaches to characterize cerebral vascular responses, Hansen et al. (1980) and Mies and Paschen (1981) were able to show using radioactively labeled iodoantipyrine that CSD is associated with major increases in CBF in anesthetized rats. Over the next two decades, other investigators using a variety of methods were able to confirm in many species, including rats (Lauritzen, 1984; Wahl et al., 1987), rabbits (Shibata et al., 1990; 1992), monkeys (Yokota et al., 2002), and cats (Wahl et al., 1987), that CSD is associated with transient dilation of cerebral arteries and arterioles and increased CBF (Table 1). Increased CBF occurs during CSD in awake as well as anesthetized animals, although variable CBF responses have been reported in non-anesthetized rats (Duckrow, 1991; Shimazawa and Hara, 1996; Sonn and Mayevsky, 2006). Depending upon the experimental conditions and species studied, a mild, very transient reduction in CBF has been reported to precede cerebral hyperemia in rodents (Duckrow, 1991; Fabricius et al., 1995; Ayata et al., 2004), and a sustained suppression of CBF or oligemia can be present afterwards in some species (Table 1). The mouse is perhaps unique in that it does not show a frank increase in CBF over baseline values during CSD, but rather demonstrates a cerebral hypoperfusion initially with a gradual return to baseline (Ayata et al., 2004). The authors of this report have suggested that enhanced sensitivity to elevated K+, which is elevated greatly during CSD in brain extracellular fluid, may be a unique contributing factor, since preconstriction with KCl of isolated arteries reduced dilation to acetylcholine in mouse but not in rat cerebral arteries. An additional finding that may support the concept that cerebrovascular control mechanisms are different in mice than in most other common laboratory animals is the report that calcitonin gene related peptide (CGRP) is an endothelium-dependent rather than a direct smooth muscle-acting dilator agent in mouse cerebral arteries (Rosenblum et al., 1993). A biphasic cerebral vascular response has also been reported in the mouse cortex during CSD, where an initial dilation is followed first by an arterial constriction and then subsequently by a secondary dilation (Brennan et al., 2007). In contrast to these two previous reports, Takano et al. (2007) reported that in mouse CSD elicited an initial dilation of pial and parenchymal arterioles followed by a sustained constriction. The reasons for the variations in findings for mice are unclear at this time, but may reflect experimental differences and technical difficulties associated with the study of these very small animals. Recently, Takano et al. (2007) have reported that severe hypoxia and neuronal swelling accompany CSD in mouse cortex, and it is possible that these features are unique to mouse and contribute to the combination of cerebral vasoconstriction and slow progression of spreading depression across the cortical surface often described in this species. In contrast, Wolf et al. (1996) failed to show development of cortical anoxia during CSD in rats. It seems unlikely that localized tissue anoxia or hypoxia is present in most species demonstrating a robust increase in CBF during CSD. Similar to studies in mice, Chuquet et al. (2007) in P15 rats were able to show pial and parenchymal arteriolar constriction, and reduction in CBF, at the onset of CSD. The cerebral vascular constriction in neontatal rats was followed by dilation of pial arterioles while parenchymal arterioles returned only to baseline diameters. We are unaware of studies of hemodynamic changes in healthy cortex of man with CSD, but a paradoxical spreading oligemia usually without a preceding hyperemia has been described with the onset of migraines with accompanying aura (Woods et al., 1994; Hadjikhani et al., 2001). While a CSD-like component as suggested by the presence of aura occurs in approximately 30% of patients experiencing migraines (Rasmussen et al., 1991), it cannot be ruled out that other acute and chronic neurological events modify the overall cerebrovascular responses to this clinical condition. Thus, all laboratory species that have been studied except the mouse show the typical pronounced cerebral hyperemia or vasodilation during CSD. However, both mice and rats have been reported to sometimes show the initial, short-lived hypoperfusion. The prolonged oligemia following CSD has been reported to regularly occur in rats and mice (Brennan et al., 2007; Tokano et al., 2007) and in some, but not all, rabbits studied (Shibata et al., 1992).
The increase in CBF with CSD shows a typical response when there is a rapid rise with little or no plateau before a rapid restoration of baseline blood flow, with the entire hyperemic phase at any given location lasting approximately 90 seconds (Figure 1) (Shibata et al., 1990). The onset of the increase in pial arteriolar diameter and rise in CBF is spatially associated with the location of the CSD, defined with DC electrodes in the rabbit (Shibata et al., 1990; 1992), but independent responses of pial arterioles and parenchymal blood flow during CSD has been described recently with confocal imaging methods in the P15 rat (Chuquet et al., 2007). Fabricius et al. (1997) showed a heterogeneous blood flow response based upon depth of the cortex in rats with CSD. Brennan et al. (2007) have suggested that the pial arteriolar dilation advances faster than the parenchymal depolarization across the cortical surface. It is possible that factors such as greater cellular density of the cortex, method of inducing CSD, enhanced metabolic rate, and/or larger baseline cortical blood flow in the rodent compared to the rabbit lead to these observed differences. Similar to our previous report in the rabbit (Shibata et al., 1990), Brennan et al. (2007) have shown in rats that CSDs repeated in quick succession can lead to elimination of pial arteriolar dilation during one or more CSDs in a series. Another feature of CSDs is that the hyperemia on the cortex ipsilateral to the spreading depression is spatially and temporally related to a modest pial arteriolar constriction on the contralateral cerebral cortex (Shibata et al., 1990). CSD apparently does not jump to adjacent gyri across a sulcus even in lissencephalic species such as the rabbit despite major increases in extracellular levels of ions and neurotransmitters in the CSD affected gyrus, and similar cerebral vascular dilations can be evoked with CSDs progressing from rostral to ventral cortex and vice versa (Shibata et al., 1990). In contrast to our findings (Shibata et al., 1990), Brennan et al. (2007) reported that pial arteriolar dilation occurs in cortical locations unaffected by CSD in mouse. The dilator responses in pial arterioles are size dependent, with smaller arterioles showing a slightly greater dilation than larger arterioles (Shibata et al., 1990; Chuquet et al., 2007). Small veins also dilate during CSD while larger ones do not, and venous responses appear to be secondary to diameter changes in resistance blood vessels (Van Harreveld and Ochs, 1957; Shibata et al., 1990; Tokano et al., 2007 Chuquet et al. 2007).
The shape and magnitude of the cerebral arteriolar or CBF responses can be consistent with repeated CSDs or vary with time. For example, Fabricius et al. (1995) have shown in the rat that each CBF response has a varying waveform, while others have shown similar CBF responses with repeated CSDs in the rat (Bari et al., 2000; Reuter et al., 1998) or rabbit (Shibata et al., 1990). Often the first CBF response in rats can be unique but the subsequent ones are very similar to each other (Bari et al., 2000; Reuter et al., 1998; Shimizu et al., 2002). The reasons for these different cerebral vascular responses are unknown, but the physiological status of the animals as well as the mode of initiation of CSD is probably a relevant factor. Read et al. (1997) reported that NO production was much greater with the initial CSD, and then fell to a lower, but constant level during the next several CSDs, and this finding is consistent with changes in cerebrovascular responses with multiple CSDs. Fabricius et al. (1995) reported variable CBF responses in halothane anesthetized rats with repeated needle stabs to the cortex. Repeated needle punctures of the cortex with or without KCl injections may damage the surrounding tissues, and thereby alter CSD characteristics. In addition, repeated CSDs may continually alter the status of the neurovascular unit because of the increased metabolic load or because of depletion of substrates or neurotransmitters. However, we found that repeated KCl injections by needle insertion leads to reproducible blood flow responses to multiple CSDs in the urethane-anesthetized rabbit (Shibata et al., 1990), possibly due to factors such as a larger brain size and lower brain metabolic rate in this species compared to the rat. In addition, we have found that application of a surgical sponge saturated with KCl over the intact dura initiates a series of CSDs which show very similar CBF waveforms over at least an hour’s duration despite differences in the numbers of CSDs due to the alterations of the depth of halothane anesthesia in rats (Figure 1) (Horiguchi et al., 2005a). Under these circumstances, there is an innate frequency of spontaneous CSDs which is dependent upon the experimental conditions. Richter and Lehmenkühler (1993) compared KCl application and needle prick and found that each approach had differential effects on the amount of cortex affected as well as the CSD characteristics. On the other hand, Brennan et al. (2007) reported that frequent, repetitive CSD induced by placing KCl crystals on the cortical surface in rats initially leads to vasodilation followed by constriction, but eventually the arterioles became persistently dilated with reduced vascular responsiveness.
Associated with the delayed oligemia in rats is a reduction in cerebrovascular dilator responsiveness to stimuli such as arterial hypercapnia, which can be demonstrated both in vivo (Lauritzen, 1987; Lacombe et al., 1992; Florence et al., 1994; Fabricius) and in isolated middle cerebral arteries removed after CSD (Seitz et al., 2004). The altered cerebrovascular responses to stimuli such as arterial hypercapnia occur in spite of continued CBF responsiveness to multiple CSDs that occur during this period (Horiguchi et al., 2005a). In people also, a reduced cerebrovascular response to arterial hypercapnia has been described during or immediately following migraine attacks (Lauritzen et al., 1983). The reduced responsiveness following CSD can be reversed in rats by stimulators of the cyclic GMP system or by administration of L-arginine (Fabricius et al., 1995; Zhang et al., 2002; Scheckenbach et al., 2006), thereby implying that impairment of the NO synthase (NOS) system is responsible for a reduction in dilator responses. This speculation is consistent with the reduced cerebral vascular responsiveness to arterial hypercapnia in rats, in which NO plays a prominent role (Fabricius and Laurizten, 1994). In the rabbit, in which some but not all animals show post-CSD cerebral vasoconstriction (Shibata et al., 1992), cerebral vascular responsiveness is intact following CSD (Busija and Meng, 1993). Additionally, in the piglet brain, which is resistant to CSD, transient depolarization by superfusion with topical KCl under the entire cranial window, followed by recovery, does not prevent normal dilator responses to several stimuli (Domoki et al., 1999a).
3.2. Comparison of CBF responses between people and animals
Findings, based upon available information, showed that responses of the human cerebral circulation to stimuli such as arterial blood hypercapnia, changes in arterial blood pressure, and changes in metabolism are similar in direction and magnitude to those determined in a variety of commonly used experimental animals (Katona et al., 2006; Meadows et al., 2005; Zhang et al., 2004; Drake and Iadecola, 2007). More specific to this discussion, the previously identified dilator agents released by other species during CSD, including CGRP, NO, and acetylcholine, apparently have similar dilator effects on human cerebral arteries (Edvinsson et al., 1987; Jansen-Olesen et al., 1996; Hansen et al., 2007; Sams et al., 2000; Zvan et al., 2002). On the other hand, the usual dramatic rise in CBF during CSD, which occurs in diverse species such as the monkey, cat, rabbit, and rat (Table 1), has been difficult to be detected consistently in the human cortex during CSD-like events (Bartonlini et al., 2005). The principal reason for this deficiency is the lack of studies due to the ethical and practical limitations of examining CBF responses during experimentally induced CSD in normal, human cerebral cortex. Initiating spreading depression in human cortex may lead to localized tissue damage, transient changes in cortical function, and alterations in gene expression (Caggiano et al., 1996a; 1996b; Shen and Gundlach, 1999; Horiguchi et al. 2005a; 2006). While spreading depression has been shown to occur in human neocortical slices (Gorji et al., 2001), responses of arteries and arterioles apparently have not been evaluated in this preparation.
A variety of electrophysiological, visual and psychological events have indicated that CSD-like events occur during the initiation of migraines (Bartolini et al., 2005; Lauritzen et al., 1983; Lauritzen, 2001; Dalkara et al., 2006; Mayevsky et al., 1996), and usually, but not always, a cerebral oligemia is the only cerebral vascular response consistently detected in these circumstances (Lauritzen et al., 1983; Olesen et al., 1981; Woods et al., 1994). From this perspective, it is possible that the human cortex displays CBF responsiveness more similar to that seen in the mouse during CSD than that observed in other species. Underlying reasons for an atypical cerebral vascular response may be due to pathological changes in the cortical thickness reported in patients with chronic migraines (DaSilva et al., 2007) and the relatively sparse presence of CGRP in perivascular nerves in cerebral arteries of people (Edvinsson et al., 1994).
Although CSDs may be initiated following strokes in people (Strong et al., 2007), the hemodynamic responses in compromised neural tissue may not reflect the normal condition as we have shown in rats (Shimizu et al., 2000b) and Strong et al. (2007) have shown in cats following transient brain ischemia. Prior CSDs are able to cause prolonged suppression of certain vascular responses in some situations in rats and rabbits, and also influence cortical levels of vaso-relevant proteins such as cyclooxygenase (COX)-2 (Caggiano et al., 1996a; 1996b; Horiguchi et al., 2006) and NO synthase (Shen and Gundlach, 1999), and so it would not be surprising to find altered cerebrovascular responsiveness to migraines in people. Another important consideration that has not been previously addressed is the effect of basal CBF and other factors on the hemodynamic response to CSD. A number of factors, such as level and type of anesthesia, resting CBF, and the presence of various pharmacological agents, are able to affect the observed changes in CBF with CSD in experimental animals as well as in people (Duckrow, 1991; Lauritzen, 1987; Sonn and Mayevsky, 2001; 2006; Shimazawa and Hara, 1996). For example, Piper and Lambert (1996) have shown anesthetic-type dependent effects on CSD initiation, and suggested the use of halothane in people following cerebral insults may interfere with detection of CSDs. Thus, in the absence of any direct experimental data, it may be misleading to overly interpret data from limited human studies in which neuronal injury has occurred due to strokes or concussive brain injury, or when the cortex has experienced previous migraines over the course of months or years. Also unstudied are the effects of gender on cerebral hemodynamic during CSD. Gender has many effects which are able to affect CBF responses to a variety of stimuli (Chrissobolis and Sobey, 2004; Li et al., 2004). Therefore, the cerebral hemodynamic consequences of CSD in normal human brain remain largely unknown.
3.3. Altered hemodynamic responses during pathological conditions
Since the cerebral hemodynamic changes during CSD are influenced by physiological factors such as basal CBF (Sonn and Mayevsky, 2001; Lauritzen, 1987), the method of inducing CSD (Fabricius et al., 1995; Shibata et al., 1990; Horiguchi et al., 2005a), and the level and type of anesthetic used (Horiguchi et al., 2005a), it is not surprising that pathological conditions also influence the response of the cerebral circulation to CSD. In one of the first such studies, Shimizu et al. (2000b) showed that the presence of cortical infarcts following transient, middle cerebral artery occlusion (MCAO) reduced or eliminated CBF increases during CSD. In these animals, the MCA was occluded for 75 minutes and blood flow responses were determined 24 hours later. Despite the presence of subcortical or cortical infarcts, normal DC deflections could be evoked suggesting energy metabolism had recovered sufficiently 1 day after MCAO to sustain multiple CSDs. However, an increase in CBF did not occur at the center of the infarct, although a reduced hyperemia occurred in the penumbral area. At both penumbral and infarcted sites the baseline CBF was greatly elevated over non-infarcted levels, and this situation may have lead to a reduced or eliminated cerebral hyperemia as the maximal CBF level was reached. Strong et al. (2007) has reported similar findings with spontaneous spreading depolarization in infarcted cortex in the cat. A recent study by Sukhotinsky et al. (2008) supports the concept that prior vasodilation adversely affects cerebral hyperemia during CSD. In addition, this group has shown that CBF in the penumbral region can be supported during cortical depolarization with mild hypertension (Shin et al., 2008). It is also possible that ischemia leads to depletion of substrates or neurotransmitters necessary for the full expression of the dilator response (Bari and Paprika, 2000). When only subcortical infarcts were present, baseline CBF was normal and cortical hemodynamic responses to CSD were intact. Similar findings were reported following occlusion of cerebral veins as a means to induce cortical infarction (Osuka et al., 2000). Kocher (1990) also found that the typical CBF responses to CSD continued to take place for the 3-6 hour reperfusion period following 30 minutes of global ischemia. In contrast to ischemic strokes, which cause the initiation of CSD into penumbral and normal tissue, with variable CBF responses (Shimizu et al., 2000b), the presence of hemoglobin, which would occur with hemorrhagic stroke, causes a spreading ischemia with cerebral vasoconstriction during pathological models of CSD, at least in rats (Petzold et al., 2003; Drierer et al., 2006).
Cerebrovascular responses during CSD have been studied in only a few other diseases. However, CSD-induced cerebrovascular dilation is unaffected by metabolic disease states, such as hyperglycemia (Wolf et al., 1997) or mild, nutritionally induced-insulin resistance (Shimizu et al., 2002), even though these diseases normally alter vascular responsiveness to dilator agents associated with CSD (Erdős et al., 2004; 2006). Cerebrovascular responses to CSD apparently are also resistant to alcohol administration in rats (Sonn and Mayevsky, 2001).
4. Mechanisms of cerebral hyperemia during CSD
4.1. Dilator Mechanisms
Given our current knowledge about the regulation of CBF in experimental animals, there are a number of dilator stimuli arising from cerebral arteries and parenchyma, including neurotransmitters from sensory and parasympathetic nerves, NO, adenosine, reactive oxygen radicals (ROS), potassium ion, glutamate, and prostanoids, which could cause or modify cerebral arterial dilation and hyperemia during CSD.
4.1.1. Nitric oxide
A role for NO was shown first by Goadsby et al. (1992) in the cat, in which a NOS inhibitor completely abolished the increase in CBF seen with this species during CSD. Additional studies in cats (Wahl et al., 1994; Read et al., 1997) and rabbits (Colonna et al., 1994b) supported these initial reports and demonstrated that NOS inhibition reduced cerebral arterial dilation during CSD by approximately 50%. However, the reason for the discrepancy between complete and partial suppression of cerebral vasodilation during CSD by NOS inhibition for the cat reported by Goadsby et al. (1992) and others (Wahl et al., 1994; Read et al,. 1997), respectively, is not clear. While it could be argued that the underlying basis for the difference was due to the measurement of CBF in one study and pial arterial diameter in the others, parallel studies evaluating the relative importance of NO involvement in cerebrovascular changes during CSD with the cranial window and microsphere methods in rabbits in the same laboratory have led to consistent conclusions (Colonna et al., 1994b; 1997). Nonetheless, the source of the NO appears to be neuronal or glial in origin, since selective inhibition of neuronal NOS had similar effects to general inhibition of NOS (Colonna et al., 1997). However, it has not been determined whether the source of the NO is from cortical neurons or astroglia, or from perivascular nerves since NOS is associated with several types of innervation of cerebral arteries (Edvinsson et al., 1998; 2001; Goadsby et al., 1996; Nozaki et al., 1993). While NOS is co-localized with CGRP in sensory nerve endings, activation of trigeminal pathways apparently releases little or no vasoactive NO despite major cerebral vasodilation (Edvinsson et al., 1998). On the other hand, astroglia may be involved in mediating the NO-dependent component of cerebrovascular dilation to arterial hypercapnia in the rat (Xu et al., 2004). The relative contribution of NO is dependent upon the physiological status of the brain, since inhibition of prostaglandin synthesis with indomethacin prevented detection of a NO component in the cerebral vascular dilator response to CSD in rabbits (Meng et al., 1995).
Despite early, preliminary indications in rats, numerous subsequent studies have failed to support an important contribution of NO to cerebral hyperemia in rats. Thus, while NO appears to be produced in significant amounts by cortex during CSD in this species (Obrenovitch et al., 2002; Horiguchi et al., 2005; Read et al., 1997), there apparently is no substantial contribution which would counter-act the initial, brief hypoperfusion (Duckrow, 1993; Fabricius et al., 1995) or promote the subsequent hyperemia (Table 1). Nonetheless, despite the lack of a significant NO contribution to CSD-induced cerebral hyperemia, administration of exogenous NO or cGMP activators are able to restore a major part of the post-CSD dilation to hypercapnia or other vasodilators (Fabricius et al., 1995; Zhang et al., 2002; Scheckenbach et al., 2006). Additionally, pharmacological blockade of NOS or degradation of NO with hemoglobin leads to a spreading ischemia rather than hyperemia following induction of a CSD-like event (Windmüller et al., 2005). Thus, the role of NO in vascular responses to CSD is complex in rats, and does not involve direct vasodilator actions that would lead to increased CBF, but rather would promote other, potentially beneficial effects in brain (see below).
4.1.1.1. Paradoxical roles of NO during CSD in rats
The lack of a significant impact of NO on cerebral hyperemia during CSD in the rat is surprising because of its potency in promoting cerebral vascular dilation and because NO is a mediator of CBF increases in this species during a variety of experimental conditions involving activation of the cerebral cortex (Dreier et al., 1995; Busija et al., 2007). Iadecola et al. (1993) and others (Estada et al., 1993) have shown that cerebral arterioles including those in the parenchyma are innervated by NOS containing nerves from the sphenopalatine ganglia. It is possible, but unexplored, that NO effects on cerebral arteries are blocked by another substance produced during CSD or because of the activation of a signaling cascade which interferes with functioning of vascular smooth muscle guanylate cyclase. For example, CO from heme oxygenase-2 in cerebral arteries has been shown to reduce vascular effects of NO (Ishikawa et al., 2005). Additionally, serotonin, which apparently is released near cerebral arteries during CSD (Gold et al., 1998), is able to suppress release of NO in rats (Read et al., 1999). Nonetheless, NO is produced in substantial quantities by brain during CSD and have other non-vascular effects on the cortex such as restoration of the physiological status of the brain (Obrenovitch et al., 2002) and the promotion of preconditioning against ischemic stress (Matsushima et al., 1996; Horiguchi et al., 2005b). Petzold et al. (2008) recently have reported NO derived from endothelial NOS maintains the threshold for initiating CSD in rodent and human brains. Additionally, NO produced by the cortex or cerebral vasculature may affect CGRP release in the dura mater (Strecker et al., 2002) and thus modulate the degree of pain associated with migraines.
In recent studies we have shown that repetitive CSDs induced in rat cortex results in NO-induced preconditioning against neural damage caused by MCAO (Horiguchi et al., 2005b; 2006). Thus, pretreatment with N(G)-nitro-l-arginine methyl ester (L – NAME) abolishes preconditioning by CSD, probably by reducing the delayed enhancement of cortical cyclooxygenase-2 (COX-2) expression. Other investigators have shown that levels of COX-2 increase after CSD in the rat (Cui et al., 2007) and in other species such as primates (Yokota et al., 2003). Rather than being a final mediator of preconditioning , enhanced COX-2 expression and production of prostaglandins of the J-series appear to be final steps in preparing the cortex to withstand potentially lethal ischemia in experimental strokes (Horiguchi et al., 2005b). Whether similar events take place in human cerebral cortex following CSD-like events such as migraines or following localized depolarizations associated with transient ischemic events is currently unknown. Also unclear is whether altered gene expression following CSD alters subsequent cortical or vascular responses to CSD. Finally, it appears that while NO does not mediate the increase in CBF during CSD in rats, the presence of NO is able to prevent cerebral vascular vasoconstriction and spreading ischemia in the rat cortex under some CSD-like conditions (Windmüller et al., 2005).
4.1.2. Perivascular nerves
The involvement of a more universally accepted dilator agent was shown simultaneously by Colonna et al. (1994a) and Wahl et al. (1994), who were able to demonstrate that pharmacological blockade of the CGRP receptors attenuated cerebral hyperemia by approximately 40-50% in rabbits and cats, respectively. Similar results were obtained in the rat from studies involving pharmacological blockade of CGRP receptors (Reuter et al., 1998), destruction of sensory nerves with capsaicin (Bari and Paprika, 2000; Bari et al., 2000), and chronic interruption of neural pathways supplying sensory nerves containing CGRP to cerebral arteries (Reuter et al., 1998). Thus, chronic sensory denervation achieved by the sectioning of the trigeminal nerves supplying cerebral arteries reduces CBF increases with CSD (Reuter et al., 1998), thereby providing strong evidence that the predominant source of dilator stimuli is from perivascular nerves. Chemical destruction of the capsaicin-sensitive trigeminal nerve endings resulted in time-dependent effects on CBF during CSD, such that reduced cerebral vasodilation soon after treatment was followed by development of vascular supersensitivity to CSD (Bari and Paprika, 2000; Bari et al., 2000). Trigeminal pathways are also important in mediating dilator responses in the cerebral circulation under other physiological and pathological conditions (Wei et al., 1992; Macfarlane et al., 1991). Thus, in all species studied, approximately one-half each of the dilation to CSD comes from CGRP (Bari et al., 2000; Colonna et al., 1994a; Reuter et al., 1998; Wahl et al., 1994), with a similar portion coming from NO (Colonna et al., 1994b; Wahl et al., 1994) in the rabbit and cat and with a similar percentage coming from perivascular parasympathetic nerves in the rat (Reuter et al., 1998). It is not surprising that dilator responses following activation of trigeminal fibers in the rat involve primarily the actions of CGRP. While NOS is present in trigeminal nerve endings, its distribution is limited and the dilator responses to trigeminal nerve stimulation appear to involve smooth muscle actions primarily of CGRP and not NO (Edvinsson et al., 1998). The involvement of additional dilator factors originating from the parasympathetic innervation in the cat and rabbit hasn’t been examined. An unusual finding is that while parasympathetic denervation and blockade of muscarinic receptors with atropine reduce cerebral hyperemia with CSD in the rat (Reuter et al., 1998) these effects did not involve production and effects of endothelial-derived NO (Shimizu et al., 2002) as seen during direct activation of parasympathetic nerves supplying cerebral arteries (Goadsby et al., 1996b). Parasympathetic activation or acetylcholine application also typically dilates arteries from peripheral circulations via production of NO from endothelial NOS (Toda and Okamura, 2003).
It appears that other dilator substances which may derive from perivascular nerves in rats, such as serotonin (Gold et al., 1998), make modest contributions to the overall cerebrovascular response to CSD. However, it is untested whether perivascular sympathetic nerves provide counteracting constrictor influences on cerebral arteries during CSD.
In addition to directly mediating much of the cerebral vasodilation by local release of CGRP, stimulation of trigeminal afferents during CSD leads to vasodilation and neurogenic inflammation in the meninges via axon-reflex activation of collateral trigeminal branches and central-reflex activation of parasympathetic nerves, respectively (Bolay et al., 2002; Dalkara et al., 2006; Edvinsson, 2007). Intravenous CGRP administration can also trigger headaches or migraines in migraine patients (Iversen, 2001; Lassen et al., 2002; 2008). It is thought that meningeal effects lead to the head pain associated with migraines with aura, and that the optical distortions represent CSD over the visual cortex. However, experimental induction of CSDs in awake rats apparently does not lead to aversive behaviors (Koroleva and Bures, 1993) and thus whether it causes headaches in this species is not known. An earlier report by Lambert and Michalicek (1994) in the cat indicated that CSD was associated with decreased meningeal blood flow, and it is unclear whether species differences or other aspects of the experiments affected the results. Although increased vascular permeability of meningeal blood vessels during migraines has been suggested to occur in some patients (reviewed by Dalkara et al., 2006), we are unaware of direct evidence that migraine-associated vasodilation in the meninges occurs in people. Antagonists against CGRP receptors have been reported to be effective against migraines in people (Olesen et al., 2004; Goadsby, 2005; Ho et al., 2008). Furthermore, other anti-migraine medicines such as dihydroergotamine and sumatriptan may provide beneficial effects by inhibiting activation or actions of the trigeminal nerves (Buzzi et al., 1991). Lastly, NO donors have been reported to induce headaches in people (Christiansen et al., 1999; Iversen et al., 2001) perhaps through the release of CGRP near cerebral arteries (Wei et al., 1992) with resultant effects on meningeal blood vessels. Nitric oxide produced by the cortex or cerebral vasculature also may directly affect CGRP release in the overlying dura mater (Strecker et al., 2002). Although usually not directly examined, it appears that removal of the dura mater in cranial window preparations, which would eliminate potential axonal and reflexive interactions between pial and dura mater nerves, has little detectable effect on cerebral hemodynamics during CSD (Shibata et al., 1990).
4.1.3. Potassium ions and glutamate
Potassium ions and glutamate are released into cortical interstitial fluid during CSD and both have been implicated in the propagation of spreading depression (Grafstein, 1956; Van Harreveld, 1959). Potassium ions exert potent vasoactive effects on cerebral arteries (Schuh-Hofer et al., 2001), but their direct participation in cerebral vascular responses during CSD is unclear since the predominant mediator of dilator effects appear to be the activation of perivascular nerves rather than the direct influence of potassium ions diffusing to vascular smooth muscle. However, potassium ions could affect CBF responses during CSD in two other ways. First, elevated potassium ion levels during CSD could activate perivascular nerves and thus indirectly induce cerebral vascular responses (see below and Figure 1). Second, elevated potassium ion levels during CSD may reduce vascular responses to dilator agents such as acetylcholine released from parasympathetic nerves (Ayata et al., 2004), at least in mice.
Glutamate and specific glutamate receptor agonists such as NMDA are potent dilator agents when applied to intact brain (Busija and Leffler, 1989; Busija et al., 2007), but there is no evidence that glutamate can exert direct vascular effects on the cerebral arteries of animal species used in CSD studies. The available information indicates that cerebral vascular dilator effects of glutamate and NMDA involve NO production from cortical neurons and/or astroglia (Busija et al., 2007) or other events not involving NO production subsequent to the activation of perivascular astroglia (Filosa et al., 2006; Straub and Nelson, 2007). The direct production of NO from cortical neurons and/or astroglia probably is not a major factor in rats because of the lack of inhibition of the CBF response by L-NAME in this species. However, glutamate-induced NO production in cortical cells may be an important vasodilator mechanism in cats and rabbits, although it is not possible to rule out perivascular nerves as the source of NO. Alternatively, Nelson and colleagues (Filosa et al., 2006; Straub and Nelson, 2007) have suggested that increased extracellular levels of glutamate will activate metabotropic glutamate receptors on astrocytes, which in turn initiates the spread of increased calcium levels from the cytosole through processes to the astrocytic endfeet. At the astrocytic endfeet, which are in close proximity to the pial and parenchymal resistance vessels, it is suggested that the increased cytosolic calcium level will: 1) Activate PLA2 with subsequent release and metabolism of arachidonic acid to prostanoids and epoxyeicosatrienoic acids (EETs), and/or 2) Promote the release of potassium from endfeet via stimulation of large conductance calcium-activated potassium (BK) channels. However, at least in the rat, the involvement of such mechanisms in promoting dilator responses is unlikely. In this species, administration of indomethacin (Shimizu et al., 2000a) as a blocker of cyclooxygenase-induced production of prostanoids, miconazole (Shimizu et al., 2002) as a blocker of epoxygenase-induced production of 11,12 and 14, 15 EETs (Alkayed et al., 1996), glibenclamide (Horiguchi et al., 2005; Shimizu et al., 2000a) as a blocker of ATP-sensitive potassium channels which may mediate dilator prostanoid effects, and charybdotoxin (Shimizu et al., 2000a) as a blocker of BK channels fails to reduce cerebrovascular dilation during CSD. Even in the rabbit, indomethacin potentiates rather than decreases CBF responses (Shibata et al., 1992). Finally, Iijima et al. (1998) have reported that increases in brain extracellular glutamate levels during CSD were not well correlated with cerebral hyperemia.
4.1.4. Reactive oxygen species
Only a few studies have examined the roles of ROS and adenosine in mediating cerebral hyperemia during CSD and the results are negative. ROS scavengers fail to alter CSD-induced hemodynamics in rat and rabbits (Duckrow and Beard, 1992; Meng and Busija, 1996). This lack of a significant role of ROS in mediating changes in cerebral vascular tone or CBF during CSD is surprising because it would be expected that the cellular disruption under these circumstances should activate ROS-generating pathways. While modest elevations in ROS levels may occur during CSD and are insufficient in magnitude to affect cerebral vascular tone, we cannot rule out the activation of ROS-dependent signaling pathways which could promote the development of neuronal preconditioning (Nagy et al., 2004).
4.2. Counteracting constrictor influences
Lauritzen (1987) was the first to examine the possible involvement of prostanoids on the cerebral arterial dilation to CSD by administering a blocker of cyclooxygenase. For example, he found that inhibition of the cyclooxygenase pathway with indomethacin enhanced dilation in rats. Furthermore, CSD in rats is associated with increased amounts of free arachidonic acid in cortex, and the availability of arachidonic acid is the rate limiting step in the production of prostanoids (Lauritzen et al., 1990). However, it is unclear whether prostanoids modulate CBF responses in rats. First, it seems likely that reduced CBF, which is a characteristic of indomethacin in rats, leads to the appearance of an enhanced response since it occurred also during arterial hypocapnia and other CBF reducing strategies (Lauritzen, 1987; Sonn and Myevsky, 2006). Second, many subsequent investigators have failed to show a significant increase in the cerebral hyperemia during CSD following indomethacin administration in rats (Table 1).
In contrast, the evidence is stronger for an important cyclooxygenase-dependent component in the rabbit capable of opposing cerebral vasodilation (Shibata et al., 1991b). In this species, indomethacin application enhances arteriolar dilation and augments CBF increases (Shibata et al., 1991b), and eliminates the post-CSD vasoconstriction (Shibata et al., 1992). In contrast to the situation in rats, baseline arteriolar diameter or CBF failed to show a significant reduction following indomethacin administration in rabbits. Furthermore, measurements of cyclooxygenase metabolites in the CSF bathing the pial surface show that levels of constrictor prostanoids, particularly PGF2α, increase with CSD, and this increase is eliminated with indomethacin administration (Shibata et al., 1991b). Thus, constrictor prostanoids appear to restrict increases in CBF during CSD and impair restoration of basal CBF in the post-CSD period in at least one species. The constrictor prostanoids appear to have access to the cerebral arteries from within the vessel wall and via the surrounding CSF, and the relative contribution from each source is time dependent. The dilator component of the pial arteriolar response to CSD is unaffected by aCSF superfusion (Shibata et al., 1991), perhaps because the prostanoids arise from within the vessel wall. The putative vascular origin of constrictor prostanoids during the immediate CSD period is supported by the virtually simultaneous effects of dilator and constrictor agents on cerebral arteries and arterioles. However, it is unclear whether the prostanoids exert a direct, counteracting constrictor influence, or whether they restrict release and/or actions of vasodilator agents. Since superfusion of the cortical surface with aCSF has similar effects as indomethacin administration on the sustained, post-CSD oligemia, it seems likely that parenchyma-derived prostanoids are able to exert a direct, constrictor effect on pial arteries under these conditions (Shibata et al., 1991b). Although it has been suggested that cytochrome P-450 monooxygenase products are produced by astroglia and affect cerebral hemodynamics, especially in the rat, we were unable to show any effect of miconazole on the increase in CBF during CSD in this species (Shimizu et al., 2002). Furthermore, endothelin does not appear to mediate the oligemia which occurs following CSD (Goadsby et al., 1996a).
4.3. Integration of multicellular contributions to cerebral hemodynamics
The final cerebrovascular responses to CSD involve additive and competing vasoactive factors derived from diverse cell types within the neurovascular unit (Figure 2). Of these, undoubtedly the perivascular nerves are the most important in promoting cerebral artery dilation. Thus, in all species studied, it appears that trigeminal fibers promote much of the cerebral artery dilation (Table 1) In addition, at least in the rat, parasympathetic nerves also contribute substantially to cerebral vascular dilation during CSD (Reuter et al., 1998). While NO is also an important component of dilation in several, but not all species, during CSD, the most that we can say at this time is that the source is neuronal rather than from the endothelium (Colonna et al., 1997; Shimizu et al., 2002), and therefore could include perivascular nerves, astrocytes, or cortical neurons. On the other hand, constrictor prostanoids are able to suppress dilation during CSD and promote post-CSD vasoconstriction in species such as rabbit. Based upon the results from the aCSF perfusion experiments and timing of vascular effects with respect to the location of the CSD, our findings show that the source of vasoactive agents during the hyperemic period appears to be from the vessel wall. Thus, we have shown that in CSD-induced cerebrovascular dilation in rabbits, neurally-derived NO, perivascular nerve-derived calcitonin gene-related peptide, and prostaglandins from parenchyma and/or blood vessels all contribute to the final, integrated blood flow response (Colonna et al., 1994a; 1994b, 1997) (Figure 1). Furthermore, as previously described the relative contribution of any of these vasoactive stimuli would be modified by physiological or pathological status.
Figure 2. Schematic demonstrating possible mechanisms involved in integrated cerebral vascular dilator responses to CSD.
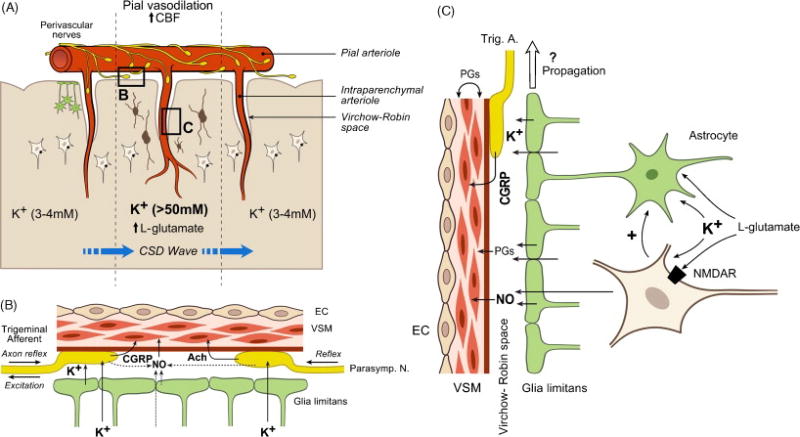
A. Overview of neurovascular unit showing perivascular innervation, astroglia projecting end feet to the pial surface, NMDA receptor neurons (lower cells), and interneurons (dark cells). NMDA receptor containing neurons may also contain neuronal NOS (nNOS) or alternatively activate nNOS containing interneurons. B. The major dilator influences on pial arteries and arterioles. The major dilator agents are calcitonin gene-related peptide (CGRP) release by trigeminal afferents and acetylcholine (Ach) released from perivascular nerves and nitric oxide (NO) derived from neural sources (perivascular nerves or parenchyma). In each species studied the relative importance of each type of vasoactive influence appears to vary. In addition, the pathophysiological and pharmacological status of the animal would affect the relative importance of each component. C. The major vasoactive influences on parenchymal arterioles. In addition to neurotransmitters such as CGRP from trigeminal afferents and neuronally-derived NO promoting dilation, vascular and perivascular prostaglandins will reduce constriction in parenchymal as well as pial resistance vessels (not shown) during CSD. In addition, parenchymal-derived prostaglandins will promote vasoconstriction during the post CSD period. Upstream propagation of arterial dilation may involve perivascular nerves or other vascular components as well as well as substances released from glial end feet, although flow dependent dilation in the “classical” case probably does not occur for reasons stated in the text. Abbreviations: EC, endothelial cell; VSM, vascular smooth muscle cell; NMDAR, NMDA receptor; Trig. A.,Trigeminal afferent; Parasymp. N., Parasympathetic nerve.
The key unresolved issue is the nature of the functional coupling between events occurring among neurons and astroglia during CSD and the subsequent transfer of this information to the pial and parenchymal arteries and arterioles. Since acute denervation of sensory and parasympathetic neural pathways, which leaves the peripheral nerve endings intact, does not substantially reduce cerebral hyperemia during CSD, cerebrovascular responses are dependent upon stimuli that arise from the cortex and do not involve centrally integrative reflexes. At least for the pial circulation, the involvement of vasoactive substances arising from large areas of the cortex during CSD appears unlikely. Superfusion of the cortical surface with aCSF does not reduce hyperemia during CSD (Shibata et al., 1992), and spreading depression on one gyrus in rabbit does not dilate pial arterioles on an adjacent gyrus. In addition, the close temporal relationship of the actions of diverse dilating and competing constricting agents with each other and with the location of the spreading depression, as well as the rapid return of blood flow to baseline values, supports the tight coupling between cortical and vascular events during CSD. Given the inclusive nature of spreading depression, in which virtually all cortical cells would be affected, actions involving astroglia and interneurons near parenchymal arterioles will contribute to CBF changes. Increases in cortical extracellular fluid levels of substances such as K+ and/or other ions and neurotransmitters may directly depolarize perivascular nerves on pial and parenchymal arteries and arterioles. These factors alternatively may indirectly activate perivascular nerves via axonal responses involving local, non-vascular projections of trigeminal fibers on the pial surface (Hildebrand et al., 1997; Risling et al., 1994). In addition, increased levels of ions or other substances can selectively activate perivascular nerves associated with parenchymal arterioles (Benagiano et al., 2000; Iadecola et al., 1993) present within the Robins-Virchow space, and result in upstream conduction of neural or vascular activation. The report that upstream propagation of arterial dilation occurs during CSD in mice (Brennan et al., 2007) supports the concept that activation of perivascular fibers on parenchymal arterioles, or other components of the vascular wall, initiates a propagated dilator response that projects to the pial arterioles. However, other evidence makes it unlikely that flow dependent dilation in the “classical” sense is involved in some animal models, since the normal delay in dilation seen in surface arterioles following parenchymal blood flow increases with a carotid occlusion model (Fujii et al., 1991) is not seen during CSD (Shibata et al., 1991b).
In support of a local, tight coupling between cortical events and dilator responses of cerebral arteries and arterioles, rather than vascular responses that are mediated by the generalized diffusion of substances from the cortex, superfusion of the cortical surface with aCSF was able to effectively suppress not only pial arteriolar dilation to arterial hypercapnia (Shibata et al., 1991a), but also the delayed and prolonged post-CSD vasoconstriction which is due to constrictor prostanoids diffusing from the parenchyma. Nonetheless, several factors, including a close anatomical association of pial arteries and arterioles to the glia limitans, the relative isolation of the parenchymal arterioles within the Virchow-Robins space, and unknown flow dynamics within the cranial window, can reduce effectiveness of superfusion with aCSF against the dramatic, but transient cerebral vascular dilation during CSD.
Glial-derived substances are able to directly interact with the cerebral arteries and arterioles on the pial surface and in the protected Virchow-Robins space. A tight, functional coupling between the glial limitans and pial arterioles, which are in close anatomical proximity to each other, has been described recently for dilator responses associated with neuronal activation (Xu et al., 2004; Xu and Pelligrino, 2007; Iadecola and Nedergaard, 2007; Jackovcevic and Harder, 2007). Cortical astroglia have been reported to swell during CSD in rodents (Anderová et al., 2001), and astroglia are activated during the processing of various ions and neurotransmitters released into extracellular fluid during CSD (Llian and Stringer, 2004). Thus, it would not be surprising that events leading to CSD would lead to astroglial activation with subsequent effects on cerebral arteries including activation of perivascular nerves. For example, neuronal activation leads to events which result in astrocyte stimulation with the release of potassium ion and/or ATP into the perivascular space to cause dilation of parenchymal arterioles (Filosa et al., 2006; Xu et al., 2007). However, the involvement of astroglia in mediating cerebrovascular responses during CSD has never been directly assessed.
While the potential role of interneurons in mediating changes of cerebral vascular tone, especially in parenchymal arterioles, has received attention recently (Hamel, 2006), their contribution to hemodynamic changes during CSD remains unclear. Cortical neurons are stimulated and depolarized during CSD, and release various vasoactive substances. In addition, similar to astroglia, neurons have been shown to swell during CSD in rodents (Takano et al., 2007). However, the majority of the changes in CBF during CSD appear to involve release of dilator neurotransmitters from perivascular nerves, although the source of NO during CSD in rabbits and cats can be from cortical neurons and/or astrocytes as well as from perivascular nerves. On the other hand, the observation that parenchymal blood flow increases are sustained even following the elimination of pial arteriolar dilation either through repeated CSDs or via chemical means may indicate that interneuron influences are present but normally masked (Chuquet et al., 2007).
5. Summary and Perspectives
Cortical spreading depression is a useful model for studying brain function and control mechanisms of the cerebral circulation. The combined contributions of many investigators especially over the last two decades have lead to a basic understanding of the factors controlling cerebrovascular responses during CSD as well as the relevance of CSD to clinical situations such as migraines and brain injury. However, more research is needed not only to verify previous, key findings, but also to clarify additional topics of interest in this field. These include investigating the:
functional linkage between events in the cortex and the subsequent cerebral vascular responses;
underlying basis for major species differences in cerebrovascular responsiveness during CSD;
reasons for lack of a prominent role of NO in mediating increased CBF especially in rodents during CSD;
possibility of gender differences in CBF responses during CSD;
normal cerebral hemodynamic responses in people during CSD; And
mechanisms of cerebral oligemia associated with CSD-like events during migraines.
We also suggest that genetically altered rats and mice, whose advantages have been shown in many studies in the cerebral circulation (Faraci, 2003) including several recent ones involving CSD (Petzold et al., 2008; Shin et al., 2007; Sukhotinsky et al. 2008), can be useful in the elucidation and validation of mechanisms of cerebral vascular control during CSD.
Acknowledgments
Supported by NIH grants HL-07731, HL-030260, and HL-065380, Y. F. Wu Research and Education Fund, WFUSM Venture Fund, and K.G. Phillips Fund for the Prevention and Treatment of Heart Disease, WFUSM Interim Funding, and by the Hungarian Science Foundation (OTKA K 63401 and IN 69967). We thank Nancy Busija, M.A., for editorial assistance.
- Ach
acetylcholine
- aCSF
artificial cerebral spinal fluid
- BBB
blood brain barrier
- CBF
Cerebral blood flow
- CGRP
calcitonin gene-related peptide
- CO
carbon monoxide
- COX
cyclooxygenase
- CSD
cortical spreading depression
- CSF
cerebrospinal fluid
- EC
endothelial cell
- EET
epoxyeicosatrienoic acid
- fNIRS
near-infrared spectroscopy
- K+
potassium ion
- KCl
potassium chloride
- L-NAME
N(G)-nitro-l-arginine methyl ester
- LPDI
laser-Doppler perfusion imaging
- MCAO
middle cerebral artery occlusion
- MMP-9
metalloproteinase-9
- 7-NI
7-nitroindazole
- NMDA
N-methyl-D-aspartate
- NMDAR
NMDA receptor
- NO
nitric oxide
- NOS
nitric oxide synthase
- Parasymp. N
Parasympathetic nerve
- ROS
reactive oxygen species
- Trig. A
Trigeminal afferent
- VSM
vascular smooth muscle cell
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akin A, Bilensoy D. Cerebrovascular reactivity to hypercapnia in migraine patients measured with near-infrared spectroscopy. Brain Res. 2006;1107:206–214. doi: 10.1016/j.brainres.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Alkayed NJ, Narayanan J, Gebremedhin D, Medhora M, Roman RJ, Harder DR. Molecular characterization of an arachidonic acid epoxygenase in rat brain astrocytes. Stroke. 1996;27:971–9. doi: 10.1161/01.str.27.5.971. [DOI] [PubMed] [Google Scholar]
- Anderová M, Kubinová S, Mazel T, Chvátal A, Eliasson C, Pekny M, Syková E. Effect of elevated K+, hypertonic stress, and cortical spreading depression on astrocyte swelling in GFAP-deficient mice. Glia. 2001;35:189–203. doi: 10.1002/glia.1084. [DOI] [PubMed] [Google Scholar]
- Ayata C, Moskowitz MA. Cortical spreading depression confounds concentration-dependent pial arteriolar dilation during N-methyl-D-aspartate superfusion. Am J Physiol Heart Circ Physiol. 2006;290:H1837–841. doi: 10.1152/ajpheart.01102.2005. [DOI] [PubMed] [Google Scholar]
- Ayata C, Shin HK, Salomone S, Ozdemir-Gursoy Y, Boas DA, Dunn AK, Moskowitz MA. Pronounced hypoperfusion during spreading depression in mouse cortex. J Cereb Blood Flow Metab. 2004;24:1172–1182. doi: 10.1097/01.WCB.0000137057.92786.F3. [DOI] [PubMed] [Google Scholar]
- Back T, Kohno K, Hossmann KA. Cortical negative DC deflections following middle cerebral artery occlusion and KCl-induced spreading depression: effect on blood flow tissue oxygenation, and electroencephalogram. J Cereb Blood Flow Metab. 1994;14:12–19. doi: 10.1038/jcbfm.1994.3. [DOI] [PubMed] [Google Scholar]
- Bari F, Paprika D. Changes in cerebrovascular reactivity after stimulation and depletion of sensory fibers in rats. In: Fukuuchi Y, Tomita M, Koto A, editors. Ischemic Blood Flow in the Brain. Tokyo: Spinger Publishers; 2000. pp. 289–295. [Google Scholar]
- Bari F, Paprika D, Jancsó G, Domoki F. Capsaicin-sensitive mechanisms are involved in cortical spreading depression-associated cerebral blood flow changes in rats. Neurosci Lett. 2000;292:17–20. doi: 10.1016/s0304-3940(00)01424-5. [DOI] [PubMed] [Google Scholar]
- Bartolini M, Baruffaldi R, Paolino I, Silvestrini M. Cerebral blood flow changes in the different phases of migraine. Funct Neurol. 2005;20:209–211. [PubMed] [Google Scholar]
- Benagiano V, Virgintino D, Rizzi A, Errede M, Bertossi M, Troccoli V, Roncali L, Ambrosi G. Cholinergic nerve fibres associated with the microvessels of the human cerebral cortex: a study based on monoclonal immunocytochemistry for choline acetyltrasferase. Eur J Histochem. 2000;44:165–169. [PubMed] [Google Scholar]
- Bolay H, Reuter U, Dunn AK, Huang Z, Boas DA, Moskowitz MA. Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med. 2002;8:136–42. doi: 10.1038/nm0202-136. [DOI] [PubMed] [Google Scholar]
- Brennan KC, Beltrán-Parrazal L, López-Valdés HE, Theriot J, Toga AW, Charles AC. Distinct vascular conduction with cortical spreading depression. J Neurophysiol. 2007;97:4143–4151. doi: 10.1152/jn.00028.2007. [DOI] [PubMed] [Google Scholar]
- Busija DW. Nervous control of the cerebral circulation. In: Bennett T, Gardiner SM, editors. Nervous control of the circulation. Amsterdam: Harwood Academic Publishers; 1996. pp. 177–206. [Google Scholar]
- Busija DW. Eicosanoids and cerebrovascular control. In: Welch KMA, Caplan LR, Reis DJ, Siesjö BK, Weir B, editors. Primer on Cerebrovascular Diseases. San Diego: Academic Press; 1997. pp. 93–96. [Google Scholar]
- Busija DW. Prostaglandins and other Eicosanoids. In: Edvinsson L, Krause D, editors. Cerebral blood flow and metabolism. Philadelphia: Lippincott Williams and Wilkins; 2002. pp. 325–338. [Google Scholar]
- Busija DW, Leffler CW. Hypothermia reduces cerebral metabolic rate and cerebral blood flow in newborn pigs. Am J Physiol Heart Circ Physiol. 1987;22:H869–H873. doi: 10.1152/ajpheart.1987.253.4.H869. [DOI] [PubMed] [Google Scholar]
- Busija DW, Leffler CW. Dilator effects of amino acid neurotransmitters on piglet pial arterioles. Am J Physiol Heart Circ Physiol. 1989;257:H1200–H1203. doi: 10.1152/ajpheart.1989.257.4.H1200. [DOI] [PubMed] [Google Scholar]
- Busija DW, Meng W. Retention of cerebrovascular dilation after cortical spreading depression in anesthetized rabbits. Stroke. 1993;24:1740–1744. doi: 10.1161/01.str.24.11.1740. [DOI] [PubMed] [Google Scholar]
- Busija DW, Bari F, Domoki F, Louis T. Mechanisms involved in the cerebrovascular dilator effects of N-methyl-D-aspartate in cerebral cortex. Brain Res Rev. 2007;56:89–100. doi: 10.1016/j.brainresrev.2007.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busija DW, Bari F, Domoki F, Louis T. Mechanisms involved in the cerebrovascular dilator effects of N-methyl-D-aspartate in cerebral cortex. Brain Res Rev. 2007;56:89–100. doi: 10.1016/j.brainresrev.2007.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busija DW, Leffler CW, Pourcyrous M. Hyperthermia increases cerebral metabolic rate and blood flow in neonatal pigs. Am J Physiol Heart Circ Physiol. 1988;24:H343–H346. doi: 10.1152/ajpheart.1988.255.2.H343. [DOI] [PubMed] [Google Scholar]
- Buzzi MG, Carter WB, Shimizu T, Heath H, Moskowitz MA. Dihydroergotamine and sumatriptan attenuate levels of CGRP in plasma in rat superior sagittal sinus during electrical stimulation of the trigeminal ganglion. Neuropharmacology. 1991;30:1193–1200. doi: 10.1016/0028-3908(91)90165-8. [DOI] [PubMed] [Google Scholar]
- Caggiano AO, Kraig RP. Eicosanoids and nitric oxide influence induction of reactive gliosis from spreading depression in microglia but not astrocytes. J Comp Neurol. 1996a;369:93–108. doi: 10.1002/(SICI)1096-9861(19960520)369:1<93::AID-CNE7>3.0.CO;2-F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caggiano AO, Breder CD, Kraig RP. Long-term elevation of cyclooxygenase-, but not lipoyxgenase,, in regions synaptically distant from spreading depression. J Comp Neurol. 1996b;376:447–462. doi: 10.1002/(SICI)1096-9861(19961216)376:3<447::AID-CNE7>3.0.CO;2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case GR, Lavond DG, Thompson RF. Cortical spreading depression and involvement of the motor cortex, auditory cortex, and cerebellum in eyeblink classical conditioning of the rabbit. Neurobiol Learn Mem. 2002;78:234–245. doi: 10.1006/nlme.2002.4061. [DOI] [PubMed] [Google Scholar]
- Cauli B, Tong XK, Rancillac A, Serluca N, Lambolez B, Rossier J, Hamel E. Cortical GABA interneurons in neurovascular coupling: relays for subcortical vasoactive pathways. J Neurosci. 2004;24:8940–8949. doi: 10.1523/JNEUROSCI.3065-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrissobolis S, Sobey CG. Influence of gender on K+-induced cerebral vasodilatation. Stroke. 2004;35:747–52. doi: 10.1161/01.STR.0000116867.28589.3A. [DOI] [PubMed] [Google Scholar]
- Christiansen I, Thomsen LL, Daugaard D, Ulrich V, Olesen J. Glyceryl trinitrite induces attacks of migraine without aura in sufferers of migraine with aura. Cephalalgia. 1999;19:660–667. doi: 10.1046/j.1468-2982.1999.019007660.x. [DOI] [PubMed] [Google Scholar]
- Chuquet J, Hollender L, Nimchinsky EA. High-resolution in vivo imaging of the neurovascular unit during spreading depression. J Neurosci. 2007;27:4036–4044. doi: 10.1523/JNEUROSCI.0721-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna DM, Meng W, Deal DD, Busija DW. Calcitonin gene-related peptide promotes cerebrovascular dilation during cortical spreading depression in rabbits. Am J Physiol Heart Circ Physiol. 1994a;266:H1095–H1102. doi: 10.1152/ajpheart.1994.266.3.H1095. [DOI] [PubMed] [Google Scholar]
- Colonna DM, Meng W, Deal DD, Busija DW. Nitric oxide promotes arteriolar dilation during cortical spreading depression in rabbits. Stroke. 1994b;25:2463–2470. doi: 10.1161/01.str.25.12.2463. [DOI] [PubMed] [Google Scholar]
- Colonna DM, Meng W, Deal DD, Gowda M, Busija DW. Neuronal NO promotes cerebral cortical hyperemia during cortical spreading depression in rabbits. Am J Physiol Heart Circ Physiol. 1997;272:H1315–H1322. doi: 10.1152/ajpheart.1997.272.3.H1315. [DOI] [PubMed] [Google Scholar]
- Cui Y, Kataoka Y, Inui T, Mochizuki T, Onoe H, Matsumura K, Urade Y, Yamada H, Watanabe Y. Up-regulated neuronal COX-2 expression after cortical spreading depressin is involved in non-REM sleep inductin in rats. J Neurosc Res. 2008;86:929–936. doi: 10.1002/jnr.21531. [DOI] [PubMed] [Google Scholar]
- Dalkara T, Zervas NT, Moskowitz MA. From spreading depression to the trigeminovascular system. Neurol Sci. 2006;27:S86–S90. doi: 10.1007/s10072-006-0577-z. [DOI] [PubMed] [Google Scholar]
- DaSilva AFM, Granziera C, Snyder J, Hadjikhani N. Thickening in the somatosensory cortex of patients with migraine. Neurology. 2007;69:1990–1995. doi: 10.1212/01.wnl.0000291618.32247.2d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauphin F, MacKenzie ET. Cholinergic and vasoactive intestinal polypeptidergic innervation of the cerebral arteries. Pharmac Ther. 1995;67:385–417. doi: 10.1016/0163-7258(95)00022-4. [DOI] [PubMed] [Google Scholar]
- Davies JA, Annels SJ, Dickie BG, Ellis Y, Knott NJ. A comparison between the stimulated and paroxysmal release of endogenous amino acids from rat cerebellar, striatal and hippocampal slices: a manifestation of spreading depression? J Neurol Sci. 1995;131:8–14. doi: 10.1016/0022-510x(95)00100-g. [DOI] [PubMed] [Google Scholar]
- De Oliveira Castro G, Martins-Ferreira H. Deformations and thickness variations accompanying spreading depression in the retina. J Neurophysiol. 1970;33:891–900. doi: 10.1152/jn.1970.33.6.891. [DOI] [PubMed] [Google Scholar]
- Domoki F, Veltkamp R, Bari F, Busija DW. Cerebrovascular reactivity remains intact after cortical depolarization in newborn piglets. Pediatr Res. 1999a;45:834–837. doi: 10.1203/00006450-199906000-00009. [DOI] [PubMed] [Google Scholar]
- Domoki F, Perciaccante JV, Veltkamp R, Bari F, Busija DW. Mitochondrial potassium channel opener diazoxide preserves neuronal-vascular function after cerebral ischemia in newborn pigs. Stroke. 1999b;30:2713–2718. doi: 10.1161/01.str.30.12.2713. [DOI] [PubMed] [Google Scholar]
- Drake CT, Iadecola C. The role of neuronal signaling in controlling cerebral blood flow. Brain Lang. 2007;102:141–152. doi: 10.1016/j.bandl.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Kleeberg J, Petzold G, Priller J, Windmüller O, Orzechowski HD, Lindauer U, Heinemann U, Einhäupl KM, Dirnagl U. Endothelin-1 potently induces Leão”s cortical spreading depression in vivo in the rat: a model for an endothelial trigger of migrainous aura? Brain. 2002;125:102–112. doi: 10.1093/brain/awf007. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Körner K, Görner A, Lindauer U, Weih M, Villringer A, Dirnagl U. Nitric oxide modulates the CBF response to increased extracellular potassium. J Cereb Blood Flow Metab. 1995;15:914–919. doi: 10.1038/jcbfm.1995.116. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Woitzik J, Fabricius M, Bhatia R, Major S, Drenckhahn C, Lehmann T, Sarrafzadeh A, Willumsen L, Hartings JA, Sakowitz OW, Seemann JH, Thieme A, Lauritzen M, Strong AJ. Delayed ischaemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations. Brain. 2006;129:3224–3237. doi: 10.1093/brain/awl297. [DOI] [PubMed] [Google Scholar]
- Duckrow RB. Regional cerebral blood flow during spreading cortical depression in conscious rats. J Cereb Blood Flow Metab. 1991;11:150–154. doi: 10.1038/jcbfm.1991.18. [DOI] [PubMed] [Google Scholar]
- Duckrow RB. A brief hypoperfusion precedes spreading depression if nitric oxide synthesis is inhibited. Brain Res. 1993;618:190–195. doi: 10.1016/0006-8993(93)91265-t. [DOI] [PubMed] [Google Scholar]
- Duckrow RB, Beard DC. Cortical hypoperfusion following spreading depression is not altered by tirilazad mesylate (U-74006F) in awake rats. Brain Res. 1992;572:296–299. doi: 10.1016/0006-8993(92)90488-u. [DOI] [PubMed] [Google Scholar]
- Edvinsson L. Novel migraine therapy with calcitonin gene-regulated peptide receptor antagonists. Expert Opin Ther Targets. 2007;11:1179–88. doi: 10.1517/14728222.11.9.1179. [DOI] [PubMed] [Google Scholar]
- Edvinsson L, Ekman R, Jansen I, McCulloch J, Uddman R. Calcitonin gene-related peptide and cerebral blood vessels: distribution and vasomotor effects. J Cereb Blood Flow Metab. 1987;7:720–728. doi: 10.1038/jcbfm.1987.126. [DOI] [PubMed] [Google Scholar]
- Edvinsson L, Elsås T, Suzuki N, Shimuzu T, Lee TJ-F. Origin and co-localization of nitric oxide synthase, CGRP, PACAP, and VIP in the cerebral circulation of the rat. Microsc Res Tech. 2001;53:221–228. doi: 10.1002/jemt.1086. [DOI] [PubMed] [Google Scholar]
- Edvinsson L, Jansen I, Cunha e Sa M, Gulbenkian S. Demonstration of neuropeptide containing nerves and vasomotor responses to perivascular peptides in human cerebral arteries. Cephalalgia. 1994;14:88–96. doi: 10.1046/j.1468-2982.1994.1402088.x. [DOI] [PubMed] [Google Scholar]
- Edvinsson L, Mulder H, Goadsby PJ, Uddman R. Calcitonin gene-related peptide and nitric oxide in the trigeminal ganglion: cerebral vasodilatation from trigeminal nerve stimulation involves mainly calcitonin gene-related peptide. J Auton Nerv Syst. 1998;70:15–22. doi: 10.1016/s0165-1838(98)00033-2. [DOI] [PubMed] [Google Scholar]
- El-Assouad D, Tayebati SK. Cholinergic innervation of pial arterioles in senescent rats: an immunohistochemical study. Mech Ageing Dev. 2002;12:529–536. doi: 10.1016/s0047-6374(01)00352-9. [DOI] [PubMed] [Google Scholar]
- Erdős B, Snipes JA, Miller AW, Busija DW. Subtype specific potassium channel dysfunction in cerebral arteries of insulin-resistant rats is mediated by reactive oxygen species. Stroke. 2004;35:964–969. doi: 10.1161/01.STR.0000119753.05670.F1. [DOI] [PubMed] [Google Scholar]
- Erdős B, Tulbert C, Snipes JA, Busija DW. Rosuvastatin reduces NAD(P)H-oxidase-dependent superoxide anion production and improves vascular function in cerebral arteries of Zucker obese rats. Am J Physiol Heart Circ Physiol. 2006;290:H1264–H1270. doi: 10.1152/ajpheart.00804.2005. [DOI] [PubMed] [Google Scholar]
- Estrada C, Mengual E, González C. Local NDPH-diaphorase neurons innervate pial arterioles and lie close of project to intracerebral blood vessels: a possible frole for nitric oxide in the regulation of cerebral blood flow. J Cereb Blood Flow Metab. 1993;13:978–984. doi: 10.1038/jcbfm.1993.122. [DOI] [PubMed] [Google Scholar]
- Fabricius M, Lauritzen M. Transient hyperemia succeeds oligemia in the wake of cortical spreading depression. Brain Res. 1994;602:350–353. doi: 10.1016/0006-8993(93)90701-n. [DOI] [PubMed] [Google Scholar]
- Fabricius M, Lauritzen M. Examination of the role of nitric oxide for the hypercapnic rise of cerebral blood flow in rats. Am J Physiol Heart Circ Physiol. 1994;266:H1457–H1464. doi: 10.1152/ajpheart.1994.266.4.H1457. [DOI] [PubMed] [Google Scholar]
- Fabricius M, Akgoren N, Lauritzen M. Arginine-nitric oxide pathway and cerebrovascular regulation in cortical spreading depression. Am J Physiol Heart Circ Physiol. 1995;269:H23–H29. doi: 10.1152/ajpheart.1995.269.1.H23. [DOI] [PubMed] [Google Scholar]
- Fabricius M, Jensen LH, Lauritzen M. Microdialysis of interstitial amino acids during spreading depression and anoxic depolarization in rat neocortex. Brain Res. 1993;612:61–69. doi: 10.1016/0006-8993(93)91644-8. [DOI] [PubMed] [Google Scholar]
- Fabricius M, Akgören N, Dirnagl U, Lauritzen M. Laminar analysis of cerebral blood flow in cortex of rats by laser-Doppler flowmetry: a pilot study. J Cereb Blood Flow Metab. 1997;17:1326–1336. doi: 10.1097/00004647-199712000-00008. [DOI] [PubMed] [Google Scholar]
- Fabricius M, Fuhr S, Bhatia R, Boutelle M, Hashemi P, Strong AJ, Lauritzen M. Cortical spreading depression and peri-infarct depolarization in acutely injured human cerebral cortex. Brain. 2006;129:778–790. doi: 10.1093/brain/awh716. [DOI] [PubMed] [Google Scholar]
- Faraci FM. Cerebral vascular function in genetically altered mice. Methods Mol Med. 2003;89:505–512. doi: 10.1385/1-59259-419-0:505. [DOI] [PubMed] [Google Scholar]
- Farkas E, Pratt R, Sengpiel F, Obrenovitch TP. Direct, live imaging of cortical spreading depression and anoxic depolarization using a fluorescent, voltage-sensitive dye. J Cereb Blood Flow Metab. 2008;28:251–262. doi: 10.1038/sj.jcbfm.9600569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellin T, Carmignoto G. Neuron-to-astrocyte signaling in the brain represents a distinct multifunctional unit. J Physiol. 2004;559:3–15. doi: 10.1113/jphysiol.2004.063214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filosa JA, Bonev AD, Nelson MT. Calcium dynamics in cortical astrocytes and arterioles during neurovascular coupling. Circ Res. 2004;95:e73–81. doi: 10.1161/01.RES.0000148636.60732.2e. [DOI] [PubMed] [Google Scholar]
- Filosa JA, Bonev AD, Straub SV, Meredith AL, Wilkerson MK, Aldrich RW, Nelson MT. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nature Neurosci. 2006;9:1397–1403. doi: 10.1038/nn1779. [DOI] [PubMed] [Google Scholar]
- Florence G, Bonvento G, Charbonne R, Seylaz J. Spreading depression reversibly impairs autoregulation of cortical blood flow. Am J Physiol Reg Int Comp Physiol. 1994;266:R1136–R1140. doi: 10.1152/ajpregu.1994.266.4.R1136. [DOI] [PubMed] [Google Scholar]
- Fujii K, Heistad DD, Faraci FM. Flow-mediated dilatation of the basilar artery in vivo. Circ Res. 1991;69:697–705. doi: 10.1161/01.res.69.3.697. [DOI] [PubMed] [Google Scholar]
- Goadsby PJ. The oligemic phase of cortical spreading depression is not blocked by tirilazad (U-74006F) Brain Res. 1992;588:140–143. doi: 10.1016/0006-8993(92)91353-g. [DOI] [PubMed] [Google Scholar]
- Goadsby PJ. Calcitonin gene-related peptide antagonists as treatment of migraine and other primary headaches. Drugs. 2005;65:2556–2567. doi: 10.2165/00003495-200565180-00002. [DOI] [PubMed] [Google Scholar]
- Goadsby PJ, Hoskin KL. Cerebral blood flow is not coupled to neuronal activity during stimulation of the facial nerve vasodilator system. Brain Res. 1994;647:192–198. doi: 10.1016/0006-8993(94)91317-x. [DOI] [PubMed] [Google Scholar]
- Goadsby PJ, Adner M, Edvinsson L. Characterization of endothelin ETA receptors in the cerebral vasculature and their lack of effect upon spreading depression. J Cereb Blood Flow Metab. 1996a;16:698–704. doi: 10.1097/00004647-199607000-00021. [DOI] [PubMed] [Google Scholar]
- Goadsby PJ, Kaube H, Hoskin KL. Nitric oxide synthesis couples cerebral blood flow and metabolism. Brain Res. 1992;595:167–170. doi: 10.1016/0006-8993(92)91470-y. [DOI] [PubMed] [Google Scholar]
- Goadsby PJ, Uddman R, Edvinsson L. Cerebral vasodilatation in the cat involves nitric oxide from parasympathetic nerves. Brain Res. 1996b;707:110–118. doi: 10.1016/0006-8993(95)01206-0. [DOI] [PubMed] [Google Scholar]
- Gold L, Back T, Arnold G, Dreier J, Einhäupl KM, Reuter U, Dirnagl U. Cortical spreading depression-associated hyperemia in rats: involvement of serotonin. Brain Res. 1998;783:188–193. doi: 10.1016/s0006-8993(97)01341-3. [DOI] [PubMed] [Google Scholar]
- Gorji A, Scheller D, Straub H, Tegtmeier F, Kohling R, Hohling JM, Tuxhorn I, Ebner A, Wolf P, Werner Panneck H, Oppel F, Speckman EJ. Spreading depression in human neocortical slices. Brain Res. 906:74–83. doi: 10.1016/s0006-8993(01)02557-4. [DOI] [PubMed] [Google Scholar]
- Grafstein B. Mechanism of spreading cortical depression. J Neurophysiol. 1956;19:154–171. doi: 10.1152/jn.1956.19.2.154. [DOI] [PubMed] [Google Scholar]
- Hadjikhani N, Sanchez del Rio M, Wu O, Schwartz D, Bakker D, Fischi B, Kwong KK, Cutrer FM, Rosen BR, Tootell RBH, Sorensen AG, Moskowitz MA. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci. 2001;98:4687–4692. doi: 10.1073/pnas.071582498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel E. Perivascular nerves and the regulation of cerebrovascular tone. J Appl Physiol. 2006;100:1059–1064. doi: 10.1152/japplphysiol.00954.2005. [DOI] [PubMed] [Google Scholar]
- Hansen AJ, Zeuthen T. Extracellular ion concentrations during spreading depression and ischemia in the rat brain cortex. Acta Physiol Scand. 1981;113:437–445. doi: 10.1111/j.1748-1716.1981.tb06920.x. [DOI] [PubMed] [Google Scholar]
- Hansen AJ, Quistorff B, Gjedde A. Relationship between local changes in cortical blood flow and extracellular K+ during spreading depression. Acta Physiol Scand. 1980;109:1–6. doi: 10.1111/j.1748-1716.1980.tb06557.x. [DOI] [PubMed] [Google Scholar]
- Hansen JM, Pedersen D, Larsen VA, Sánchez-del-Rio M, Alvarez Linera JR, Olesen J, Ashina M. Magnetic resonance angiography shows dilatation of the middle cerebral artery after infusion of glyceryl trinitrate in healthy volunteers. Cephalalgia. 2007;27:118–127. doi: 10.1111/j.1468-2982.2006.01257.x. [DOI] [PubMed] [Google Scholar]
- Hawkins BR, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- Herreras O, Somjen GG. Propagation of spreading depression among dendrites and somata of the same cell population. Brain Res. 1993;610:276–282. doi: 10.1016/0006-8993(93)91411-k. [DOI] [PubMed] [Google Scholar]
- Hildebrand C, Karlsson M, Risling M. Ganglionic axons in motor roots and pia mater. Prog Neurobiol. 1997;51:89–128. doi: 10.1016/s0301-0082(96)00052-4. [DOI] [PubMed] [Google Scholar]
- Ho T, Mannix LK, Fan X, Assaid C, Furtek C, Jones CJ, Lines CR, Rapoport AM. Randomized controlled trial of an oral CGRP receptor antagonist, MK-0974, in acute treatment of migraine. Neurology. 2008;70:1304–1312. doi: 10.1212/01.WNL.0000286940.29755.61. [DOI] [PubMed] [Google Scholar]
- Horiguchi T, Kis B, Rajapakse N, Shimizu K, Busija DW. Cortical spreading depression (CSD)-induced tolerance to transient focal cerebral ischemia in halothane anesthetized rats is affected by anesthetic level but not ATP-sensitive potassium channels. Brain Res. 2005a;1062:127–133. doi: 10.1016/j.brainres.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Horiguchi T, Snipes JA, Kis B, Shimizu S, Busija DW. The role of nitric oxide in the development of cortical spreading depression-induced tolerance to transient focal cerebral ischemia in rats. Brain Res. 2005b;1039:84–89. doi: 10.1016/j.brainres.2005.01.047. [DOI] [PubMed] [Google Scholar]
- Horiguchi T, Snipes JA, Kis B, Shimizu K, Busija DW. Cyclooxygenase-2 mediates the development of cortical spreading depression (CSD) – induced tolerance to transient focal cerebral ischemia in rats. Neuroscience. 2006;140:723–730. doi: 10.1016/j.neuroscience.2006.02.025. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Nedergaard M. Glial regulation of the cerebral vasculature. Nat Neurosci. 2007;10:1369–1376. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Beitz AJ, Renno W, Xu X, Mayer B, Zhang F. Nitric oxide synthase-containing neural processes on large cerebral arteries and cerebral microvessels. Brain Res. 1993;606:148–155. doi: 10.1016/0006-8993(93)91583-e. [DOI] [PubMed] [Google Scholar]
- Iijima T, Shimase C, Iwao Y, Sankawa H. Relationships between glutamate release, blood flow and spreading depression: real-time monitoring using an electroenzymatic dialysis electrode. Neurosci Res. 1998;32:201–7. doi: 10.1016/s0168-0102(98)00090-x. [DOI] [PubMed] [Google Scholar]
- Ishikawa M, Kajimura M, Adachi T, Maruyama K, Makino N, Goda N, Yamaguchi T, Sekizuka E, Suematsu M. Carbon monoxide from heme oxygenase-2 is a tonic regulator against NO-dependent vasodilatation in the adult rat cerebral microcirculation. Circ Res. 2005;97:104–114. doi: 10.1161/01.RES.0000196681.34485.ec. [DOI] [PubMed] [Google Scholar]
- Iversen HK. Human migraine models. Cephalalgia. 2001;21:781–785. doi: 10.1111/j.1468-2982.2001.00250.x. [DOI] [PubMed] [Google Scholar]
- Jakovcevic D, Harder DR. Role of astrocytes in matching blood flow to neuronal activity. Curr Top Dev Biol. 2007;79:75–97. doi: 10.1016/S0070-2153(06)79004-4. [DOI] [PubMed] [Google Scholar]
- Jansen-Olesen I, Mortensen A, Edvinsson L. Calcitonin gene-related peptide is released from capsaicin-sensitive nerve fibres and induces vasodilation of human cerebral arteries concomitant with activation of adenylyl cyclase. Cephalalgia. 1996;16:310–316. doi: 10.1046/j.1468-2982.1996.1605310.x. [DOI] [PubMed] [Google Scholar]
- Kaube H, Knight YEE, Storer RJ, Hoskin KL, May A, Goadsby PJ. Vasodilator agents and supracollicular transaction fail to inhibit cortical spreading depression in the cat. Cephalalgia. 1999;19:592–597. doi: 10.1046/j.1468-2982.1999.019006592.x. [DOI] [PubMed] [Google Scholar]
- Katona E, Settakis G, Varga Z, Juhász M, Paragh G, Bereczki D, Fulesdi B, Páll D. Both nitric oxide and endothelin-1 influence cerebral blood flow velocity at rest and after hyper- and hypocapnic stimuli in hypertensive and healthy adolescents. Kidney Blood Press Res. 2006;29:152–158. doi: 10.1159/000095348. [DOI] [PubMed] [Google Scholar]
- Kawahara N, Ruetzler C, Klatzo I. Protective effect of spreading depression against neuronal damage following cardiac arrest cerebral ischaemia. Neurol Res. 1995;17:9–16. doi: 10.1080/01616412.1995.11740281. [DOI] [PubMed] [Google Scholar]
- Kiss C, Shepard PD, Bari F, Schwarcz R. Cortical spreading depression augments kynurenate levels and reduces malonate toxicity in the rat cortex. Brain Res. 2004;1002:129–135. doi: 10.1016/j.brainres.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Harris VA, Welsh FA. Spreading depression induces tolerance of cortical neurons to ischemia in rat brain. J Cereb Blood Flow Metab. 1995;15:721–727. doi: 10.1038/jcbfm.1995.92. [DOI] [PubMed] [Google Scholar]
- Kocher M. Metabolic and hemodynamic activation of postischemic rat brain by cortical spreading depression. J Cereb Blood Flow Metab. 1990;10:564–71. doi: 10.1038/jcbfm.1990.99. [DOI] [PubMed] [Google Scholar]
- Koehler RC, Gebremedhin D, Harder DR. Role of astrocytes in cerebrovascular regulation. J Appl Physiol. 2006;100:307–317. doi: 10.1152/japplphysiol.00938.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koroleva VI, Bures J. Rats do not experience cortical or hippocampal spreading depression as aversive. Neurosci Lett. 1993;149:153–156. doi: 10.1016/0304-3940(93)90759-e. [DOI] [PubMed] [Google Scholar]
- Kunkler PE, Kraig RP. Calcium waves precede electrophysiological changes of spreading depression in hippocampal organ cultures. J Neurosci. 1998;18:3416–3425. doi: 10.1523/JNEUROSCI.18-09-03416.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacombe P, Sercombe R, Correze JL, Springhetti V, Seylaz J. Spreading depression induces prolonged reduction of cortical blood flow reactivity in the rat. Exp Neurol. 1992;117:278–286. doi: 10.1016/0014-4886(92)90137-f. [DOI] [PubMed] [Google Scholar]
- Lalo U, Pankratov Y, Kirchhoff F, North RA, Verkhratsky A. NMDA receptors mediate neuron-to-glia signaling in mouse cortical astrocytes. J Neurosci. 2006;26:2673–2683. doi: 10.1523/JNEUROSCI.4689-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert GA, Michalicek J. Cortical spreading depression reduces dural blood flow—a possible mechanism for migraine pain? Cephalalgia. 1994;14:430–436. doi: 10.1046/j.1468-2982.1994.1406430.x. [DOI] [PubMed] [Google Scholar]
- Lassen LH, Haderslev PA, Jacobsen VB, Iversen HK, Sperling B, Olesen J. CRP may play a causative role in migraine. Cephalalgia. 2002;22:54–61. doi: 10.1046/j.1468-2982.2002.00310.x. [DOI] [PubMed] [Google Scholar]
- Lassen LH, Jacobsen VB, Haderslev PA, Ssperling B, Iversen HK, Olesen J, Tfelt-Hasen P. Involvement of calcitonin gene-related peptide in migraine: regional cerebral blood flow and blood flow velocity in migraine patients. J Headache Pain. 2008 doi: 10.1007/s10194-008-0036-8. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritzen M. Long-lasting reduction of cortical blood flow of the brain after spreading depression with preserved autoregulation and impaired CO2 response. J Cereb Blood Flow Metab. 1984;4:546–554. doi: 10.1038/jcbfm.1984.79. [DOI] [PubMed] [Google Scholar]
- Lauritzen M. Regional cerebral blood flow during cortical spreading depression in rat brain: increased reactive hyperperfusion in low-flow states. Acta Neurol Scand. 1987a;75:1–8. doi: 10.1111/j.1600-0404.1987.tb07881.x. [DOI] [PubMed] [Google Scholar]
- Lauritzen M. Cerebral blood flow in migraine and cortical spreading depression. Acta Neurol Scand. 1987b;113(Suppl):1–40. [PubMed] [Google Scholar]
- Lauritzen M. Cortical spreading depression in migraine. Cephalalgia. 2001;21:757–760. doi: 10.1111/j.1468-2982.2001.00244.x. [DOI] [PubMed] [Google Scholar]
- Lauritzen M, Diemer NH. Uncoupling of cerebral blood flow and metabolism after single episode of cortical spreading depression in the rat brain. Brain Res. 1986;370:405–408. doi: 10.1016/0006-8993(86)90504-4. [DOI] [PubMed] [Google Scholar]
- Lauritzen M, Fabricius M. Real time laser-Doppler perfusion imaging of cortical spreading depression in rat neocortex. Neuroreport. 1995;19:1271–12173. doi: 10.1097/00001756-199506090-00010. [DOI] [PubMed] [Google Scholar]
- Lauritzen M, Hansen AJ, Kronborg D, Wieloch T. Cortical spreading depression is associated with arachidonic acid accumulation and preservation of energy charge. J Cereb Blood Flow Metab. 1990;10:115–122. doi: 10.1038/jcbfm.1990.14. [DOI] [PubMed] [Google Scholar]
- Lauritzen M, Jørgensen MB, Diemer NH, Gjedde A, Hansen AJ. Persistent oligemia of rat cerebral cortex in the wake of spreading depression. Ann Neurol. 1982;12:469–474. doi: 10.1002/ana.410120510. [DOI] [PubMed] [Google Scholar]
- Lauritzen M, Olsen TS, Lassen NA, Paulson OB. Regulation of regional cerebral blood flow during and between migraine attacks. Ann Neurol. 1983a;14:569–572. doi: 10.1002/ana.410140512. [DOI] [PubMed] [Google Scholar]
- Lauritzen M, Skyhøj Olsen T, Lassen NA, Paulson OB. Changes in regional cerebral blood flow during the course of classic migraine attacks. Ann Neurol. 1983b;13:633–641. doi: 10.1002/ana.410130609. [DOI] [PubMed] [Google Scholar]
- Leão AAP. Pial circulation and spreading depression of activity in the cerebral cortex. J Neurophysiol. 1944;7:391–396. doi: 10.1152/jn.1947.10.6.409. [DOI] [PubMed] [Google Scholar]
- Leffler CW, Busija DW. Prostanoids in cortical subarachnoid cerebrospinal fluid and pial arterial diameter in newborn pigs. Circ Res. 1985;57:689–694. doi: 10.1161/01.res.57.5.689. [DOI] [PubMed] [Google Scholar]
- Leffler CW, Balabanova L, Fedinec AL, Parfenova H. Nitric oxide increases carbon monoxide production by piglet cerebral microvessels. Am J Physiol Heart Circ Physiol. 2005;289:H1442–H2447. doi: 10.1152/ajpheart.00464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZJ, Matsuda H, Asada T, Ohnishi T, Kanetaka H, Imabayashi E, Tanaka F. Gender difference in brain perfusion 99mTc-ECD SPECT in aged healthy volunteers after correction for partial volume effects. Nucl Med Commun. 2004;25:999–1005. doi: 10.1097/00006231-200410000-00003. [DOI] [PubMed] [Google Scholar]
- Lian XY, Stringer JL. Astrocytes contribute to regulation of extracellular calcium and potassium in the rat cerebral cortex during spreading depression. Brain Res. 2004;25:177–184. doi: 10.1016/j.brainres.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Macfarlane R, Tasdemiroglu E, Moskowitz MA, Uemura Y, Wei EP, Kontos HA. Chronic trigeminal ganglionectomy or topical capsaicin application to pial vessels attenuates postocclusive cortical hyperemia but does not influence postischemic hypoperfusion. J Cereb Blood Flow Metab. 1991;11:261–71. doi: 10.1038/jcbfm.1991.58. [DOI] [PubMed] [Google Scholar]
- Martins-Ferreira H, de Oliveira Castro G. Light-scattering changes accompanying spreading depression in isolated retina. J Neurophysiol. 1966;29:715–26. doi: 10.1152/jn.1966.29.4.715. [DOI] [PubMed] [Google Scholar]
- Martins-Ferreira H, de Oliveira Castro G. Spreading depression in isolated chick retina. Vision Res. 1971;3(Suppl):171–84. doi: 10.1016/0042-6989(71)90038-1. [DOI] [PubMed] [Google Scholar]
- Matsushima K, Hogan MJ, Hakim AM. Cortical spreading depression protects against subsequent focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 1996;16:221–226. doi: 10.1097/00004647-199603000-00006. [DOI] [PubMed] [Google Scholar]
- Mayevsky A, Weiss HR. Cerebral blood flow and oxygen consumption in cortical spreading depression. J Cereb Blood Flow Metab. 1991;11:829–836. doi: 10.1038/jcbfm.1991.142. [DOI] [PubMed] [Google Scholar]
- Meadows GE, Kotajima F, Vazir A, Kostikas K, Simonds AK, Morrell MJ, Corfield DR. Overnight changes in the cerebral vascular response to isocapnic hypoxia and hypercapnia in healthy humans: protection against stroke. Stroke. 2005;36:2367–2372. doi: 10.1161/01.STR.0000185923.49484.0f. [DOI] [PubMed] [Google Scholar]
- Mayevsky A, Doron A, Manor T, Meilin S, Zarchin N, Quaknine GE. Cortical spreading depression recorded from the human brain using a mutiparametric monitoring system. Brain Res. 1996;740:268–274. doi: 10.1016/s0006-8993(96)00874-8. [DOI] [PubMed] [Google Scholar]
- Meng W, Busija DW. Oxygen radicals do not play a role in arteriolar dilation during cortical spreading depression. J Cereb Blood Flow Metab. 1996;16:175–179. doi: 10.1097/00004647-199601000-00021. [DOI] [PubMed] [Google Scholar]
- Meng W, Colonna DM, Tobin JR, Busija DW. Nitric oxide and prostaglandins interact to mediate arteriolar dilation during cortical spreading depression. Am J Physiol Heart Circ Physiol. 1995;269:H176–H181. doi: 10.1152/ajpheart.1995.269.1.H176. [DOI] [PubMed] [Google Scholar]
- Mercier F, Hatton GI. Immunocytochemical basis for a meningeo-glial network. J Comp Neurol. 2000;420:445–465. doi: 10.1002/(sici)1096-9861(20000515)420:4<445::aid-cne4>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Miettinen S, Fusco FR, Yriänheikki J, Keinänen R, Hirvonen T, Toivainen R, Närhi M, Hökfelt T, Koistinaho J. Spreading depression and focal brain ischemia induce cyclooxygenase-2 in cortical neurons through N-methyl-D-aspartic acid-repectors and phsopholipase A2. Proc Natl Acad Sci USA. 1997;94:6500–6505. doi: 10.1073/pnas.94.12.6500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mies G, Niebuhr I, Hossmann KA. Simultaneous measurement of blood flow and glucose metabolism by autoradiographic techniques. Stroke. 1981;12:581–588. doi: 10.1161/01.str.12.5.581. [DOI] [PubMed] [Google Scholar]
- Mraovitch S, Calando Y, Goadsby PJ, Seylaz J. Subcortical cerebral blood flow and metabolic changes elicited by cortical spreading depression in rat. Cephalalgia. 1992;12:137–141. doi: 10.1046/j.1468-2982.1992.1203137.x. [DOI] [PubMed] [Google Scholar]
- Mulligan SJ, MacVicar BA. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature. 2004;431:195–199. doi: 10.1038/nature02827. [DOI] [PubMed] [Google Scholar]
- Nam MJ, Thore CR, Busija DW. Effects of protein kinase C activation on prostaglandin production and cyclooxygenase mRNA levels in ovine astroglia. Prostaglandins. 1996;51:203–213. doi: 10.1016/0090-6980(96)00004-4. [DOI] [PubMed] [Google Scholar]
- Murphy S, Rich G, Orgren KI, Moore SA, Faraci FM. Astrocyte-derived lipoxygenase product evokes endothelium-dependent relaxation of the basilar artery. J Neurosci Res. 1994;38:314–318. doi: 10.1002/jnr.490380309. [DOI] [PubMed] [Google Scholar]
- Nagy K, Kis B, Rajapakse NC, Bari F, Busija DW. Diazoxide preconditioning protects against neuronal cell death by attenuation of oxidative stress upon glutamate stimulation. J Neurosci Res. 2004;76:697–704. doi: 10.1002/jnr.20120. [DOI] [PubMed] [Google Scholar]
- Nedergaard M, Hansen AJ. Spreading depression is not associated with neuronal injury in the normal brain. Brain Res. 1988;449:395–398. doi: 10.1016/0006-8993(88)91062-1. [DOI] [PubMed] [Google Scholar]
- Nielsen AN, Fabricius M, Lauritzen M. Scanning laser-Doppler flowmetry of rat cerebral circulation during cortical spreading depression. J Vasc Res. 2000;37:513–522. doi: 10.1159/000054084. [DOI] [PubMed] [Google Scholar]
- Niermann H, Amiry-Moghaddam M, Holthoff K, Witte OW, Ottersen OP. A novel role of vasopressin in the brain: modulation of activity-dependent water flux in the neocortex. J Neurosci. 2001;21:3045–3051. doi: 10.1523/JNEUROSCI.21-09-03045.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki K, Moskowitz MA, Maynard KI, Koketsu N, Dawson TM, Bredt DS, Snyder SH. Possible origins and distribution of immunoreactive nitric oxide synthase-containing nerve fibers in cerebral arteries. J Cereb Blood Flow Metab. 1993;13:70–79. doi: 10.1038/jcbfm.1993.9. [DOI] [PubMed] [Google Scholar]
- Obrenovitch TP, Urenjak J, Wang M. Nitric oxide formation during cortical spreading depression is critical for rapid subsequent recovery of ionic homeostasis. J Cereb Blood Flow Metab. 2002;22:680–688. doi: 10.1097/00004647-200206000-00006. [DOI] [PubMed] [Google Scholar]
- Olesen J. Cerebral and extracranial circulatory disturbances in migraine: pathophysiological implications. Cerebrovasc Brain Metab Rev. 1991;3:1–28. [PubMed] [Google Scholar]
- Olesen J, Larsen B, Lauritzen M. Focal hyperemia followed by spreading oligemia and impaired activation of rCBF in classic migraine. Ann Neurol. 1981;9:344–352. doi: 10.1002/ana.410090406. [DOI] [PubMed] [Google Scholar]
- Olesen J, Diener HC, Husstedt IW, Goadsby PJ, Hall D, Meier U, Pollentier S, Lesko LM. Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acture treatment of migraine. N Engl J Med. 2004;350:1104–1110. doi: 10.1056/NEJMoa030505. [DOI] [PubMed] [Google Scholar]
- Otsuka H, Ueda K, Heimann A, Kempski O. Effects of cortical spreading depression on cortical blood flow, impedance, DC potential, and infarct size in a rat venous infarct model. Exp Neurol. 2000;162:201–214. doi: 10.1006/exnr.1999.7326. [DOI] [PubMed] [Google Scholar]
- Otori T, Greenberg JH, Welsh FA. Cortical spreading depression causes a long-lasting decrease in cerebral blood flow and induces tolerance to permanent focal ischemia in rat brain. J Cereb Blood Flow Metab. 2003;23:43–50. doi: 10.1097/01.WCB.0000035180.38851.38. [DOI] [PubMed] [Google Scholar]
- Petzold GC, Einhäupl KM, Dirnagl U, Dreier JP. Ischemia triggered by spreading neural activation is induced by endothelin-1 and hemoglobin in the subarachnoid space. Ann Neurol. 2003;54:591–598. doi: 10.1002/ana.10723. [DOI] [PubMed] [Google Scholar]
- Petzold GC, Haack S, von Bohlen, Und Halbach O, Priller J, Lehmann TN, Heinemann U, Dirnagl U, Dreier JP. Nitric oxide modulates spreading depolarization threshold in the human and rodent cortex. Stroke. 2008;39:1292–1299. doi: 10.1161/STROKEAHA.107.500710. [DOI] [PubMed] [Google Scholar]
- Phillis JW. Adenosine and adenine nucleotides as regulators of cerebral blood flow: roles of acidosis, cell swelling, and KATP channels. Crit Rev Neurobiol. 2004;16:237–270. doi: 10.1615/critrevneurobiol.v16.i4.20. [DOI] [PubMed] [Google Scholar]
- Piper RD, Lambert GA. Inhalational anesthetics inhibit spreading depression: relevance to migraine. Cephalalgia. 1996;16:87–92. doi: 10.1046/j.1468-2982.1996.1602087.x. [DOI] [PubMed] [Google Scholar]
- Rasmussen BK, Jensen R, Schroll M. Epidemiology of headache in the general population: a prevalence study. J Clin Epidemiol. 1991;44:1147–1157. doi: 10.1016/0895-4356(91)90147-2. [DOI] [PubMed] [Google Scholar]
- Read SJ, Manning P, McNeil CJ, Hunter AJ, Parsons AA. Effects of sumatriptan on nitric oxide and superoxide balance during glyceryl trinitrate infusion in the rat. Implications for antimigraine mechanisms. Brain Res. 1999;847:1–8. doi: 10.1016/s0006-8993(99)01985-x. [DOI] [PubMed] [Google Scholar]
- Read SJ, Smith MI, Hunter AJ, Parsons AA. The dynamics of nitric oxide release measured directly and in real time following repeated waves of cortical spreading depression in the anaesthetized cat. Neurosci Lett. 1997;232:127–130. doi: 10.1016/s0304-3940(97)00604-6. [DOI] [PubMed] [Google Scholar]
- Read SJ, Smith MI, Hunter AJ, Upton N, Parsons AA. SB-220453, a potential novel antimigraine agent, inhibits nitric oxide release following induction of cortical spreading depression in the anaesthetized cat. Cephalalgia. 2000;20:92–99. doi: 10.1046/j.1468-2982.2000.00022.x. [DOI] [PubMed] [Google Scholar]
- Reuter U, Weber JR, Gold L, Arnold G, Wolf T, Dreier J, Lindauer U, Dirnagl U. Perivascular nerves contribute to cortical spreading depression-associated hyperemia in rats. Am J Physiol Heart Circ Physiol. 1998;43:H1979–H1987. doi: 10.1152/ajpheart.1998.274.6.H1979. [DOI] [PubMed] [Google Scholar]
- Richter F, Lehmenkühler A. Spreading depression can be restricted to distinct depths of the rat cerebral cortex. Neurosci Lett. 1993;152:65–68. doi: 10.1016/0304-3940(93)90484-3. [DOI] [PubMed] [Google Scholar]
- Richter F, Lehmenkühler A, Fechner R, Manveljan L, Haschke W. Postnatal conditioning for spreading cortical depression in the rat brain. Brain Res Dev Brain Res. 1998;106:217–221. doi: 10.1016/s0165-3806(98)00018-2. [DOI] [PubMed] [Google Scholar]
- Risling M, Dalsgaard CJ, Frisén J, Sjögren AM, Fried K. Substance P-,calcitonin gene-related peptide, growth-associated protein-43, and neurotrophin receptor-like immunoreactivity associated with unmyelinated axons in feline ventral roots and pia mater. J Comp Neurol. 1994;339:365–386. doi: 10.1002/cne.903390306. [DOI] [PubMed] [Google Scholar]
- Rosenblum WI, Shimizu T, Nelson GH. Endothelium-dependent effects of substance P and calcitonin gene-related peptide on mouse pial arterioles. Stroke. 1993;24:1043–1047. doi: 10.1161/01.str.24.7.1043. [DOI] [PubMed] [Google Scholar]
- Roy C, Sherrington C. On the regulation of the blood supply of the brain. J Physiol. 1890;11:85–108. doi: 10.1113/jphysiol.1890.sp000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sams A, Knyihár-Csillik E, Engberg J, Szok D, Tajti J, Bodi I, Edvinsson L, Vécsei L, Jansen-Olesen I. CGRP and adrenomedullin receptor populations in human cerebral arteries: in vitro pharmacology and molecular investigations in different artery sizes. Eur J Pharmacol. 2000;408:183–193. doi: 10.1016/s0014-2999(00)00781-0. [DOI] [PubMed] [Google Scholar]
- Schuh-Hofer S, Lobsien E, Brodowsky R, Vogt J, Dreier JP, Klee R, Dirnagl U, Lindauer U. The cerebrovascular response to elevated potassium--role of nitric oxide in the in vitro model of isolated rat middle cerebral arteries. Neurosci Lett. 2001;306:61–4. doi: 10.1016/s0304-3940(01)01878-x. [DOI] [PubMed] [Google Scholar]
- Seitz I, Dirnagl U, Lindauer U. Impaired vascular reactivity of isolated rat middle cerebral artery after cortical spreading depression in vivo. J Cereb Blood Flow Metab. 2004;24:526–530. doi: 10.1097/00004647-200405000-00006. [DOI] [PubMed] [Google Scholar]
- Shen PJ, Gundlach AL. Prolonged induction of neuronal NOS expression and activity following cortical spreading depression (SD): Implications for SD- and NO-mediated neuroprotection. Exp Neurol. 1999;160:317–332. doi: 10.1006/exnr.1999.7218. [DOI] [PubMed] [Google Scholar]
- Shibata M, Leffler CW, Busija DW. Cerebral hemodynamics during cortical spreading depression in rabbits. Brain Res. 1990;530:267–274. doi: 10.1016/0006-8993(90)91294-q. [DOI] [PubMed] [Google Scholar]
- Shibata M, Leffler CW, Busija DW. Evidence against parenchymal metabolites directly promoting pial arteriolar dilation during cortical spreading depression in rabbits. Brain Res Bull. 1991a;26:753–758. doi: 10.1016/0361-9230(91)90171-f. [DOI] [PubMed] [Google Scholar]
- Shibata M, Leffler CW, Busija DW. Prostanoids attenuate pial arteriolar dilation induced by cortical spreading depression in rabbits. Am J Physiol Reg Int Comp Physiol. 1991b;261:R828–R834. doi: 10.1152/ajpregu.1991.261.4.R828. [DOI] [PubMed] [Google Scholar]
- Shibata M, Leffler CW, Busija DW. Pial arteriolar constriction following cortical spreading depression is mediated by prostanoids. Brain Res. 1992;572:190–197. doi: 10.1016/0006-8993(92)90469-p. [DOI] [PubMed] [Google Scholar]
- Shimizu K, Bari F, Busija DW. Glibenclamide enhances cortical spreading depression-associated hyperemia in the rat. Neuroreport. 2000a;11:2103–2106. doi: 10.1097/00001756-200007140-00009. [DOI] [PubMed] [Google Scholar]
- Shimizu K, Veltkamp R, Busija DW. Characteristics of induced spreading depression after transient focal ischemia in the rat. Brain Res. 2000b;861:316–324. doi: 10.1016/s0006-8993(00)02032-1. [DOI] [PubMed] [Google Scholar]
- Shimizu K, Miller AW, Erdős B, Bari F, Busija DW. Role of endothelium in hyperemia during cortical spreading depression. Brain Res. 2002;928:40–49. doi: 10.1016/s0006-8993(01)03352-2. [DOI] [PubMed] [Google Scholar]
- Shimazawa M, Hara H. An experimental model of migraine with aura: cortical hypoperfusion following spreading depression in the awake and freely moving rat. Clin Exp Pharmacol Physiol. 1996;23:890–892. doi: 10.1111/j.1440-1681.1996.tb01139.x. [DOI] [PubMed] [Google Scholar]
- Shin HK, Jones PB, Garcia-Alloza M, Borrelli L, Greenberg SM, Bacskai BJ, Frosch MP, Hyman BT, Moskowitz MA, Ayata C. Age-dependent cerebrovascular dysfunction in a transgenic mouse model of cerebral amyloid angiopathy. Brain. 2007;130:2310–2319. doi: 10.1093/brain/awm156. [DOI] [PubMed] [Google Scholar]
- Shin HK, Nishimura M, Jones PB, Ay H, Boas DA, Moskowitz MA, Ayata C. Mild induced hypertension improves blood flow and oxygen metabolism in transient focal cerebral ischemia. Stroke. 2008;39:1548–1555. doi: 10.1161/STROKEAHA.107.499483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sietz I, Dirnagl U, Landauer U. Impaired vascular reactivity of isolated rat middle cerebral artery after cortical spreading depression in vivo. J Cereb Blood Flow Metab. 2004;24:526–530. doi: 10.1097/00004647-200405000-00006. [DOI] [PubMed] [Google Scholar]
- Smith JM, Bradley DP, James MF, Huang CL. Physiological studies of cortical spreading depression. Biol Rev Camb Philos Soc. 2006;81:457–481. doi: 10.1017/S1464793106007081. [DOI] [PubMed] [Google Scholar]
- Somjen GG. Mechanisms of spreading depression and hypoxic spreading depression-like depolarizations. Physiol Rev. 2001;81:1065–1096. doi: 10.1152/physrev.2001.81.3.1065. [DOI] [PubMed] [Google Scholar]
- Somjen GG. Is spreading depression bad for you? Focus on “repetitive normoxic spreading depression-like events result in cell damage in juvenile hippocampal slice cultures.”. J Neurophysiol. 2006;95:16–17. doi: 10.1152/jn.01008.2005. [DOI] [PubMed] [Google Scholar]
- Sonn J, Mayevsky A. The effect of ethanol on metabolic, hemodynamic and electrical responses to cortical spreading depression. Brain Res. 2001;908:174–186. doi: 10.1016/s0006-8993(01)02643-9. [DOI] [PubMed] [Google Scholar]
- Sonn J, Mayevsky A. Effects of anesthesia on the responses to cortical spreading depression in the rat brain in vivo. Neurol Res. 2006;28:206–219. doi: 10.1179/016164105X49445. [DOI] [PubMed] [Google Scholar]
- Strecker T, Dux M, Messlinger K. Nitric oxide releases calcitonin gene-related peptide from rat dura mater encephali promoting increases in meningeal blood flow. J Vasc Res. 2002;39:89–496. doi: 10.1159/000067206. [DOI] [PubMed] [Google Scholar]
- Strong AJ, Hartings JA, Dreier JP. Cortical spreading depression: an adverse but treatable factor in intensive care? Curr Opin Crit Care. 2007;13:126–33. doi: 10.1097/MCC.0b013e32807faffb. [DOI] [PubMed] [Google Scholar]
- Strong AJ, Anderson PJ, Watts HR, Virley DJ, Lloyd A, Irving EA, Nagafuji T, Ninomiya M, Nakamura H, Dunn AK, Graf R. Peri-infarct depolarizations lead to loss of perfusion in ischaemic gyrencephalic cerebral cortex. Brain. 2007;130:995–1008. doi: 10.1093/brain/awl392. [DOI] [PubMed] [Google Scholar]
- Sukhotinsky I, Dilekoz E, Moskowitz MA, Ayata C. Hypoxia and hypotension transform the blood flow response to cortical spreading depression from hyperemia into hypoperfusion in the rat. J Cereb Blood Flow Metab. 2008 doi: 10.1038/jcbfm.2008.35. in press. [DOI] [PubMed] [Google Scholar]
- Takano T, Tian G-F, Peng W, Lou N, Lovatt D, Hansen AJ, Kasischke KA, Nedergaard M. Cortical spreading depression causes and coincides with tissue hypoxia. Nat Neurosci. 2007;10:754–762. doi: 10.1038/nn1902. [DOI] [PubMed] [Google Scholar]
- Thompson CS, Hakim AM. Cortical spreading depression modifies components of the inflammatory cascade. Mol Neurobiol. 2005;32:51–57. doi: 10.1385/MN:32:1:051. [DOI] [PubMed] [Google Scholar]
- Thore CR, Nam M, Busija D. Phorbol ester-induced prostaglandin production in piglet cortical astroglia. Am J Physiol Reg Int Physiol. 1994;267:R34–R37. doi: 10.1152/ajpregu.1994.267.1.R34. [DOI] [PubMed] [Google Scholar]
- Thore CR, Nam MJ, Busija DW. Immunofluorescent localization of constitutive and inducible prostaglandin H synthase in ovine astroglia. J Comp Neurol. 1996;367:1–9. doi: 10.1002/(SICI)1096-9861(19960325)367:1<1::AID-CNE1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Toda N, Okamura T. The pharmacology of nitric oxide in the peripheral nervous system of blood vessels. Pharmacol Rev. 2003;55:271–324. doi: 10.1124/pr.55.2.3. [DOI] [PubMed] [Google Scholar]
- Tomita M, Schiszler I, Tomita Y, Tanahashi N, Takeda H, Osada T, Suzuki N. Initial oligemia with capillary flow stop followed by hyperemia during K+-induced cortical spreading depression in rats. J Cereb Blood Flow Metab. 2005;25:742–747. doi: 10.1038/sj.jcbfm.9600074. [DOI] [PubMed] [Google Scholar]
- Umemura A, Yamada K. Contribution of cerebrovascular parasympathetic and sensory innervation to the development of cerebral edema in rat focal ischemia and reperfusion. Neurosci Lett. 1996;215:134–136. [PubMed] [Google Scholar]
- Van Harreveld A. Compounds in brain extracts causing spreading depression of cerebral cortical activity and contraction of crustacean muscle. J Neurochem. 1959;3:300–315. doi: 10.1111/j.1471-4159.1959.tb12636.x. [DOI] [PubMed] [Google Scholar]
- Van Harreveld A, Ochs S. Electrical and vascular concomitants of spreading depression. Am J Physiol. 1957;189:159–66. doi: 10.1152/ajplegacy.1957.189.1.159. [DOI] [PubMed] [Google Scholar]
- Van Harreveld A, Stamm JS. Vascular concomitants of spreading cortical depression. J Neurophysiol. 1952;15:487–496. doi: 10.1152/jn.1952.15.6.487. [DOI] [PubMed] [Google Scholar]
- Van Harreveld A, Stamm JS, Christensen E. Spreading depression in rabbit, cat, and monkey. Am J Physiol. 1956;184:312–320. doi: 10.1152/ajplegacy.1956.184.2.312. [DOI] [PubMed] [Google Scholar]
- Veltkamp R, Domoki F, Bari F, Busija DW. Potassium channel activators protect the N-Methyl-D-Aspartate—induced cerebral vascular dilation after combined hypoxia and ischemia in piglets. Stroke. 1998;29:837–843. doi: 10.1161/01.str.29.4.837. [DOI] [PubMed] [Google Scholar]
- Vaucher E, Tong XK, Cholet N, Lantin S, Hamel E. GABA neurons provide a rich input to microvessels but not nitric oxide neurons in the rat cerebral cortex: A means for direct regulation of local cerebral blood flow. J Comp Neurol. 2000;421:161–171. [PubMed] [Google Scholar]
- Verhaegen MJ, Todd MM, Warner DW, James B, Weeks JB. The role of electrode size on the incidence of spreading depression and on cortical cerebral blood flow as measured by H2 clearance. J Cereb Blood Flow Metab. 1992;12:230–237. doi: 10.1038/jcbfm.1992.33. [DOI] [PubMed] [Google Scholar]
- von Baumgarten L, Trabold R, Thal S, Back T, Plesnila N. Role of cortical spreading depressions for secondary brain damage after traumatic brain injury in mice. J Cereb Blood Flow Metab. 2008 doi: 10.1038/jcbfm.2008.30. in press. [DOI] [PubMed] [Google Scholar]
- Wagerle LC, Busija DW. Effect of thromboxane A2/Endoperoxide antagonist SQ29548 on the contractile response to acetylcholine in the newborn piglet cerebral arteries. Circ Res. 1990;6:824–831. doi: 10.1161/01.res.66.3.824. [DOI] [PubMed] [Google Scholar]
- Wahl M, Schilling L, Parsons AA, Kaumann A. Involvement of calcitonin gene-related peptide (CGRP) and nitric oxide (NO) in the pial artery dilation elicited by cortical spreading depression. Brain Res. 1994;637:204–210. doi: 10.1016/0006-8993(94)91234-3. [DOI] [PubMed] [Google Scholar]
- Wang M, Urenjak J, Fedele E, Obrenovitch TP. Effects of phosphodiesterase inhibition on cortical spreading depression and associated changes in extracellular cyclic GMP. Biochem Pharmacol. 2004;67:1619–1627. doi: 10.1016/j.bcp.2003.12.029. [DOI] [PubMed] [Google Scholar]
- Wei EP, Moskowitz MA, Boccalini P, Kontos HA. Calcitonin gene-related peptide mediates nitroglycerin and sodium nitroprusside-induced vasodilation in feline cerebral arteries. Circ Res. 1992;70:1313–1319. doi: 10.1161/01.res.70.6.1313. [DOI] [PubMed] [Google Scholar]
- Wolf T, Landauer U, Obrig H, Dreier J, Back T, Villringer A, Dirnagl U. Systemic nitric oxide synthase inhibition does not affect brain oxygenation during cortical spreading depression in rats: a noninvasive near-infrared spectroscopy and laser-Doppler flowmetry study. J Cereb Blood Flow Metab. 1996;16:1100–1107. doi: 10.1097/00004647-199611000-00003. [DOI] [PubMed] [Google Scholar]
- Wolf T, Lindauer U, Villringer A, Dirnagl U. Excessive oxygen or glucose supply does not alter the blood flow response to somatosensory stimulation or spreading depression in rats. Brain Res. 1997;761:290–299. doi: 10.1016/s0006-8993(97)00354-5. [DOI] [PubMed] [Google Scholar]
- Woods RP, Iacoboni M, Mazziotta JC. Brief report: bilateral spreading cerebral hypoperfusion during spontaneous migraine headache. N Engl J Med. 1994;331:1689–1692. doi: 10.1056/NEJM199412223312505. [DOI] [PubMed] [Google Scholar]
- Windmüller O, Lindauer U, Foddis M, Einhäupl KM, Dirnagl U, Heinemann U, Dreier JP. Ion changes in spreading ischaemia induce rat middle cerebral artery constriction in the absence of NO. Brain. 2005;128:2042–2051. doi: 10.1093/brain/awh545. [DOI] [PubMed] [Google Scholar]
- Xu HL, Koenig HM, Ye S, Feinstein DL, Pelligrino DA. Influence of the glia limitans on pial arteriolar relaxation in the rat. Influence of the glia limitans on pial arteriolar relaxation in the rat. Am J Physiol Heart Circ Physiol. 2004;287:H331–H339. doi: 10.1152/ajpheart.00831.2003. [DOI] [PubMed] [Google Scholar]
- Xu HL, Pelligrino DA. ATP release and hydrolysis contribute to rat pial arteriolar dilatation elicited by neuronal activation. Exp Physiol. 2007;92:647–651. doi: 10.1113/expphysiol.2006.036863. [DOI] [PubMed] [Google Scholar]
- Yokota C, Inoue H, Kuge Y, Abumiya T, Tagaya M, Hasegawa Y, Ejima N, Tamaki N, Minematsu K. Cyclooxygenase-2 expression associated with spreading depression in a primate model. J Cereb Blood Flow Metab. 2003;23:395–398. doi: 10.1097/01.WCB.0000055293.67563.2E. [DOI] [PubMed] [Google Scholar]
- Yokota C, Kuge Y, Hasegawa Y, Tagaya M, Abumiya T, Ejima N, Tamaki N, Yamaguchi T, Minematsu K. Unique profile of spreading depression in a primate model. J Cereb Blood Flow Metab. 2002;22:835–842. doi: 10.1097/00004647-200207000-00008. [DOI] [PubMed] [Google Scholar]
- Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, Carmignoto G. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci. 2003;6:43–50. doi: 10.1038/nn980. [DOI] [PubMed] [Google Scholar]
- Zhang ZG, Chopp M, Maynard KI, Moskowitz MA. Cerebral blood flow changes during cortical spreading depression are not altered by inhibition of nitric oxide synthesis. J Cereb Blood Flow Metab. 1994;14:939–943. doi: 10.1038/jcbfm.1994.125. [DOI] [PubMed] [Google Scholar]
- Zhang F, Sprague SM, Farrokhi F, Henry MN, Son MG, Vollmer DG. Reversal of attenuation of cerebrovascular reactivity to hypercapnia by a nitric oxide donor after controlled cortical impact in a rat model of traumatic brain injury. J Neurosurg. 2002;97:963–969. doi: 10.3171/jns.2002.97.4.0963. [DOI] [PubMed] [Google Scholar]
- Zhang R, Wilson TE, Witkowski S, Sui J, Crandall GG, Levine BD. Inhibition of nitric oxide synthase does not alter dynamic cerebral autoregulation in humans. Am J Physiol Heart Circ Physiol. 2004;286:H863–H869. doi: 10.1152/ajpheart.00373.2003. [DOI] [PubMed] [Google Scholar]
- Zvan B, Zaletel M, Pogacnik T, Kiauta T. Testing of cerebral endothelium function with L-arginine after stroke. Int Angiol. 2002;21:256–259. [PubMed] [Google Scholar]