

Abstract
DC maturation is known to be a necessary step in the generation of an effective immune response. We have used vaccinia virus (VACV) as a model to investigate the regulation of DC subsets in vivo following infection. While a number of in vitro studies have shown that DC infected with VACV fail to undergo maturation, the effect of VACV infection on the maturation of and cytokine production by DC subsets in vivo remains less defined. We have found that following systemic infection with vaccinia virus, both CD8+ and CD8− dendritic cells are infected. The number of infected DC peaked at 6 hours and was highly decreased by 24h postinfection. In both subsets, there was evidence of generalized upregulation of costimulatory molecules. Surprisingly, this included vaccinia infected DC, suggesting the regulation of DC maturation in vivo is much more complex and likely influenced by DC extrinsic signals. However, while we observed generalized upregulation of costimulatory molecules, IL-12 production was restricted to a subset of non-infected cells in both the CD8+ and CD8− DC populations. Importantly, the control of IL-12 production was differentially dependent on MyD88 signaling. IL-12 production was ablated in the absence of MyD88 in CD8-DC, while it was unchanged in CD8+ DC. These findings provide new insights into the control of DC maturation in vivo and demonstrate that the regulation of maturation in vivo following virus infection can be differentially controlled in distinct types of DC.
Keywords: Vaccinia virus, Dendritic cell, antigen presentation, maturation, costimulatory molecule
INTRODUCTION
Dendritic cells are a critical player in the activation of naive T cells. Numerous studies support the view that maturation is required for DC to become efficient activators of T cells (for review see (Macagno et al., 2007)). Maturation involves the upregulation of costimulatory molecules, like CD80 and CD86, as well as the production of cytokines, especially IL-12. Given the critical importance of DC maturation it is perhaps not surprising that viruses have developed mechanisms to prevent this process. One example of such a virus is vaccinia virus (VACV), which has a large arsenal of immune evasion proteins that counteract many of the anti-viral mechanisms of the innate and adaptive immune response (Alcami and Koszinowski, 2000; Seet et al., 2003). The VACV proteins encoded by the K3L and E3L genes specifically inhibit dsRNA resulting in a failure to produce and respond to type I IFN, a potent cytokine for DC maturation (Beattie et al., 1991; Chang et al., 1992; Paez and Esteban, 1984; Xiang et al., 2002). In addition, the protein encoded by the A46R gene can block TLR signaling by interacting with TLR adaptor proteins MyD88, Toll-IL-1 receptor domain-containing adaptor-inducing IFN-beta (TRIF), and TRIF-related adapter molecule (TRAM) (Stack et al., 2005), while the A52R gene encodes a protein that has been implicated in blocking TLR signaling by blocking activation of NF-κB, a key transcription factor involved in the induction of inflammatory gene transcription (Bowie et al., 2000; Harte et al., 2003; Sen and Baltimore, 1986b; Sen and Baltimore, 1986a; Baeuerle and Henkel, 1994). Each of these proteins has the potential to impact dendritic cell maturation and therefore to affect the ability of DC to contribute to the generation of an adaptive immune response.
Based on the myriad of immunoregulatory proteins produced by vaccinia virus, it was not surprising that our previous studies found that in vitro infection of murine bone marrow derived dendritic cells (BMDC) did not result in maturation (Yates and Alexander-Miller, 2007). A similar finding has been reported for human DC(Drillien et al., 2000; Engelmayer et al., 1999). However the failure of vaccinia virus infection to induce maturation was seemingly at odds with the reports that direct presentation contributes significantly to the generation of a CD8+ T cell response in vivo (Basta et al., 2002; Shen et al., 2002), a process which presumably would require a mature DC. Not surprisingly, cross-presentation mediated by CD8+ DC, has also been shown to contribute to the generation of a T cell response following infection with VACV (Basta et al., 2002; Shen et al., 2002).
Understanding the contributions of distinct DC subsets in the generation of an anti-viral response necessitates identification of how these populations respond following in vivo infection. For example, are distinct subsets equally susceptible to infection and how does their maturation state differ with regard to the expression of costimulatory molecules and cytokines important for T cell activation? In the present study, we addressed these issues using the model of systemic vaccinia virus infection. We found that all DC exhibited increased expression of the costimulatory markers CD80 and CD86. This included infected DC, a result counter to that found with in vitro studies. However, in contrast to the generalized upregulation of costimulatory molecules in infected as well as non-infected DC, IL-12 production was restricted to non-infected cells. While IL-12 production was apparent in both CD8+ and CD8− DC populations, surprisingly the mechanism by which IL-12 production was controlled differed substantially between the subsets. CD8− DC produced this cytokine in an MyD88 dependent manner while CD8+ DC were independent of this signaling pathway. These data suggest that while both subsets have the ability express the signals necessary to directly activate naïve CD8+ T cells, the mechanism by which they achieve the requisite maturation state is distinctly controlled.
RESULTS
Both CD8+ and CD8− dendritic cells in the spleen are infected following intravenous administration of vaccinia virus
In a previous study, CD8+ dendritic cells were reported to be the relevant subset for the activation of naïve CD8+ T cells following infection with vaccinia virus (Belz et al., 2004). This finding suggested that CD8− DC either lacked viral antigen or the appropriate costimulatory molecules necessary for T cell activation. To test this first possibility, we employed a recombinant vaccinia virus that expressed a fusion protein wherein the Ova 257–264 epitope had been inserted between the influenza NP protein and eGFP (VACV-NP-S-EGFP) (Norbury et al., 2002). Following intravenous administration of virus, we determined the extent to which CD8+ and CD8− DC were infected as measured by the expression of eGFP. At 6, 15, 24 or 48 hours postinfection, splenic DC were isolated by enrichment with CD11c magnetic beads. Isolated cells were stained with antibodies to CD11c and CD8 and analyzed by flow cytometry for the presence of eGFP. These studies showed that both CD8+ and CD8− DC were susceptible to infection and for both subsets, infection was maximal at 6 hours (Fig. 1). At this time, on average approximately 1.7% of CD8− DC and 1.8% of CD8+ DC were eGFP+. The percentage of positive cells decreased significantly by 15h, with very limited to undetectable numbers by 48h postinfection, depending on the animal examined. The changes in the absolute number of eGFP+ cells mirrored the percentages, peaking at 6h and declining in a time-dependent fashion thereafter (Fig. 1D).
Figure 1. The percentage of eGFP+ cells peaks at 6h postinfection in both CD8+ and CD8− subsets.

C57BL/6 mice were intravenously infected with 1×107 pfu of VACV-NP-S-EGFP. At 6, 15, 24 and 48h p.i., splenic DC were enriched using CD11c magnetic beads. Cells were stained with antibodies to CD11c and CD8. A) Representative FSC/SSC and CD11c vs. CD8 plots. B) Representative plots following gating on CD11c+ CD8− (top row) or CD11c+ CD8+ (bottom row) populations. C) Average percent eGFP+ DC from 4 independent experiments. D) Average number of eGFP+ DC in the CD8+ or CD8− subsets over time.
To determine whether eGFP positivity was a readout for infected cells, we performed microscopic analysis to determine the localization of the eGFP protein. The VACV-NP-S-EGFP construct expresses eGFP as part of a chimeric protein wherein the SIINFEKL epitope and eGFP are appended to the C terminus of the NP protein. Thus virally-expressed eGFP localizes to the nucleus as a result of the karyophilic properties of NP (Norbury et al., 2002). As such, nuclear localization of eGFP can be used as an indicator of viral synthesis in the cell. Analysis of eGFP expression in infected dendritic cells isolated at 6 hours p.i. showed nuclear localization in both DC subsets, consistent with infection (Fig. 2). There was no evidence of punctate cytoplasmic staining that would be expected if cells were eGFP+ as a result of uptake of exogenous eGFP. Together these data show that both CD8+ and CD8− DC were susceptible to infection with a transient early wave of infected dendritic cells that is highly diminished by 24–48 p.i.
Figure 2. EGFP+ CD8+ and CD8− DC show evidence of direct infection.

C57BL/6 mice were intravenously infected with 1×107 pfu of VACV-NP-S-EGFP. At 6h p.i., splenic DC were enriched using CD11c magnetic beads. Cells were stained with antibodies to CD11c and CD8. Nuclei are visualized by DAPI staining. Co-localization of DAPI staining and eGFP is indicative of direct infection. Representative eGFP+ CD8+ and eGFP+ CD8− DC are shown.
Dendritic cells infected with vaccinia virus upregulate CD80 and CD86
It has been recently reported that DC in the spleen of vaccinia virus infected cells exhibit generalized upregulation of CD86 (Zhu et al., 2007). However, given the small percentage of cells that were infected our analysis (Fig 1.), it was feasible that infected cells failed to upregulate CD86 and that this was not evident in the analysis of the overall population. Thus the question of whether DC infected in vivo underwent maturation remained unknown. To address this issue, CD11c+ cells were isolated from mice at 6, 15, 24, and 48 hours postinfection with VACV-NP-S-EGFP. As a measure of maturation, expression of CD80 and CD86 on eGFP+ CD11c+ cells was analyzed. In contrast to our previous in vitro studies, where infected cells failed to undergo maturation (Yates and Alexander-Miller, 2007), the expression of CD80 and CD86 was significantly increased on eGFP+ DC isolated from infected mice compared to DC obtained from mock infected mice (Fig. 3). This was not the result of virus-isolate specific differences as this construct failed to induce DC maturation in vitro (data not shown), a finding in agreement with our previous studies using the VV-Ova virus (Yates and Alexander-Miller, 2007). Costimulatory molecule expression increased over the 24 hours that infected cells were routinely detectable following infection. On average CD8− dendritic cells exhibited a 3.0-fold increase in CD80 expression and an 15.7-fold increase in CD86 expression, while CD8+ DC increased CD80 expression by 5.7-fold and CD86 expression by 9.2-fold over the first 24 hours of infection (Fig 3A right two columns and 3B, gray bars). Thus DC infected in vivo efficiently upregulate the expression of costimulatory molecules.
Figure 3. EGFP+ and eGFP− DC undergo similar upregulation of CD80 and CD86.

C57BL/6 mice were intravenously infected with 1×107 pfu of VACV-NP-S-EGFP. At 6, 15, 24 and 48h p.i., splenic DC were enriched using CD11c magnetic beads. Cells were stained with antibodies to CD11c, CD8, CD80, and CD86. A) Representative CD80 and CD86 profiles over time for eGFP+ and eGFP− cells following gating on CD11c+ CD8− and CD11c+ CD8+ populations. B) Average increases in CD80 and CD86 expression in eGFP− (black bars) and eGFP+ (gray bars) cells. Results are shown as fold change over DC from mock infected animals to allow comparison across the four experiments.
We compared the level of expression of CD80 and CD86 on the eGFP− cells to that on eGFP+ cells to determine whether these two populations exhibited similar upregulation of these molecules. Non-infected cells, both CD8+ and CD8− populations, showed similar increases in CD80 and CD86 expression compared to eGFP+ cells (Fig. 3). On average CD8− dendritic cells exhibited a 3.1-fold increase in CD80 expression and an 10.6-fold increase in CD86 expression, while CD8+ DC increased CD80 expression by 4.5-fold and CD86 expression by 7.4-fold over the first 24 hours of infection (Fig 3A, left two columns and 3B, black bars). This bystander maturation suggested the presence of a signal that can induce generalized upregulation of costimulatory molecules. This effect was restricted to early times post-infection as DC present in the spleen at 48 hours showed little evidence of maturation.
A subpopulation of eGFP− DC produce IL-12p40
The upregulation of maturation markers on the majority of cells suggested that many of the dendritic cells present in the spleen, including infected cells, had the potential to deliver costimulatory signals for T cell activation. However, optimal CD8+ T cell activation requires a cytokine signal, IL-12 or IFNαβ (Curtsinger et al., 2003; Hernandez et al., 2002; Curtsinger et al., 2005). In this regard, IL-12 has been shown to be critical for the generation of lytic function in vaccinia-specific effectors (van den Broek et al., 2000). As a recent study has reported that the requirements for the production of IL-12 differ from those required for the upregulation of costimulatory molecules (Schulz et al., 2000), we tested whether cells that had upregulated costimulatory molecules also produced IL-12. At 6 or 24h p.i., splenocyte populations isolated following collagenase digestion were cultured overnight in the presence of GolgiPlug to promote cytokine accumulation. The presence of IL-12p40 was then determined by ICCS. These analyses showed that while upregulation of costimulatory molecules was relatively promiscuous, IL-12 production was restricted to a small subset cells in both the CD8+ and CD8− populations (Fig. 4). Further IL-12p40 producing cells were restricted to the eGFP− populations (Fig. 4A). The production of IL-12 peaked early and was reduced in DC isolated 24h p.i. These data suggested that 1) a fraction of the DC in each subset was receiving an additional signal that promoted IL-12 production and that 2) infected DC were either resistant to or did not encounter this signal.
Figure 4. IL-12 production is limited to a subpopulation of eGFP− DC.

C57BL/6 mice were intravenously infected with 1×107 pfu of VACV-NP-S-EGFP. At 6 or 24h p.i., splenocytes were cultured overnight in the presence GolgiPlug. Following culture, cells were stained with CD11c and CD8-specific antibodies followed detection of IL-12 production by ICCS. Representative data from 6hr p.i. are shown in A and averaged data from 6 and 24hr p.i. in B. * p= <0.005.
IL-12 production is differentially dependent on MyD88 in CD8+ versus CD8− DC
The production of IL-12 is often a consequence of TLR engagement. TLR receptors, with the exception of TLR3, utilize MyD88 for signal transduction (Akira et al., 2000). To determine whether IL-12 production following VACV infection was dependent on MyD88 we administered virus to MyD88-deficent mice or WT C57BL/6 mice. At 6h p.i. DC were isolated and cultured overnight in the presence of GolgiPlug as above. In agreement with our hypothesis, there was a complete loss of IL-12 production in the absence of MyD88 in CD8− DC, with the percentage of IL-12p40+ cells reduced to that found in mock infected animals (Fig. 5). However, in stark contrast to the results obtained for CD8− DC, the production of IL-12p40 by CD8+ DC was similar in DC obtained from WT and MyD88-deficient mice. These data suggest that IL-12 production is differentially controlled in the CD8+ versus CD8− DC subsets.
Figure 5. CD8− and CD8+ DC differ in their dependence on MyD88 for the production of IL-12.

MyD88-deficient C57BL/6 or WT C57BL/6 mice were infected intravenously with 1×107 pfu of VACV-NP-S-EGFP. Six hours p.i., splenocytes were isolated and cultured overnight in the presence of GolgiPlug. Cells were then stained with CD11c and CD8-specific antibodies followed detection of IL-12 production by ICCS. Data are from 6 animals run over three experiments. * p= <0.001.
Both CD8+ and CD8− DC are capable of activating naïve T cells
The upregulation of costimulatory molecules on both infected CD8+ and CD8− DC and the presence of IL-12 producing DC within these two populations suggested that both subsets would be competent for activation of CD8+ T cells. To test this possibility mice were intravenously infected with VACV-NP-S-EGFP and at 6 and 24 hours postinfection, mice were euthanized and splenic DC isolated. CD11c+ CD8+ and CD11c+ CD8− DC were sorted and co-cultured with OT-I cells from naïve mice. On d3, cells were restimulated with Ova257–264 peptide and the production of IFNγ determined by intracellular staining. The results in figure 6 demonstrate that both CD8+ and CD8− DC were capable of activating OT-I T cells to become IFNγ-producing effectors. At 6h postinfection the two subsets appeared similar in their activating potential. However, by 24h p.i. CD8− DC were inferior to CD8+ cells, showing a 45% reduction in the percentage of cells that could produce IFNγ. Thus both CD8+ and CD8− DC have the capacity to contribute to the generation of the CD8+ T cell response.
Figure 6. Both CD8+ and CD8− DC can activate naïve T cells.

Splenic DC were isolated from C57BL/6 mice at 6 or 24h p.i. with 1×107 pfu of VACV-NP-S-EGFP. Cells were stained with CD11c and CD8-specific antibodies, followed by sorting into CD11c+ CD8− and CD11c+ CD8+ populations. 2.5×104 OT-I cells and 1×105 DC were co-cultured for 3 days after which cultures were restimulated with 1µM Ova257–264 peptide and IFNγ production determined by ICCS. The percentage of CD8+ T cells that produced IFNγ is shown. Results are the average of 4–5 independent experiments. * p≤0.05, ** p≤0.005.
Transfer of naïve CD8+ T cells at 48 hours postinfection results in a reduced effector population compared that obtained when cells are transferred at earlier timepoints
A prediction of the analysis of costimulatory molecule expression is that DC present at 48 hours would be reduced in their ability to activate naïve T cells, i.e. T cells that encounter DC at this time would be predicted to be less efficiently activated. To test this possibility, OT-I cells were transferred into CD8-deficient mice infected with VACV-NP-S-EGFP 6, 24, or 48h prior.
Infection was staggered such that OT-I cells were transferred simultaneously for all conditions, thus ensuring an identical pool of responders (Fig 7A). Analysis of IFNγ producing cells revealed that the peak number of Ova257–264-specific effectors was obtained when OT-1 cells were transferred at 24h following infection (Fig 7B). Transfer of OT-I cells at 6h postinfection resulted in a somewhat reduced (40%) Ova257–264-specific CD8+ T cell population. Although unknown at this time, a number of possibilities could explain this result including initial encounter with a DC that was incompletely matured or activation by infected DC at early times versus cross-presenting DC at later times, which may differ in their efficiency of activation. Importantly, when transfer of OT-I cells was delayed until 48h p.i., the number of IFNγ-producing cells decreased by 4-fold compared to that observed at 24h p.i. This was not due to the failure of these cells to enter into the effector pool because of an ongoing endogenous response, as the recipient mice were devoid of CD8+ T cells. These data strongly support our analysis of DC maturation markers and suggest that optimal priming of CD8+ T cells occurs within the first 24h following infection.
Figure 7. Transfer of OT-I cells at 48h p.i. results in reduced activation compared to transfer at 6 or 24h p.i.
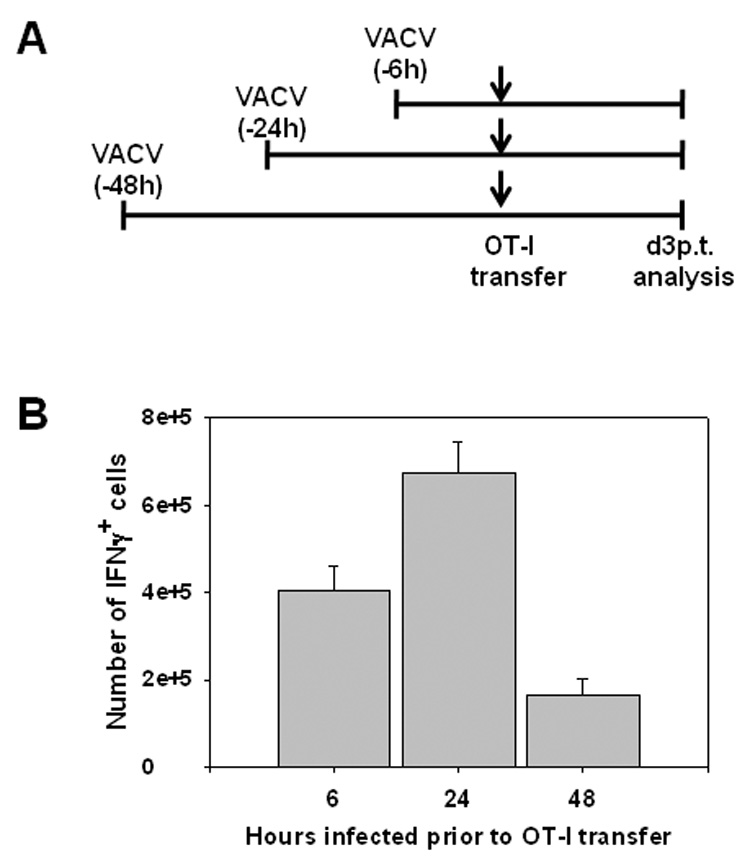
CD8-deficient recipient mice were infected with VACV-NP-S-EGFP at 6, 24, or 48h prior to transfer of OT-I splenocytes (1×105) (A). On d3 post-transfer, splenocytes were isolated and IFNγ production determined by ICCS following stimulation with Ova257–264 peptide. The total number of IFNγ producing cells from each condition is shown (B). Data are the average of 5 mice/condition compiled from two independent experiments.
DISCUSSION
While there are now a relatively large number of reports assessing the regulation of DC maturation as a result of viral infection in vitro, in vivo studies to address this question are much more limited. The latter are of importance as they allow analysis of DC subsets that may not be adequately reproduced through in vitro differentiation cultures. Further, and perhaps more significantly, in vivo studies permit the contribution of the complex vivo environment, including additional cell populations and cytokines that are absent in the culture conditions used in vitro. In our studies, we have used vaccinia virus as a model to probe the regulation of DC subsets in vivo-focusing on CD8+ vs. CD8− populations. This division is based on previous findings suggesting the critical role of CD8+ DC in the activation of naïve virus-specific CD8+ T cells (Belz et al., 2004).
Our data show that vaccinia virus infected DC undergo maturation in vivo, as measured by the upregulation of CD80 and CD86. Further the majority of these infected cells are not undergoing apoptosis (data not shown). That said, it remains possible that some of the infected, dying cells are quickly cleared in vivo and thus are not observed in our analysis. Nonetheless, the presence of mature, infected DC in vivo is in sharp contrast to what we and others have observed in vitro, where bone marrow derived DC do not increase costimulatory marker expression following infection with vaccinia virus (Drillien et al., 2000; Engelmayer et al., 1999; Yates and Alexander-Miller, 2007). One possibility to explain the mature phenotype of infected DC observed in our study was that vaccinia virus infection targeted cells that were already mature. However, our kinetic data argue against this interpretation as CD80 and CD86 expression was only modestly upregulated at 6h p.i. In addition infection of previously matured DC in vitro was highly abortive, suggesting mature DC are resistant to infection (Yates and Alexander-Miller, 2007).
We noted that the level of CD80 and CD86 expressed by infected DC was similar to the generalized DC maturation observed in the non-infected population. This would be consistent with a model of a shared signal for the induction of CD80 and CD86 in infected versus non-infected DC. In a previous report by Zhu et al., the authors found that the increased costimulatory molecule expression following infection with vaccinia virus was the result of a combination of an MyD88-dependent TLR2 signal and an MyD88-independent signal (Zhu et al., 2007). Further the increase was independent of type I IFN (Zhu et al., 2007). We have reproduced this latter finding and in addition have determined that maturation is also independent of TNFα (data not shown). However, it remains possible that other soluble factors present in the infected environment induce maturation.
Both DC subsets were competent for T cell activation as judged by the acquisition of IFNγ producing capabilities in the responding T cells. Proliferation induced by CD8+ versus CD8− DC was relatively similar, although the amount of proliferation was somewhat variable between experiments (data not shown). We note that these analyses were complicated by higher than expected proliferation when DC from mock-infected mice were utilized. Although very few cells were recovered under this condition, those that were present had undergone high amounts of proliferation, however, importantly they did not produce IFNγ. Thus acquisition of IFNγ producing capability appears to be the more stringent readout of activation in this approach.
We did note that at while CD8− and CD8+ DC isolated at 6h p.i. were similar in their activating potential, at 24h p.i. CD8+ DC displayed an increased capacity to induce effector function. This could be the result of a number of factors, including the level of presented antigen or the number of cells presenting antigen. For example, at later times p.i., presentation may be increased in CD8+ DC as a result of cross-presentation, whereas in CD8− DC, presentation remains limited to the small number of infected cells present at this time. The restriction of IL-12 production, a factor shown in many instances to be important for effector cell function, to eGFP− DC would suggest that either IFNγ production by effector T cells generated in the context of a vaccinia virus infection does not require IL-12 or that non-infected DC supply this factor in trans to T cells in conjugate with peptide-presenting infected cells. Certainly it seems reasonable that T cells could utilize this cytokine if produced by neighboring DC in close proximity. We point out that in the case of CD8+ DC, non-infected, IL-12-producing cells may be efficient activators of CD8+ T cells as a result of cross-presentation of viral antigen, although this remains untested at this time.
An attractive possibility for the increased activating potential of CD8+ DC at later times is that it is the result of the increased CD80 expression on CD8+ vs. CD8− DC. CD80 increased by 3-fold in eGFP+ CD8− DC compared to 5.7-fold in eGFP+ CD8+ DC. CD80 expression in eGFP− DC was also increased to a higher level in CD8+ vs. CD8− DC. We have recently reported the important and non-redundant role played by CD80 versus CD86 in the activation of CD8+ T cells (Pejawar-Gaddy and Alexander-Miller, 2006). CD80 is critical for the maximal upregulation of CD25 expression following activation of naïve CD8+ T cells. Thus the higher levels of CD80 expressed by CD8+ DC may contribute to increased proliferation and acquisition of effector function. Another possibility is that it is a result of the higher percentage of CD8+ DC which produce IL-12, a factor known to be involved in the acquisition of effector function (Curtsinger et al., 2003). These models are not mutually exclusive and it is possible that multiple factors contribute to the increased efficacy of CD8+ DC.
The ability of both CD8+ and CD8− DC to serve as APC for the activation of naïve T cells was surprising to us given the previous report from Belz and co-workers showing that only CD8+ DC were capable of activating T cells. One potential difference to explain this discrepancy is the lower dose of virus used in their study. Given that the analysis was performed at 24h, a timepoint where a large percentage of infected cells has been lost (Fig 1 and (Belz et al., 2004)), it is possible that the number of infected CD8− DC was not at sufficient levels to be detected in the assay. The timepoint at which DC are maximally contributing to T cell activation in our model is unknown. However a recent report from Hickman et al. suggested that the majority of priming following subcutaneous administration of VACV occurred within 12 hours following infection (Hickman et al., 2008). Thus DC presenting antigen at the early times, i.e. 6h p.i., may be the most relevant for CD8+ T cell activation.
The finding that both CD8− and CD8+ populations were infected by vaccinia virus and undergo maturation supports the previously reported role of direct and cross-presentation in the generation of the anti-viral response in vivo. Our understanding of these subsets would suggest that CD8+ DC may contribute through both direct and cross-priming, while CD8− DC are restricted to the direct presentation pathway. The use of both pathways of presentation for T cell activation is likely advantageous as a previous report has suggested that direct infection results in the presentation of VACV-derived epitopes that are not generated by cross-presentation (Basta et al., 2002). Thus direct presentation of viral antigens may promote a more comprehensive immune response, ensuring that major epitopes that are presented as a result of infection can be recognized by the T cell response. The development by the immune system of multiple approaches for T cell activation ensures efficient presentation and T cell activation in the face of poxvirus infection.
The restriction of IL-12 production to a subset of uninfected DC was unexpected. This finding suggested that IL-12 production was not triggered by infection, at least not infection that proceeded to the level of detectable eGFP expression. Previous studies have indicated that in some cases the production of IL-12 requires conditioning by a microbial signal followed by a T cell derived CD40L signal (Schulz et al., 2000). Thus it was possible that CD40 engagement on a small subset of DC was responsible for IL-12 production we observed. However, infection of CD40-deficient mice with vaccinia virus did not result in the loss of IL-12 production (data not shown). Given the commonly observed role of TLR signaling in the control of IL-12 production (Bekeredjian-Ding et al., 2006), we tested whether the production of IL-12 was dependent on signaling through MyD88, an adapter molecule involved in all TLR signaling except TLR3 (Akira et al., 2000). Surprisingly we found that the requirement for signaling through MyD88 differed in the CD8+ versus CD8− subset, with CD8+ DC production of IL-12 being independent of this pathway. Importantly, these data suggest that the two DC subsets respond to infection via distinct mechanisms. While the signals responsible for IL-12 production in CD8+ vs. CD8− DC are unknown, one possibility is differential TLR receptor expression in the two DC types. CD8+ DC and CD8− DC have been reported to diverge in their expression of TLR receptors (Edwards et al., 2003). CD8+ DC lack expression of TLR7, but have 28-fold higher expression of TLR3 (Edwards et al., 2003). Given that TLR3 signal transduction is mediated through TRIF, this receptor may promote MyD88-independent signals that result in IL-12 production. In support of the ability of VACV to utilize this pathway, peritoneal macrophages from mice with a defective TRIF protein were decreased in their ability to limit vaccinia replication (Hoebe et al., 2003). Studies are underway to address whether VACV signals through distinct TLR receptors in these two DC populations. Alternatively, it is possible that cytoplasmic receptors, e.g. RIG-I or DAI (Takaoka et al., 2007; Onomoto et al., 2007), are differentially expressed or regulated in these cells. DAI is a recently reported sensor of cytoplasmic DNA (Takaoka et al., 2007), while RIG-I recognizes dsRNA (Onomoto et al., 2007). However, the role of these receptors in the innate response to VACV infection has not been investigated.
In conclusion, the studies presented here provide a model for the regulation of CD8+ and CD8− DC in vivo following infection with VACV. We find that both subsets supported synthesis of viral protein following infection and expressed high levels of costimulatory molecules. Although IL-12 was not produced by these cells, IL-12 producing DC that could serve as a source of signal 3 were found within both the CD8+ and CD8− populations. Surprisingly the mechanism by which IL-12 production was controlled differed between CD8+ and CD8− DC populations. To our knowledge this is the first report of differential control of cytokine production in these two populations following viral infection in vivo and suggests diverse types of DC are sensing viral infection in distinct ways. In the larger picture these data suggest that in vivo targeting of distinct DC subsets for maturation and cytokine production may require independent strategies, i.e. triggering of different TLR or cytoplasmic sensors. Finally, on a practical level, our studies suggest caution in applying in vitro findings with regard to DC regulation to the in vivo setting. This does not necessarily mean that the nature of the interaction between the virus and DC differs in vivo, although it may, but certainly that signals from neighboring cells are likely to have a profound effect on the control of DC maturation.
MATERIALS AND METHODS
Mice and infections
C57BL/6 mice were purchased from the Frederick Cancer Research and Development Center (Frederick, MD). OT-I/RAG-1 (004175) mice were obtained through the NIAID Exchange Program, NIH: (Hogquist et al., 1994; Mombaerts et al., 1992). CD8-deficient mice on a C57BL/6 background (B6.129S2-cd8atm1Mak/J) (Fung-Leung et al., 1991) were purchased from Jackson Laboratories (Bar Harbor, ME). The MyD88-deficient mice (on a C57BL/6 background) were provided by Dr Jason Hoth (Wake Forest University School of Medicine) with permission from Dr Shizuo Akira (Kawai et al., 1999). The recombinant vaccinia virus VACV-NP-S-EGFP was a kind gift from Jack Bennink (NIH) (Princiotta et al., 2003). VACV was sucrose gradient purified prior to infection of mice. Mice were infected intravenously with 1×107 PFU of VACV. All research performed on mice in this study complied with federal and institutional guidelines set forth by the Wake Forest University Animal Care and Use Committee.
Isolation of splenic DC
Minced spleens from C57BL/6 mice were incubated in the presence 100 µg/ml Collagenase D (Roche, Indianapolis, IN) for 30 min at 37°C. The tissue was then passed through a 70 µm filter. CD11c+ cells were enriched by positive selection using the Miltenyi microbeads as per the manufacturer's instructions (Miltenyi Biotec, Auburn, CA).
Cell staining and flow cytometry
DC were stained with the following antibodies: allophycocyanine-conjugated anti-CD11c, phycoerythrin-conjugated anti-CD80, and PerCpCy5.5 conjugated anti-CD8 (all from BD, San Diego, CA) and Pacific Blue conjugated anti-CD86 antibodies (BioLegend, San Diego, CA). Flow cytometric analysis was performed using a FACSAria and data analyzed with Diva software (both from BD, San Diego, CA).
Fluorescent Microscopy
Cells were isolated from the spleens of mock or infected mice (6h p.i.) by collagenase digestion. DC were subsequently enriched over a column using CD11c microbeads. Isolated DC were allowed to adhere to poly-l-lysine coated cover slips for 15 minutes and then fixed in 4% paraformaldehyde for 30 minutes followed by washing with PBS. PFA was quenched by washing with 50mM ammonium chloride. Cells were then stained with Alexa Fluor 647 conjugated anti-mouse CD8α (BioLegend, San Diego, CA) and PE-conjugated CD11c (PharMingen, San Diego, CA) and mounted in ProLong Gold antifade reagent with DAPI (Invitrogen,Eugene,OR). Isotype matched antibodies were used as a control for non-specific binding and mock infected cells were used to set exposure times for eGFP expression by infected cells. Cells were visualized using a Nikon Eclipse TE300 fluorescent microscope using a 60X/0.70 objective. Images were collected using a Retiga EX QImaging camera and analyzed with Adobe Photoshop software.
IL-12p40 production by DC
Splenocytes were cultured overnight in the presence of GolgiPlug in RPMI 1640 medium (Invitrogen Life Technologies) supplemented with 10% FCS (Sigma), HEPES, gentamicin sulfate (BioWhittaker), and 5 × 10−5 M 2-ME . 20 ng/ml rGM-CSF (Biosource International) was also added to enhance survival of the DC population. Following culture, cells were stained with anti-CD11c-PE and anti-CD8-PerCpCy5.5 antibodies. Cells were then washed, fixed, permeabilized and stained with anti-IL-12p40-APC antibody. Samples were acquired on a FacsCalibur and data analyzed with CellQuest software (both from BD, San Diego, CA).
OT-I activation Assay
At 6 or 24h p.i. DC were enriched from the spleen followed by staining with CD11c and CD8-antibodies. DC subsets were sorted based on CD8 expression. OT-I transgenic T cells were co-cultured with sorted DC for 3 days at a ratio of 1:2.5 at 37°C in a 96-well plate (4×104 T cells and 1×105 DC). Following culture, cells were harvested and re-stimulated with 10−6 M Ova257–264 peptide for 5 h in the presence of GolgiPlug (BD, San Diego, CA). Cells were then stained with anti-CD8-PerCpCy5.5 antibody, fixed, permeabilized, and IFN production assessed by intracellular staining with anti IFNγ-PE antibodies.
In vivo activation of OT-I cells
Splenocytes (105) isolated from OT-I mice were intravenously administered to CD8-deficient C57BL/6 mice at 6, 24, or 48h post-infection with VACV-NP-S-EGFP. Three days post-transfer, splenocytes were harvested and Ova-specific responses determined by analysis of IFNγ production using ICCS as described above. Timing of infections were such that OT-I transfers were performed on the same day and Ova257–264-specific responses from all conditions measured in parallel.
Statistical analyses
All p values were calculated using the Student’s t test unless otherwise indicated.
ACKNOWLEDGEMENTS
We are grateful to Dr. Jack Bennink (NIH) for provision of VACV-NP-S-EGFP and to Dr. Jason Hoth (WFUSM) for provision of MyD88 deficient mice with the permission of Dr. Shizuo Akira.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Akira S, Hoshino K, Kaisho T. The role of Toll-like receptors and MyD88 in innate immune responses. J. Endotoxin Res. 2000;6:383–387. [PubMed] [Google Scholar]
- Alcami A, Koszinowski UH. Viral mechanisms of immune evasion. Trends Microbiol. 2000;8:410–418. doi: 10.1016/S0966-842X(00)01830-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeuerle PA, Henkel T. Function Amd Activation of Nf-Kappa-B in the Immune-System. Annu. Rev. Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- Basta S, Chen W, Bennink JR, Yewdell JW. Inhibitory effects of cytomegalovirus proteins US2 and US11 point to contributions from direct priming and cross-priming in induction of vaccinia virus-specific CD8+ T cells. J. Immunol. 2002;168:5403–5408. doi: 10.4049/jimmunol.168.11.5403. [DOI] [PubMed] [Google Scholar]
- Beattie E, Tartaglia J, Paoletti E. Vaccinia virus-encoded eIF-2 alpha homolog abrogates the antiviral effect of interferon. Virology. 1991;183:419–422. doi: 10.1016/0042-6822(91)90158-8. [DOI] [PubMed] [Google Scholar]
- Bekeredjian-Ding I, Roth SI, Gilles S, Giese T, Ablasser A, Hornung V, Endres S, Hartmann G. T cell-independent, TLR-induced IL-12p70 production in primary human monocytes. J Immunol. 2006;176:7438–7446. doi: 10.4049/jimmunol.176.12.7438. [DOI] [PubMed] [Google Scholar]
- Belz GT, Smith CM, Eichner D, Shortman K, Karupiah G, Carbone FR, Heath WR. Cutting edge: conventional CD8 alpha+ dendritic cells are generally involved in priming CTL immunity to viruses. J. Immunol. 2004;172:1996–2000. doi: 10.4049/jimmunol.172.4.1996. [DOI] [PubMed] [Google Scholar]
- Bowie A, Kiss-Toth E, Symons JA, Smith GL, Dower SK, O'Neill LAJ. A46R and A52R from vaccinia virus are antagonists of host IL-1 and toll-like receptor signaling. Proc. Natl. Acad. Sci. USA. 2000;97:10162–10167. doi: 10.1073/pnas.160027697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HW, Watson JC, Jacobs BL. The E3L gene of vaccinia virus encodes an inhibitor of the interferon- induced, double-stranded RNA-dependent protein kinase. Proc. Natl. Acad. Sci. USA. 1992;89:4825–4829. doi: 10.1073/pnas.89.11.4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtsinger JM, Johnson CM, Mescher MF. CD8 T cell clonal expansion and development of effector function require prolonged exposure to antigen, costimulation, and signal 3 cytokine. J. Immunol. 2003;171:5165–5171. doi: 10.4049/jimmunol.171.10.5165. [DOI] [PubMed] [Google Scholar]
- Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. Cutting edge: Type IIFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J. Immunol. 2005;174:4465–4469. doi: 10.4049/jimmunol.174.8.4465. [DOI] [PubMed] [Google Scholar]
- Drillien R, Spehner D, Bohbot A, Hanau D. Vaccinia virus-related events and phenotypic changes after infection of dendritic cells derived from human monocytes. Virology. 2000;268:471–481. doi: 10.1006/viro.2000.0203. [DOI] [PubMed] [Google Scholar]
- Edwards AD, Diebold SS, Slack EM, Tomizawa H, Hemmi H, Kaisho T, Akira S, Reis e Sousa Toll-like receptor expression in murine DC subsets: lack of TLR7 expression by CD8 alpha+ DC correlates with unresponsiveness to imidazoquinolines. Eur J Immunol. 2003;33:827–833. doi: 10.1002/eji.200323797. [DOI] [PubMed] [Google Scholar]
- Engelmayer J, Larsson M, Subklewe M, Chahroudi A, Cox WI, Steinman RM, Bhardwaj N. Vaccinia virus inhibits the maturation of human dendritic cells: a novel mechanism of immune evasion. J. Immunol. 1999;163:6762–6768. [PubMed] [Google Scholar]
- Fung-Leung WP, Schilham MW, Rahemtulla A, Kundig TM, Vollenweider M, Potter J, van Ewijk W, Mak TW. CD8 is needed for development of cytotoxic T cells but not helper T cells. Cell. 1991;65:443–449. doi: 10.1016/0092-8674(91)90462-8. [DOI] [PubMed] [Google Scholar]
- Harte MT, Haga IR, Maloney G, Gray P, Reading PC, Bartlett NW, Smith GL, Bowie A, O'Neill LAJ. The poxvirus protein A52R targets toll-like receptor signaling complexes to suppress host defense. J. Exp. Med. 2003;197:343–351. doi: 10.1084/jem.20021652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez J, Aung S, Marquardt K, Sherman LA. Uncoupling of proliferative potential and gain of effector function by CD8+ T cells responding to self-antigens. J. Exp. Med. 2002;196:323–333. doi: 10.1084/jem.20011612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman HD, Takeda K, Skon CN, Murray FR, Hensley SE, Loomis J, Barber GN, Bennink JR, Yewdell JW. Direct priming of antiviral CD8+ T cells in the peripheral interfollicular region of lymph nodes. Nat Immunol. 2008;9:155–165. doi: 10.1038/ni1557. [DOI] [PubMed] [Google Scholar]
- Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, Crozat K, Sovath S, Han J, Beutler B. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 2003;424:743–748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- Macagno A, Napolitani G, Lanzavecchia A, Sallusto F. Duration, combination and timing: the signal integration model of dendritic cell activation. Trends Immunol. 2007;28:227–233. doi: 10.1016/j.it.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. Rag-1-Deficient Mice Have No Mature Lymphocytes-B and Lymphocytes-T. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- Norbury CC, Malide D, Gibbs JS, Bennink JR, Yewdell JW. Visualizing priming of virus-specific CD8+ T cells by infected dendritic cells in vivo. Nat. Immunol. 2002;3:265–271. doi: 10.1038/ni762. [DOI] [PubMed] [Google Scholar]
- Onomoto K, Yoneyama M, Fujita T. Regulation of antiviral innate immune responses by RIG-I family of RNA helicases. Curr. Top. Microbiol. Immunol. 2007;316:193–205. doi: 10.1007/978-3-540-71329-6_10. [DOI] [PubMed] [Google Scholar]
- Paez E, Esteban M. Nature and Mode of Action of Vaccinia Virus Products That Block Activation of the Interferon-Mediated Ppp(A2'P)Na-Synthetase. Virology. 1984;134:29–39. doi: 10.1016/0042-6822(84)90269-1. [DOI] [PubMed] [Google Scholar]
- Pejawar-Gaddy S, Alexander-Miller MA. Ligation of CD80 is critical for high-level CD25 expression on CD8+ T lymphocytes. J Immunol. 2006;177:4495–4502. doi: 10.4049/jimmunol.177.7.4495. [DOI] [PubMed] [Google Scholar]
- Princiotta MF, Finzi D, Qian SB, Gibbs J, Schuchmann S, Buttgereit F, Bennink JR, Yewdell JW. Quantitating protein synthesis, degradation, and endogenous antigen processing. Immunity. 2003;18:343–354. doi: 10.1016/s1074-7613(03)00051-7. [DOI] [PubMed] [Google Scholar]
- Schulz O, Edwards AD, Schito M, Aliberti J, Manickasingham S, Sher A, Reis e Sousa CD40 triggering of heterodimeric IL-12 p70 production by dendritic cells in vivo requires a microbial priming signal. Immunity. 2000;13:453–462. doi: 10.1016/s1074-7613(00)00045-5. [DOI] [PubMed] [Google Scholar]
- Seet BT, Johnston JB, Brunetti CR, Barrett JW, Everett H, Cameron C, Sypula J, Nazarian SH, Lucas A, McFadden G. Poxviruses and immune evasion. Annu. Rev Immunol. 2003;21:377–423. doi: 10.1146/annurev.immunol.21.120601.141049. [DOI] [PubMed] [Google Scholar]
- Sen R, Baltimore D. Inducibility of Kappa-Immunoglobulin Enhancer-Binding Protein Nf-Kappa-B by A Posttranslational Mechanism. Cell. 1986a;47:921–928. doi: 10.1016/0092-8674(86)90807-x. [DOI] [PubMed] [Google Scholar]
- Sen R, Baltimore D. Multiple Nuclear Factors Interact with the Immunoglobulin Enhancer Sequences. Cell. 1986b;46:705–716. [PubMed] [Google Scholar]
- Shen XF, Wong SBJ, Buck CB, Zhang JW, Siliciano RF. Direct priming and cross-priming contribute differentially to the induction of CD8+ CTL following exposure to vaccinia virus via different routes. J. Immunol. 2002;169:4222–4229. doi: 10.4049/jimmunol.169.8.4222. [DOI] [PubMed] [Google Scholar]
- Stack J, Haga IR, Schroder M, Bartlett NW, Maloney G, Reading PC, Fitzgerald KA, Smith GL, Bowie AG. Vaccinia virus protein Toll-like-interleukin-1 A46R targets multiple receptor adaptors and contributes to virulence. J. Exp. Med. 2005;201:1007–1018. doi: 10.1084/jem.20041442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, Ohba Y, Taniguchi T. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- van den Broek MF, Bachmann MF, Kohler G, Barner M, Escher R, Zinkernagel R, Kopf M. IL-4 and IL-10 antagonize IL-12-mediated protection against acute vaccinia virus infection with a limited role of IFN-gamma and nitric oxide synthetase 2. J. Immunol. 2000;164:371–378. doi: 10.4049/jimmunol.164.1.371. [DOI] [PubMed] [Google Scholar]
- Xiang Y, Condit RC, Vijaysri S, Jacobs B, Williams BRG, Silverman RH. Blockade of interferon induction and action by the E3L double-stranded RNA binding proteins of vaccinia virus. J. Virol. 2002;76:5251–5259. doi: 10.1128/JVI.76.10.5251-5259.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates NL, Alexander-Miller MA. Vaccinia virus infection of mature dendritic cells results in activation of virus-specific naive CD8+ T cells: a potential mechanism for direct presentation. Virology. 2007;359:349–361. doi: 10.1016/j.virol.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Martinez J, Huang X, Yang Y. Innate immunity against vaccinia virus is mediated by TLR2 and requires TLR-independent production of IFN-beta. Blood. 2007;109:619–625. doi: 10.1182/blood-2006-06-027136. [DOI] [PMC free article] [PubMed] [Google Scholar]