

Abstract
Sarcomeric organization of thin and thick filaments in striated muscle is important for efficient generation of contractile forces. Sarcomeric actin filaments are uniform in their lengths and regularly arranged in a striated pattern. Tropomodulin caps the pointed end of actin filaments and is a critical regulator of sarcomere assembly. Here, we report unexpected synergistic functions of tropomodulin with enhancers of actin filament dynamics in Caenorhabditis elegans striated muscle. Pointed-end capping by tropomodulin inhibited actin filament depolymerization by ADF/cofilin in vitro. However, in vivo, depletion of tropomodulin strongly enhanced disorganization of sarcomeric actin filaments in ADF/cofilin mutants, rather than antagonistically suppressing the phenotype. Similar phenotypic enhancements by tropomodulin depletion were also observed in mutant backgrounds for AIP1 and profilin. These in vivo effects cannot be simply explained by antagonistic effects of tropomodulin and ADF/cofilin in vitro. Thus, we propose a model in which tropomodulin and enhancers of actin dynamics synergistically regulate elongation and shortening of actin filaments at the pointed end.
Keywords: Actin dynamics, myofibrils, striated muscle, pointed end
Introduction
In striated muscle, contractile proteins are assembled into sarcomeres that contain highly ordered arrangements of actin thin filaments and myosin thick filaments. Sarcomeric actin filaments are straight and uniform in length. This is in contrast to in vitro polymerized actin filaments that can be much longer and more variable in length than muscle thin filaments. Therefore, polymerization and depolymerization of actin must be precisely regulated during assembly and maintenance of striated myofibrils. However, the regulatory mechanism of assembly and maintenance of sarcomeric actin filaments is not clearly understood.
Several actin-associated proteins have been implicated in organized assembly of actin filaments in striated muscle (Littlefield and Fowler, 1998; Obinata, 1993). Nebulin is important for sarcomere assembly in vertebrate muscle (Bang et al., 2006; McElhinny et al., 2005; Witt et al., 2006). These cell biological and genetic studies revealed that nebulin deficiency alters thin filament length but not uniformity of the length, which is inconsistent with the proposed function of nebulin as a ruler for thin filaments (Labeit et al., 1991). Rather, more recent studies suggest that nebulin regulates actin filament dynamics by interacting with capping proteins (Fowler et al., 2006; McElhinny et al., 2001; Pappas et al., 2008). Tropomodulin (Tmod) and capping protein cap pointed and barbed ends of actin filaments, respectively, and limit polymerization and depolymerization from filament ends (Gregorio et al., 1995; Schafer et al., 1995). Actin depolymerizing factor (ADF)/cofilin and actin interacting protein 1 (AIP1) enhance disassembly of actin filaments and are required for organized assembly of actin filaments in Caenorhabditis elegans muscle (Ono, 2001; Ono et al., 1999). Tropomyosin protects actin filaments from severing by ADF/cofilin and AIP1 and stabilizes sarcomeric actin organization (Ono and Ono, 2002; Yu and Ono, 2006). Recently, Drosophila SALS (Bai et al., 2007) and vertebrate leiomodin (Chereau et al., 2008) have been identified as novel regulators of actin polymerization in muscle. A number of mutations in genes coding for these actin-regulatory proteins, including nebulin, tropomyosin, and cofilin, cause human hereditary myopathies (Laing, 2007), indicating that proper regulation of sarcomeric actin organization is critical for normal function of striated muscle. In vivo, these actin regulators may function together to promote sarcomeric actin organization, but the functional relationship among these proteins is not completely understood.
Although both ends of sarcomeric actin filaments are capped, actin is still dynamically polymerized and depolymerized at these ends (Littlefield et al., 2001). To maintain constant filament length, capping of pointed ends by Tmod is particularly important (Gregorio et al., 1995; Littlefield et al., 2001; Mardahl-Dumesnil and Fowler, 2001). Tmod caps the pointed end of actin filaments and inhibits both polymerization and depolymerization. Tropomyosin binds to Tmod and enhances capping activity (Fischer and Fowler, 2003). Gene knockout of tropomodulin 1 (Tmod 1) in mice results in impaired assembly of myofibrils in the embryonic heart (Chu et al., 2003; Fritz-Six et al., 2003). However, the phenotypic consequences of Tmod inhibition are somewhat complex. Inhibition of Tmod in cardiac myocytes by antibody injection causes either elongation of actin filaments from their pointed ends (Gregorio et al., 1995) or disassembly of thin filaments (Mudry et al., 2003). Concentrations of free actin monomers and activity of actin filament severing proteins could influence the dynamics of actin filaments when Tmod is inhibited. Therefore, we were motivated to investigate functional relationship between Tmod and enhancers of actin filament dynamics.
The body wall muscle of the nematode C. elegans is obliquely striated muscle and has organized sarcomeres (Waterston, 1988). ADF/cofilin (UNC-60B) (Ono et al., 2003; Ono et al., 1999) and AIP1 (UNC-78) (Ono, 2001) enhance actin filament dynamics and are required for assembly of sarcomeric actin filaments. Tropomyosin (Ono and Ono, 2002), kettin (Ono et al., 2006), and UNC-87 (a calponin-like protein) (Yamashiro et al., 2007) bind to the side of actin filaments and stabilize them. PFN-3 is a muscle-specific profilin isoform, but its function is still unclear because a pfn-3 null mutant shows only minor muscle defects (Polet et al., 2006). Recently, unc-94/tmd-1 has been demonstrated to encode Tmod that is expressed in the body wall muscle (Stevenson et al., 2007). Thus, C. elegans is an excellent model system to examine how Tmod might functionally interact with other regulators of actin filament dynamics. In addition, the C. elegans genome does not have genes encoding homologs of nebulin, SALS, or leiomodin, suggesting that the C. elegans muscle utilizes a conserved core mechanism to regulate sarcomeric actin organization. In this study, we found that Tmod promotes sarcomeric actin organization synergistically with enhancers of actin dynamics including ADF/cofilin, AIP1, and profilin. This in vivo effect cannot be simply explained by the antagonistic activity of Tmod against ADF/cofilin-mediated actin depolymerization observed in vitro. We propose that Tmod synergistically functions with enhancers of actin dynamics for organized assembly of sarcomeric actin filaments in vivo.
Results
Anunc-94/tmd-1 mutation causes severe disorganization of actin filaments in the body wall muscle
Previous studies demonstrated that point mutations in the unc-94/tmd-1 gene or unc-94/tmd-1(RNAi) result in disorganization of sarcomeric actin filaments in body wall muscle (Stevenson et al., 2007; Zengel and Epstein, 1980). However, the reported phenotypes are relatively mild, and the mutant or RNAi-treated worms retain substantially organized sarcomeric actin filaments. This is in contrast to very severe defects in the cardiac muscle of tropomodulin-knockout mice (Chu et al., 2003; Fritz-Six et al., 2003). We found that a new unc-94/tmd-1 allele, tm724, causes very severe defects in actin filament organization in the body wall muscle (Fig. 1). unc-94/tmd-1(tm724) has a deletion of 685 bp that removes most of the fourth exon that is common to the two splice variants (Fig. 1A). Even if the third exon splices onto the fifth exon, it is predicted to cause a frame shift such that the resultant mRNA encodes only the N-terminal 126 amino acids, which lacks the leucine-rich repeats and the C-terminal actin capping site. By Western blot, the TMD-1 protein was significantly reduced in unc-94/tmd-1(tm724) (Fig. 1B), although weak bands around 50−60 kDa were inconsistently detected (our unpublished data). unc-94/tmd-1(tm724) was recessive, and unc-94/tmd-1(tm724)/+ heterozygotes were indistinguishable from wild-type (data not shown). These results suggest that this is a null or strong loss-of-function allele, although the possibility that this allele has a neomorphic effect cannot be excluded.
Figure 1.

An unc-94/tmd-1 mutation causes severe disorganization of actin filaments in body wall muscle. (A) Genomic organization of the unc-94/tmd-1 gene. Exons are indicated by boxes. Two alternatively spliced isoforms are generated as described (Stevenson et al., 2007). Position of the unc-94/tmd-1(tm724) deletion is indicated. (B) Western blot analysis of the TMD-1 protein in wild-type and unc-94/tmd-1(tm724) adult worms. The bands at approximately 68 kDa (arrow) are not related to the TMD-1 protein (Stevenson et al., 2007). Anti-actin antibody was used to monitor equal loading of the protein samples. (C) Actin filaments in embryos (a and b) and adults (c and d) were visualized by staining with tetramethylrhodamine-phalloidin in wild-type (a and c) and unc-94/tmd-1(tm724) (b and d). Bar, 10 μm. (D) Organization of α-actinin (a-f) and myosin (g-l) in body wall muscle. Wild-type (a-c and g-i) or unc-94/tmd-1(tm724) (d-f and j-l) were immunostained for actin (a, d, g, and j) and α-actinin (b and e) or MyoA myosin heavy chain (h and k). Merged images are shown in c, f, i, and l (actin in red and α-actinin or MyoA in green). Arrows indicate positions of actin aggregates where α-actinin or myosin did not localize. Bar, 10 μm.
unc-94/tmd-1(tm724) homozygotes had severe defects in actin filament organization in the body wall muscle from embryonic to adult stages (Fig. 1C). In wild-type embryos, actin filaments are assembled into myofibrils, and continuous accumulations of actin filaments were clearly detected in the body wall muscle (Fig. 1Ca). However, in unc-94/tmd-1(tm724), the actin arrays were often discontinuous and a number of aggregates were detected (Fig. 1Cb). Adult unc-94/tmd-1(tm724) worms appeared flaccid and moved more slowly than wild-type (Table 1). In addition, unc-94/tmd-1(tm724) hermaphrodites produced a number of arrested embryos and early larva (38 % lethal, n = 407). Time-lapse Nomarski observation revealed that 45 % of arrested embryos (n = 22) were paralyzed at about the 3.5-fold stage and failed to hatch, which is consistent with severe defects in the assembly of body wall muscle (Williams and Waterston, 1994). Actin filaments in the adult body wall muscle of unc-94/tmd-1(tm724) were severely disorganized (Fig. 1Cd) as compared with striated actin organization in wild-type muscle (Fig. 1Cc). In unc-94/tmd-1(tm724), the striated pattern of actin filaments was difficult to discern, and abnormal accumulations of actin filaments often formed near both ends of spindle-shaped muscle cells (Fig. 1Cd). Disorganized filament organization in unc-94/tmd-1(tm724) was also observed by electron microscopy (our unpublished data).
Table 1.
Motility of adult worms in various mutant backgrounds after RNAi treatment
| Genotype and RNAi treatment | Worm motility (beating/30 sec ± s.d., n=10) |
|---|---|
| Wild-type control RNAi | 118 ± 3.55 |
| Wild-type unc-94/tmd-1 RNAi | 103 ± 3.08 |
| unc-94/tmd-1 (tm 724) | 73.3 ± 9.98 |
| unc-60B(r398) control RNAi | 72.0 ± 10.1 |
| unc-60B(r398) unc-94/tmd-1 RNAi | 16.4 ± 7.06 |
| unc-60B(s1307) control RNAi | 66.3 ± 15.4 |
| unc-60B(s1307) unc-94/tmd-1 RNAi | 18.5 ± 12.7 |
| unc-60B(s1309) control RNAi | 19.3 ± 10.5 |
| unc-60B(s1309) unc-94/tmd-1 RNAi | 12.4 ± 8.42 |
| unc-60B(m35) control RNAi | 17.4 ± 7.15 |
| unc-60B(m35) unc-94/tmd-1 RNAi | 12.0 ± 7.83 |
| unc-78(gk27) control RNAi | 89.2 ± 4.49 |
| unc-78(gk27) unc-94/tmd-1 RNAi | 24.2 ± 10.3 |
| pfn-2(ok458) control RNAi | 114 ± 6.12 |
| pfn-2(ok458) unc-94/tmd-1 RNAi | 105 ± 3.22 |
| pfn-3(tm1362) control RNAi | 114 ± 4.54 |
| pfn-3(tm1362) unc-94/tmd-1 RNAi | 102 ± 3.34 |
| pfn-3(tm1362) pfn-2(ok458) control RNAi | 111 ± 2.60 |
| pfn-3(tm1362) pfn-2(ok458) unc-94/tmd-1 RNAi | 98.7 ± 3.95 |
| unc-60B(r398); pfn-3(tm1362) pfn-2(ok458) control RNAi | 72.5 ± 13.3 |
| unc-60B(r398); pfn-3(tm1362) pfn-2(ok458) unc-94/tmd-1 RNAi | 15.7 ± 8.29 |
| unc-60B(s1309); pfn-3(tm1362) pfn-2(ok458) control RNAi | 19.0 ± 10.5 |
| unc-60B(s1309); pfn-3(tm1362) pfn-2(ok458) unc-94/tmd-1 RNAi | 12.2 ± 4.26 |
Other components of myofibrils were only mildly disorganized in unc-94/tmd-1(tm724). Localization of α-actinin, a component of dense bodies, was only slightly affected in unc-94/tmd-1(tm724) (Fig. 1Dd-f). In wild-type, α-actinin localizes to dense bodies in a punctate pattern (Fig. 1Da-c). In unc-94/tmd-1(tm724), a similar pattern of α-actinin was detected, although some neighboring dots often appeared continuous (Fig. 1Ee). α-actinin did not localize to the actin aggregates (Fig. 1D, d-f, arrows), which is similar to the observation in Tmod1-knockout mice (Fritz-Six et al., 2003). Striation of myosin thick filaments was less organized in unc-94/tmd-1(tm724) than wild-type (Fig. 1D, g-l). In unc-94/tmd-1(tm724), bands of myosin heavy chain were often wider than those in wild-type, and spacing between myosin bands was irregular (Fig. 1Dk). Again, myosin was not found in the actin aggregates (Fig. 1D, j-l, arrows). These results suggest that mild disorganization of dense bodies and thick filaments is a secondary effect of severely disorganized actin filaments.
C. elegans tropomodulin protects against actin depolymerization from the pointed end mediated by ADF/cofilin in vitro
Tmod caps the pointed end of actin filament and regulates actin dynamics at the pointed ends in muscle cells (Fischer and Fowler, 2003). On the other hand, ADF/cofilin enhances actin depolymerization at the pointed ends (Carlier et al., 1997; Maciver et al., 1998; Yamashiro et al., 2005) and is a critical regulator of actin dynamics in muscle cells (Ono, 2003a; Ono, 2007). Therefore, we hypothesized that Tmod and ADF/cofilin have opposite roles in regulating actin dynamics at the pointed end.
We tested whether Tmod has a protective role against pointed-end depolymerization induced by ADF/cofilin in vitro. Bacterially expressed UNC-94/TMD-1 protein has typical Tmod-like pointed-end capping activity (E. Cox, S. Yamashiro, R. Zaidel-Bar, S. Ono, and J. Hardin, manuscript in preparation). CapZ-capped F-C. elegans (Ce-) actin (CapZ : actin = 1:100) was incubated for 15 min with Latrunculin A (Lat-A) in the presence of TMD-1, Ce-tropomyosin (CeTM), UNC-60B, or combinations of these proteins, and ultracentrifuged to separate monomers and short oligomers from long filaments (Fig. 2A). In the presence of Lat-A, which sequesters G-actin, ∼50 % of actin spontaneously depolymerzed (Fig. 2A, lanes 1 and 5, and B). TMD-1 strongly inhibited depolymerization to ∼20 % (Fig. 2A, lanes 2 and 6, and B), while CeTM had only a weak inhibitory effect (Fig. 2A, lanes 3 and 7, and B). The combination of TMD-1 and CeTM had a similar effect to TMD-1 alone (Fig. 2A, lanes 4 and 8, and B). In contrast, UNC-60B enhanced depolymerization to ∼70 % (Fig. 2A, lanes 9 and 13, and B). This effect was moderately inhibited by either TMD-1 alone (Fig. 2A, lanes 10 and 14, and B) or CeTM alone (Fig. 2A, lanes 11 and 15, and B) and strongly inhibited in the presence of both TMD-1 and CeTM (Fig. 2A, lanes 12 and 16, and B). Thus, the combination of TMD-1 and CeTM strongly antagonizes UNC-60B-induced actin depolymerization in vitro.
Figure 2.

TMD-1 inhibits UNC-60B (ADF/cofilin)-dependent depolymerization of actin filaments in vitro. (A, B) Depolymerization of CapZ-capped actin filaments in the presence of Latrunculin A (Lat-A) was examined by a pelleting assay. CapZ-capped F-Ce-actin (5 μM actin, CapZ : actin = 1:100) was incubated with various combinations of TMD-1, CeTM, and UNC-60B in the presence of 10 μM Lat-A for 15 min. The mixtures were ultracentrifuged and fractionated into supernatants (sup) and pellets (ppt) and analyzed by SDS-PAGE (A) and densitometric quantification of depolymerized actin (%) in the supernatants (B). Data are means ± SD of three experiments. (C) Kinetic measurements of depolymerization of CapZ-capped actin filaments. CapZ-capped F-Ce-actin (CapZ : actin = 1:100, 10 % labeled with pyrene) was diluted to 0.5 μM actin with 1 μM Lat-A in the presence of indicated combinations of UNC-60B, CeTM, and TMD-1. Depolymerization of actin was monitored by decrease in pyrene fluorescence.
Effects of UNC-60B, TMD-1 and CeTM on actin depolymerization were further examined by a kinetic assay. CapZ-capped F-Ce-actin (CapZ : actin = 1:100) was diluted to 0.5 μM actin in the presence of Latrunculin A and these proteins, and depolymerization was monitored by measurement of pyrene fluorescence. UNC-60B enhanced depolymerization (Fig. 2B, red curve) as previously reported (Yamashiro et al., 2005). TMD-1 alone (0.125 − 1 μM) did not inhibit UNC-60B-induced depolymerization (Fig. 2C), while CeTM alone showed a relatively strong inhibitory effect on UNC-60B-induced depolymerization (Fig. 2C, light blue curve). The combination of TMD-1 and CeTM inhibited depolymerization more strongly than either TMD-1 or CeTM alone (Fig. 2C). 0.125 μM TMD-1 plus 1 μM CeTM (Fig. 2C, grey curve) had a stronger effect than 1 μM CeTM alone (Fig. 2C, light blue curve), and 1 μM TMD-1 plus 1 μM CeTM (Fig. 2C, dark yellow curve) inhibited depolymerization to a rate similar to controls (capped F-actin alone and Latrunculin A). These results indicate that TMD-1 and CeTM synergistically inhibit actin depolymerization induced by UNC-60B in vitro.
Tropomodulin synergistically functions with ADF/cofilin and AIP1 in actin filament organization in the C. elegans body wall muscle
Our in vitro data in Fig. 2 indicate that Tmod and ADF/cofilin antagonistically regulate actin dynamics at the pointed end. On the other hand, Tmod and ADF/cofilin can cause similar effects in limiting the length of actin filaments: Tmod inhibits elongation at the pointed end to keep filaments short (Littlefield et al., 2001; Mardahl-Dumesnil and Fowler, 2001), and ADF/cofilin severs and depolymerizes actin filaments to generate short filaments (Ono, 2007). To understand the in vivo functional relationship between Tmod and ADF/cofilin in regulation of the actin cytoskeleton, and whether they are antagonistic or cooperative, we examined phenotypic consequences of single and double mutant or knockdown of TMD-1 and UNC-60B in C. elegans body wall muscle. If TMD-1 and UNC-60B are antagonistic, double knockdown of these proteins is expected to suppress the phenotype. However, if they are cooperative, double knockdown is expected to enhance the phenotype.
We used RNA interference (RNAi) to knock down TMD-1 because unc-94/tmd-1(RNAi) causes a mild phenotype as compared with the unc-94/tmd-1(tm724) mutant and thus is suitable for detecting phenotypic enhancement or suppression when combined with other mutations. Western blot analysis indicated that unc-94/tmd-1(RNAi) caused 70−80 % reduction of the TMD-1 protein (Fig. 3A). Therefore, the RNAi treatment did not completely deplete TMD-1, which explains why the RNAi phenotype was much weaker than the unc-94/tmd-1(tm724) mutant phenotype.
Figure 3.

unc-94/tmd-1(RNAi) strongly enhances actin disorganization in unc-60B mutant backgrounds. (A) Effect of unc-94/tmd-1(RNAi) on the TMD-1 protein level. Wild-type worms were treated by control RNAi or unc-94/tmd-1(RNAi) and the TMD-1 protein level was determined by Western blot using anti-TMD-1 antibody. Anti-actin antibody was used to monitor equal loading of the protein samples. (B) Localization of TMD-1 and UNC-60B in unc-60B mutant and unc-94/tmd-1(RNAi) muscles, respectively. TMD-1 (a and d) and actin (b and e) were immunolocalized in the body wall muscle of wild-type (a-c) and unc-60B(r398) (d-f). Merged images are shown in c and f (TMD-1 in green and actin in red). UNC-60B (g and j) and actin (h and k) were immunolocalized in the body wall muscle of wild-type (g-i) and unc-94/tmd-1(RNAi) (j-l). Merged images are shown in i and l (UNC-60B in green and actin in red). Bar, 10 μm. (C) Effects of unc-94/tmd-1(RNAi) on worm motility in unc-60B backgrounds. Wild-type and four unc-60B alleles were examined. m35 and s1309 are strong loss-of-function, while r398 and s1307 are weak loss-of-functinon. Data are means ± SD, n=10. Asterisks indicate p<0.005 by t-test comparing control RNAi and unc-94/tmd-1(RNAi) for each strain. (D) Appearance of wild-type (a and f) and unc-60B (b-e, g-j) worms treated with control RNAi (a-e) or unc-94/tmd-1(RNAi) (f-j) on agar plates. Bar, 0.5 mm. (E) Actin filament organization in the body wall muscle of wild-type (a and b) and unc-60B (c-j) treated with control RNAi (a, c, e, g, and i) or unc-94/tmd-1(RNAi) (b, d, f, h, and j). Arrowheads in b indicate actin aggregates. Arrows in c, e, g, and i indicate striated organization of actin filaments. Bar, 10 μm.
First, we found that mutation or knockdown of TMD-1 or UNC-60B drastically alters the other's localization in muscle cells. In wild-type, TMD-1 localizes to edges of the actin bands where the pointed ends are concentrated (Fig. 3B, a-c). However, in unc-60B(r398), a weak loss-of-function mutant, TMD-1 was enriched in the actin aggregates, while localization of TMD-1 to the striated myofibrils was greatly diminished (Fig. 3B, d-f). On the other hand, UNC-60B colocalized with actin to myofibrils (Fig. 3B, g-i) as reported previously (Ono et al., 1999). unc-94/tmd-1(RNAi) caused only moderate disorganization of actin filaments with formation of actin aggregates (Fig. 3B, k). Nonetheless, UNC-60B was highly concentrated at the core of the actin aggregates and greatly reduced from the myofibrils (Fig. 3B, j-l). The strong influence of TMD-1 and UNC-60B on each other's localization may be a secondary effect of actin filament disorganization. However, α-actinnin and myosin did not accumulate in the actin aggregates in an unc-94/tmd-1 mutant (Fig. 1D) suggesting that not all actin-binding proteins are enriched in the actin aggregates and that TMD-1 and UNC-60B are involved in the same actin-regulatory process.
To better understand how TMD-1 and UNC-60B function, we characterized phenotypes when both TMD-1 and UNC-60B were impaired. We found that combination of unc-60B mutation and unc-94/tmd-1(RNAi) synergistically enhanced the phenotype. unc-60B mutants moved much slower than wild-type (Fig. 3C). Strong loss-of-function alleles m35 and s1309 showed much more severe motility defects than the weak loss-of-function alleles, r398 and s1307 (Fig. 3C) as reported previously (Ono et al., 1999). unc-94/tmd-1(RNAi) slightly affected motility in the wild-type background (Fig. 3C). However, in unc-60B mutant backgrounds, unc-94/tmd-1(RNAi) greatly enhanced the motility defects (Fig. 3C). Strong loss-of-function alleles, unc-60B(m35) and unc-60B(s1309), were apparently only slightly affected by unc-94/tmd-1(RNAi) in the motility assay (Fig. 3C), but the unc-94/tmd-1(RNAi)-treated unc-60B mutants were nearly immobile on agar plates (Fig. 3D, g and h). Because the worm motility assay was performed in solution, worms may be less mobile on the surface of an agar plate than in fluid. Weak loss-of-function alleles, unc-60B(r398) and unc-60B(s1307), were more profoundly affected by unc-94/tmd-1(RNAi) (Fig. 3C). Worm motility was remarkably reduced by unc-94/tmd-1(RNAi) in these mutant backgrounds (Fig. 3C), and the worms were nearly paralyzed on agar plates (Fig. 3D, i and j). Examination of actin filament organization in the body wall muscle revealed that unc-94/tmd-1(RNAi) enhanced disorganization of actin filaments in unc-60B mutants (Fig. 3E). In the wild-type background, unc-94/tmd-1(RNAi) induced only moderate disorganization of actin filaments with formation of actin aggregates (Fig. 3E, compare a and b, arrowheads). In unc-60B mutants, although actin filaments were severely disorganized in control RNAi-treated worms (Fig. 3E, panels c, e, g, and i), some actin filaments were still organized in a striated manner (Fig. 3E, panels c, e, g, and I, arrows). unc-94/tmd-1(RNAi) decreased striated organization of actin filaments and enhanced formation of large wavy actin aggregates (Fig. 3E, panels d, f, h, and j). Thus, mutations of UNC-60B and knockdown of TMD-1 synergistically enhanced defects in muscle contractility by increasing disorganization of actin filaments. These results suggest that TMD-1 and UNC-60B are synergistic rather than antagonistic in regulation of actin organization in vivo.
Actin-interacting protein 1 (AIP1) cooperates with ADF/cofilin to enhance severing of actin filaments (Ono, 2003b). In C. elegans, unc-78 encodes AIP1 and cooperates with unc-60B to promote organized assembly of actin filaments in body wall muscle (Mohri et al., 2006; Mohri and Ono, 2003; Ono, 2001). Therefore, we also tested whether TMD-1 and UNC-78 collaborate in sarcomeric actin organization. Similarly to unc-60B mutants, TMD-1 mislocalized to actin aggregates in an unc-78 null mutant [unc-78(gk27)] (Fig. 4A, d-f). In contrast, UNC-78 localizes to the diffuse cytoplasm and the myofibrils in wild-type (Fig. 4A, g-i), and this pattern was not significantly affected by unc-94/tmd-1(RNAi) (Fig. 4A, j-l). Worm motility was only slightly affected by unc-94/tmd-1(RNAi) in wild-type but remarkably reduced by unc-94/tmd-1(RNAi) in the unc-78 null background (Fig. 4B). As a result, unc-94/tmd-1(RNAi); unc-78 worms appeared nearly paralyzed on an agar plate (Fig. 4C, panel d, compare with Fig. 4C, a-c). Disorganization of actin filaments was also enhanced by unc-94/tmd-1(RNAi): aggregates of actin were larger and more extensive in unc-94/tmd-1(RNAi); unc-78(gk27) (Fig. 4D, panel d) than in unc-78(gk27) with control RNAi (Fig. 4D, panel c). These results strongly suggest that TMD-1 and UNC-78/AIP1 are also synergistic in regulation of actin organization in vivo.
Figure 4.

unc-94/tmd-1(RNAi) strongly enhances actin disorganization in an unc-78 (AIP1) null background. (A) Localization of TMD-1 and UNC-78 in unc-78 mutant and unc-94/tmd-1(RNAi) muscles, respectively. TMD-1 (a and d) and actin (b and e) were immunolocalized in the body wall muscle of wild-type (a-c) and unc-78(gk27) (d-f). Merged images are shown in c and f (TMD-1 in green and actin in red). UNC-78 (g and j) and actin (h and k) were immunolocalized in the body wall muscle of wild-type (g-i) and unc-94/tmd-1(RNAi) (j-l). Merged images are shown in i and l (UNC-78 in green and actin in red). Bar, 10 μm. (B) Effects of unc-94/tmd-1(RNAi) on worm motility in an unc-78 null background. Wild-type and unc-78(gk27) were examined. Data are means ± SD, n=10. Asterisks indicate p<0.005 by t-test comparing control RNAi and unc-94/tmd-1(RNAi) for each strain. (C) Appearance of wild-type (a and c) and unc-78(gk27) (b and d) worms treated with control RNAi (a and b) or unc-94/tmd-1(RNAi) (c and d)on agar plates. Bar, 0.5 mm. (D) Actin filament organization in the body wall muscle of wild-type (a and b) and unc-78 (c and d) treated with control RNAi (a and c) or unc-94/tmd-1(RNAi) (b and d). Bar, 10 μm.
Tropomodulin synergistically functions with profilin in actin filament organization in C. elegans body wall muscle
Profilin is another regulator of actin dynamics that can functionally interact with tropomodulin at the pointed end. Profilin prevents association of actin monomers with the pointed end and inhibits elongation when the barbed end is capped (Pantaloni and Carlier, 1993). Therefore, profilin is expected to have a similar effect to tropomodulin on actin elongation at the pointed end. We tested functional relationship between profilin and tropomodulin by examining phenotypic consequences on actin organization when both profilin and tropomodulin were depleted.
C. elegans has three profilin genes, pfn-1, pfn-2, and pfn-3 (Polet et al., 2006). Although PFN-3 is the only profilin isoform that was detected in the body wall muscle, PFN-2 may also have a function in muscle because depletion of both PFN-2 and PFN-3 causes mild disorganization of sarcomeric actin, which is more severe than phenotypes when a single profilin isoform was depleted (Polet et al., 2006). In control RNAi, no actin aggregates were detected in pfn-2 or pfn-3 single mutant or pfn-3 pfn-2 double mutant (Fig. 5A and B, a, c, e, and g) (note that both pfn-3 and pfn-2 are on the X chromosome, and that pfn-3 is located left to pfn-2. In such a case, a double mutant is described as pfn-3 pfn-2 in a standard practice in C. elegans genetics). When TMD-1 was depleted by RNAi, actin aggregates were found in ∼50 % of treated worms in wild-type (Fig. 5A and Bb), and the pfn-2 mutation did not enhance this phenotype (Fig. 5A and Bd). However, in the pfn-3 null background, actin aggregates were found in 100 % of unc-94/tmd-1 (RNAi) worms (Fig. 5A), although the appearance of actin aggregates was similar to that in wild-type or pfn-2 (Fig. 5Bf). Thus, pfn-3 mutation enhances the unc-94/tmd-1 (RNAi) phenotype. In the pfn-3 pfn-2 double mutant, more intense and larger actin aggregates were formed in 100 % of unc-94/tmd-1 (RNAi) worms (Fig. 5Bh) as compared with unc-94/tmd-1 (RNAi)-treated pfn-3 single mutant worms (Fig. 5Bf). Therefore, pfn-2 mutation enhances the unc-94/tmd-1 (RNAi) phenotype in pfn-3 mutants but not in wild-type, suggesting that pfn-2 has a partially redundant function with pfn-3, while pfn-3 appears to play a major role. Despite the fact that actin disorganization was enhanced in the profilin mutants, we did not detect significant differences in motility of unc-94/tmd-1 (RNAi)-treated worms compared with wild-type and profilin mutants (Table 1). This suggests that the functional interaction between Tmod and profilin is only moderately important in muscle. Taken together, these results suggest that tropomodulin and profilin collaborate in regulation of actin filament organization in C. elegans body wall muscle, which is consistent with a model in which they have similar effects on actin elongation at the pointed end.
Figure 5.

unc-94/tmd-1(RNAi) enhances formation of actin aggregates in profilin mutant backgrounds. (A) Frequency of adult worms that have actin aggregates in the body wall muscle. Adult worms were stained with tetramethylrhodamine-phalloidin, and worms with actin aggregates in their muscle were scored. Wild-type, pfn-2(null) and pfn-3(null) single mutants, and a pfn-3 pfn-2 double mutant were treated with control RNAi or unc-94/tmd-1(RNAi). (B) Actin filament organization in the body wall muscle of wild-type (a and b), pfn-2(null) (c and d), pfn-3(null) (e and f) and pfn-3(null) pfn-2(null) (g and h) treated with control RNAi (a, c, e, and g) or unc-94/tmd-1(RNAi) (b, d, f, and h). Bar, 10 μm.
Profilin mutation does not enhance the ADF/cofilin mutant phenotype
Profilin and ADF/cofilin synergistically enhance actin filament turnover in vitro (Didry et al., 1998), and their cooperative function in vivo has been demonstrated in yeast (Nakano and Mabuchi, 2006; Wolven et al., 2000). Under these biochemical and cellular conditions, free actin barbed ends are constantly generated, so that treadmilling of actin filaments is efficiently enhanced by profilin and ADF/cofilin. However, in striated muscle, sarcomeric actin filaments are straight without branching, and their barbed ends are tightly capped by capping protein. Functional relationship between profilin and ADF/cofilin under such conditions where free barbed ends are limited has not been investigated.
We examined phenotypes of unc-60B; pfn-3 pfn-2 triple mutant and found that the profilin mutation does not enhance the unc-60B mutant phenotype (Fig. 6). When unc-60B(r398) (weak loss-of-function) or unc-60B(s1309) (strong loss-of-function) was combined with pfn-3(null) pfn-2(null), the extent of actin disorganization was nearly identical to each of the unc-60B single mutants (Fig. 6, compare E with G and I with K). The presence of the pfn-3 pfn-2 mutation did not alter worm motility of unc-60B mutants (Table 1). In addition, the pfn-3 pfn-2 mutation neither enhanced nor suppressed the effects of unc-94/tmd-1(RNAi) in the unc-60B mutant background (Fig. 6F, H, J, and L, and Table 1). These results suggest that a synergistic function between profilin and ADF/cofilin is not detectable in C. elegans striated muscle and does not have a significant impact on sarcomeric actin organization.
Figure 6.

Profilin mutation does not enhance the unc-60B phenotype. Actin filament organization in the body wall muscle of wild-type (+; +) (A and B), +; pfn-3(null) pfn-2(null) (C and D), unc-60B(r398); + (E and F), unc-60B(r398); pfn-3(null) pfn-2(null) (G and H), unc-60B(s1309); + (I and J) and unc-60B(s1309); pfn-3(null) pfn-2(null) (K and L) treated with control RNAi (A, C, E, G, I, and K) or unc-94/tmd-1(RNAi) (B, D, F, H, J, and L). Bar, 10 μm.
Discussion
In this study, we characterized phenotypes of an unc-94/tmd-1mutant phenotype and functional relationships of tropomodulin with enhancers of actin filament dynamics in C. elegans striated muscle. The unc-94/tmd-1(tm724) mutant phenotype was more severe than previously reported unc-94/tmd-1 loss-of-function or RNAi phenotypes (Stevenson et al., 2007). Actin filaments in the unc-94/tmd-1(tm724) worms were severely disorganized in the body wall muscle, indicating that Tmod is a critical regulator of actin filament organization in C. elegans striated muscle. Our genetic analysis suggests that Tmod is cooperative, rather than antagonistic, with ADF/cofilin, AIP1, and profilin in regulating actin filament organization. These in vivo functional relationships cannot be explained simply by their antagonistic effects on the rate of actin turnover. Rather, Tmod, ADF/cofilin, AIP1, and profilin may collaborate to regulate lengths of actin filaments and promote organized assembly of sarcomeric thin filaments.
Tmod is a conserved regulator of sarcomeric actin organization
The severe defects in striated myofibril organization in the unc-94/tmd-1(tm724) mutant are consistent with myofibril defects in the heart of Tmod1 null mice (Chu et al., 2003; Fritz-Six et al., 2003). Tmod1 is predominantly expressed in the mouse heart during embryogenesis, and knockout of Tmod1 results in embryonic lethality due to a severe defect in heart development (Chu et al., 2003; Fritz-Six et al., 2003). Aggregates of F-actin are formed in Tmod1-null muscle, and they do not contain α−actinin, a component of the Z-line (Fritz-Six et al., 2003). Similarly, aggregates of F-actin are formed in unc-94/tmd-1-mutant C. elegans muscle and do not contain α−actinin, suggesting that disorganization of actin is caused by similar mechanisms in Tmod-null muscle in mice and worms. One possible cause of actin aggregate formation is excessive elongation of actin filaments from the pointed end. In striated muscle, actin dynamics at the pointed end is important for regulation of thin filament lengths (Littlefield et al., 2001; Mardahl-Dumesnil and Fowler, 2001). Inhibition of Tmod allows actin elongation from the pointed end (Gregorio et al., 1995), and excessively long thin filaments may become unstable, dissociate from myofibrils and form aggregates. The other possibility is detachment of actin filaments when TMD-1 is depleted. TMD-1 localizes to cell-cell junction in the body wall muscle (Stevenson et al., 2007) and has a role in formation of adherens junction in epidermal cells (E. A. Cox, S. Yamashiro, R. Zaidel-Bar, S. Ono, and J. Hardin, unpublished data). Although the precise mechanism of actin aggregate formation is not clear, the phenotype strongly suggests that TMD-1 is a critical factor for assembly and maintenance of sarcomeric actin organization.
Tmod synergistically functions with ADF/cofilin and AIP1
The synergistic effects of TMD-1 and UNC-60B (ADF/cofilin) on sarcomeric actin organization are paradoxical, given their antagonistic effects on actin depolymerization at the pointed end in vitro. However, if one assumes that the major functions of TMD-1 and UNC-60B are to regulate thin filament lengths, they could have similar effects. Tmod caps the pointed end and limits elongation, thereby keeping the filament short. ADF/cofilin severs and depolymerize actin filaments and shortens the filaments. Therefore, TMD-1 and UNC-60B could cooperate to prevent formation of excessively long thin filaments. This model is consistent with our prediction that the actin aggregates are formed from excessively long thin filaments as mentioned above. However, because the actin filaments are severely disorganized in unc-94/tmd-1 and unc-60B mutants, investigation of length distribution of thin filaments is technically difficult. Perhaps, temperature-sensitive or conditional activation or inactivation of TMD-1 and UNC-60B will be useful in the future to determine their roles in regulating thin filament lengths.
The synergistic effects of TMD-1 and UNC-78/AIP1 also support our model that Tmod and ADF/cofilin-AIP1 do not simply compete for actin pointed end dynamics in vivo. AIP1 is a conserved WD-repeat protein that promotes actin filament disassembly in the presence of ADF/cofilin (Ono, 2003b; Ono, 2007). AIP1 enhances fragmentation rather than pointed-end depolymerization by ADF/cofilin (Ono et al., 2004). A severing-defective mutant of UNC-60B (ADF/cofilin) can still induce actin depolymerization from filament ends, but this activity is not enhanced by UNC-78/AIP1 (Mohri and Ono, 2003), indicating that UNC-78/AIP1 does not affect actin depolymerization from filament ends. Therefore, TMD-1 and UNC-78/AIP1 are not likely to interact at the pointed end. Thus, our results support a model in which ADF/cofilin and AIP1 promote actin filament severing to maintain thin filament length in cooperation with the inhibitory effect of Tmod on actin elongation.
Tmod synergistically functions with profilin
The synergy between Tmod and profilin revealed a new function of profilin in muscle and suggests that profilin acts as an inhibitor of pointed-end elongation in vivo, which had previously only been demonstrated in vitro. Profilin binds to G-actin and inhibits its association with the actin pointed end, while it promotes barbed-end elongation especially in the presence of formin (Kovar et al., 2003; Pantaloni and Carlier, 1993; Romero et al., 2004). When the barbed end is capped, profilin simply inhibits actin polymerization. In striated muscle cells, the actin barbed end is tightly capped by capping protein, and constant generation of free barbed ends by Arp2/3-mediated branching or severing is expected to be minor. Therefore, profilin is likely to sequester G-actin in muscle cells and inhibit actin elongation from the pointed end. When Tmod is depleted, actin can elongate from the pointed end, but profilin can inhibit monomer association with the pointed end. However, if both Tmod and profilin are depleted, actin can elongate excessively from the pointed end, which may cause disorganization of the sarcomeric structure. Nonetheless, the synergy between Tmod and profilin is weaker than that between Tmod and ADF/cofilin. In striated muscle, the cellular concentration of G-actin is low, near the critical concentration level (Shimizu and Obinata, 1986). Therefore, the rate of actin polymerization should be slow, and the inhibitory effect of profilin on actin polymerization may be only a supportive mechanism to limit actin filament length.
Functional relationship between ADF/cofilin and profilin in striated muscle
We observed that profilin null mutations did not enhance the ADF/cofilin mutant phenotype. However, in vitro, profilin and ADF/cofilin synergistically enhance actin turnover (Blanchoin and Pollard, 1998; Didry et al., 1998), and their synergistic roles in vivo have been demonstrated in yeast (Nakano and Mabuchi, 2006; Wolven et al., 2000). ADF/cofilin preferentially disassembles ADP-actin from or near the pointed end, and profilin catalytically enhances exchange of ADP with ATP and promotes assembly at the barbed end. Therefore, profilin and ADF/cofilin can synergistically enhance actin treadmilling when the barbed end is free or when new barbed ends are constantly generated. However, as mentioned above, most of the actin barbed ends are expected to be capped in striated muscle, and profilin and ADF/cofilin may not function synergistically under these conditions. When the activity of ADF/cofilin is weakened by mutation, actin monomers will not be supplied by depolymerization, so that the presence or absence of profilin may not have a major impact on pointed-end elongation and actin filament organization.
A model of synergistic functions of Tmod and enhancers of actin dynamics in sarcomeric actin organization
Taken together, we propose a model in which Tmod collaborates with ADF/cofilin, AIP1, and profilin to promote organized assembly of sarcomeric actin filaments (Fig. 7). When a thin filament is decorated by tropomyosin and capped by Tmod, the filament is protected from severing and depolymerization by ADF/cofilin and AIP1 (Fig. 7A). However, Tmod in myofibrils is dynamic and allows elongation from the pointed end (Littlefield et al., 2001) (Fig. 7A and B). This elongation generates a tropomyosin-free part of the filament (Fig. 7C), where ADF/cofilin and AIP1 can sever and inhibit excessive growth of the filament (Fig. 7D). Profilin supports this action by sequestering monomeric actin from the pointed end (Fig. 7E). A defect in this machinery can cause excessive elongation of the filament leading to loss of integrity and stability of the thin filament. Such an unstable filament may be released from myofibrils and contribute to formation of actin aggregates (Fig. 7F). Although this model still needs to be tested rigorously, Tmod, tropomyosin, ADF/cofilin, AIP1, and profilin are strong candidates for regulators of a conserved core mechanism of sarcomeric actin organization in striated muscle.
Figure 7.
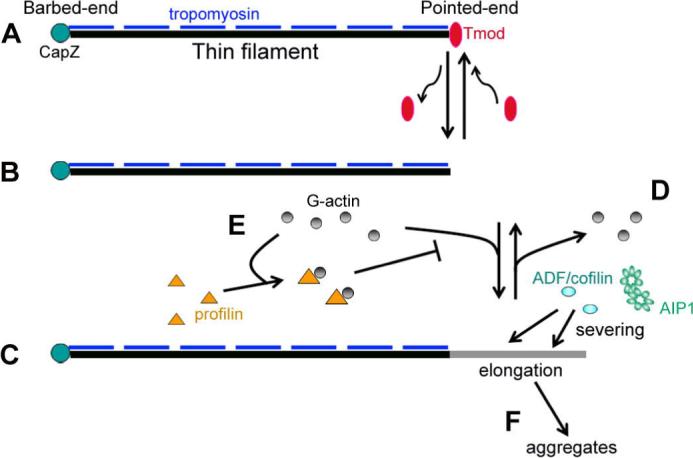
A model of synergistic regulation of sarcomeric actin organization by Tmod, ADF/cofilin, AIP1, and profilin. A stable thin filament has CapZ at the barbed end, Tmod at the pointed end, and tropomyosin on the side (A). Dynamic behavior of Tmod at the pointed end transiently generates a free pointed end (B) and allows elongation (C). This tropomyosin-free region of the filament is subjected to severing by ADF/cofilin and AIP1 (D), and depolymerized actin is captured by profilin, which prevents elongation from the pointed end (E). When this machinery is impaired, excessively elongated filaments may become unstable and form aggregates (F).
Materials and Methods
Nematode strains
Nematodes were grown at 20 °C as described previously (Brenner, 1974). Wild-type strain N2 was obtained from the Caenohabditis Genetic Center (Minneapolis, Minnesota, USA). unc-94/tmd-1(tm724)I was obtained from Shohei Mitani (National BioResource Project, Tokyo Women's Medical University School of Medicine, Tokyo, Japan) and outcrossed six times. unc-60B(r398)V, unc-60B(s1309)V, unc-60B(m35)V, and unc-60B(s1307)V were reported previously (McKim et al., 1988). unc-78(gk27)X was reported previously (Ono, 2001). pfn-2(ok458)X and pfn-3(tm1362)X were reported previously (Polet et al., 2006). The pfn-3(tm1362) pfn-2(ok458) double mutant was generated by crossing pfn-3(tm1362) and pfn-2(ok458) single mutants and isolating a recombinant that carry both mutations on the same chromosome. All mutants were used as homozygotes in this study.
Western blot
Western blot analysis of TMD-1 was performed essentially as described (Ono and Ono, 2002). Briefly, 20 adult worms were lysed in SDS-lysis buffer, and subjected to SDS-PAGE and blotting. Rabbit anti-TMD-1 antibody (Stevenson et al., 2007) was diluted at 1:2000 in Signal Enhancer HIKARI (Nacalai USA, Inc.) and used to detect TMD-1 protein. Mouse anti-actin monoclonal antibody (C4, MP Biomedicals) was used to demonstrate equal loading of the samples.
Proteins
C. elegans actin (Ce-actin) (Ono, 1999) and C. elegans tropomyosin (CeTM) (Ono and Ono, 2002) were purified from wild-type N2 strain as described previously. Bacterially expressed recombinant UNC-60B was prepared as described (Ono and Benian, 1998). Bacterially expressed recombinant chicken CapZ (a gift of Takashi Obinata, Chiba University) was prepared as described (Soeno et al., 1998). Pyrene-labeled rabbit muscle G-actin was prepared as described previously (Kouyama and Mihashi, 1981). Recombinant TMD-1 was expressed in E. coli with an N-terminal 6×His-tag. Details of the purification procedure for TMD-1 will be reported elsewhere (E. Cox, S. Yamashiro, R. Zaidel-Bar, S. Ono, and J. Hardin, manuscript in preparation).
Assays for F-actin depolymerization from pointed ends by pelleting
G-Ce-actin (10 μM) in G-buffer (2 mM Tris-HCl, 0.2 mM CaCl2, 0.2 mM ATP, 0.2 mM DTT) was mixed with 100 nM CapZ, and then polymerization was initiated by adding salts and buffers to final concentrations of 2 mM MgCl2, 0.1 M KCl and 20 mM Hepes-NaOH, pH 7.5. After polymerization for overnight, the CapZ-capped actin filaments (5 μM actin) was incubated with combinations of 2.5 μM each of TMD-1, CeTM, and UNC-60B in the presence of 10 μM Latrunculin A (Lat-A) (Biomol) in F-buffer (0.1 M KCl, 2 mM MgCl2, 1 mM DTT, and 20 mM Hepes-NaOH, pH 7.5) for 15 min. The mixtures were ultracentrifuged in a Beckman TLA100 at 446,000 × g for 15 min. The supernatants and pellets were adjusted to the same volumes and analyzed by SDS-PAGE. Protein bands were stained with Coomassie Brilliant Blue R-250 (National Diagnostics), and band intensity was quantified by ImageJ.
Monitoring kinetics of actin depolymerization from pointed ends
Unlabeled G-Ce-actin (4.5 μM) was mixed with pyrene-labeled rabbit muscle G-actin (0.5 μM) in G-buffer with 50 nM CapZ, and then polymerization was initiated by adding salts and buffers to final concentrations of 2 mM MgCl2, 0.1 M KCl, and 20 mM Hepes-NaOH, pH 7.5. After incubation overnight, the CapZ-capped actin filaments were diluted to 0.5 μM actin with F-buffer with 1 μM UNC-60B, 1 μM CeTM and various concentrations of TMD-1 in the presence of 1 μM Lat-A. Changes in the pyrene fluorescence (excitation at 366 nm and emission at 384 nm) were monitored with a Perkin-Elmer LS50B fluorescence spectrophotometer.
RNA interference experiments
RNAi against unc-94/tmd-1 was performed by feeding with E. coli expressing double stranded RNA (MRC GeneService clone I-2F12) as described previously (Ono and Ono, 2002). L4 larvae were treated with RNAi, and phenotypes were characterized in the F1 generation.
Fluorescence microscopy
Immunofluorescent staining of adult nematodes was performed as described previously (Finney and Ruvkun, 1990). Primary antibodies used were rabbit anti-TMD-1 (Stevenson et al., 2007), mouse monoclonal anti-actin (C4, MP Biomedicals), rabbit anti-UNC-60B (Ono et al., 1999), rabbit anti-UNC-78 (Mohri and Ono, 2003), mouse monoclonal anti-α-actinin (MH40) (Francis and Waterston, 1985) and mouse monoclonal anti-MyoA (5−6) (Miller et al., 1983). Secondary antibodies used were Alexa488-conjugated goat anti-mouse IgG (Invitrogen) and Cy3-conjugated donkey anti-rabbit IgG (Jackson ImmunoResearch Laboratories). Staining of adult worms using tetramethylrhodamine-phalloidin was performed as described previously (Ono, 2001).
Samples were observed by epifluorescence using a Nikon Eclipse TE2000 inverted microscope with CFI Plan Fluor ELWD 40X (dry, NA 0.60) objective. Images were captured by a SPOT RT monochrome CCD camera (Diagnostic Instruments) and processed by IPLab imaging software (BD Biosciences) and Adobe Photoshop CS3.
Motility assay
Worm motility was quantified as described previously (Epstein and Thomson, 1974). Briefly, adult worms were placed in M9 buffer. Then, one beat was counted when a worm swung its head to either the right or left. The total number of beats in 30 s was recorded.
Acknowledgements
The authors thank Paul Sims for performing ultrastructural observation of the unc-94/tmd-1 mutant phenotype. Monoclonal antibody 5−6 was developed by Dr. Henry Epstein (University of Texas Medical Branch, Galveston, TX) and was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biological Sciences, Iowa City, IA 52242. Some C. elegans strains were provided by the Caenorhabditis Genetics Center, which is funded by the National Institute of Health National Center for Research Resources. D. L. B. is a Canadian Research Chair in Genomics. This work was supported by grants from NSERC (Canada) and CIHR (Canada) to D. L. B., National Institute of Health grant GM058038 and a grant from Muscular Dystrophy Association to J. D. H., and National Institute of Health grant AR48615 to S. O.
References
- Bai J, Hartwig JH, Perrimon N. SALS, a WH2-domain-containing protein, promotes sarcomeric actin filament elongation from pointed ends during Drosophila muscle growth. Dev. Cell. 2007;13:828–842. doi: 10.1016/j.devcel.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Bang ML, Li X, Littlefield R, Bremner S, Thor A, Knowlton KU, Lieber RL, Chen J. Nebulin-deficient mice exhibit shorter thin filament lengths and reduced contractile function in skeletal muscle. J. Cell Biol. 2006;173:905–916. doi: 10.1083/jcb.200603119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchoin L, Pollard TD. Interaction of actin monomers with Acanthamoeba actophorin (ADF/cofilin) and profilin. J. Biol. Chem. 1998;273:25106–25111. doi: 10.1074/jbc.273.39.25106. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlier MF, Laurent V, Santolini J, Melki R, Didry D, Xia GX, Hong Y, Chua NH, Pantaloni D. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin-based motility. J. Cell Biol. 1997;136:1307–1322. doi: 10.1083/jcb.136.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chereau D, Boczkowska M, Skwarek-Maruszewska A, Fujiwara I, Hayes DB, Rebowski G, Lappalainen P, Pollard TD, Dominguez R. Leiomodin is an actin filament nucleator in muscle cells. Science. 2008;320:239–243. doi: 10.1126/science.1155313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu X, Chen J, Reedy MC, Vera C, Sung KL, Sung LA. E-Tmod capping of actin filaments at the slow-growing end is required to establish mouse embryonic circulation. Am. J. Physiol. Heart Circ. Physiol. 2003;284:H1827–H1838. doi: 10.1152/ajpheart.00947.2002. [DOI] [PubMed] [Google Scholar]
- Didry D, Carlier MF, Pantaloni D. Synergy between actin depolymerizing factor/cofilin and profilin in increasing actin filament turnover. J. Biol. Chem. 1998;273:25602–25611. doi: 10.1074/jbc.273.40.25602. [DOI] [PubMed] [Google Scholar]
- Epstein HF, Thomson JN. Temperature-sensitive mutation affecting myofilament assembly in Caenorhabditis elegans. Nature. 1974;250:579–580. doi: 10.1038/250579a0. [DOI] [PubMed] [Google Scholar]
- Finney M, Ruvkun G. The unc-86 gene product couples cell lineage and cell identity in C. elegans. Cell. 1990;63:895–905. doi: 10.1016/0092-8674(90)90493-x. [DOI] [PubMed] [Google Scholar]
- Fischer RS, Fowler VM. Tropomodulins: life at the slow end. Trends Cell Biol. 2003;13:593–601. doi: 10.1016/j.tcb.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Fowler VM, McKeown CR, Fischer RS. Nebulin: does it measure up as a ruler? Curr. Biol. 2006;16:R18–R20. doi: 10.1016/j.cub.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Francis GR, Waterston RH. Muscle organization in Caenorhabditis elegans: localization of proteins implicated in thin filament attachment and I-band organization. J. Cell Biol. 1985;101:1532–1549. doi: 10.1083/jcb.101.4.1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz-Six KL, Cox PR, Fischer RS, Xu B, Gregorio CC, Zoghbi HY, Fowler VM. Aberrant myofibril assembly in tropomodulin1 null mice leads to aborted heart development and embryonic lethality. J. Cell Biol. 2003;163:1033–1044. doi: 10.1083/jcb.200308164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorio CC, Weber A, Bondad M, Pennise CR, Fowler VM. Requirement of pointed-end capping by tropomodulin to maintain actin filament length in embryonic chick cardiac myocytes. Nature. 1995;377:83–86. doi: 10.1038/377083a0. [DOI] [PubMed] [Google Scholar]
- Kouyama T, Mihashi K. Fluorimetry study of N-(1-pyrenyl)iodoacetamide-labelled F-actin. Local structural change of actin protomer both on polymerization and on binding of heavy meromyosin. Eur. J. Biochem. 1981;114:33–38. [PubMed] [Google Scholar]
- Kovar DR, Kuhn JR, Tichy AL, Pollard TD. The fission yeast cytokinesis formin Cdc12p is a barbed end actin filament capping protein gated by profilin. J. Cell Biol. 2003;161:875–887. doi: 10.1083/jcb.200211078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labeit S, Gibson T, Lakey A, Leonard K, Zeviani M, Knight P, Wardale J, Trinick J. Evidence that nebulin is a protein-ruler in muscle thin filaments. FEBS Lett. 1991;282:313–316. doi: 10.1016/0014-5793(91)80503-u. [DOI] [PubMed] [Google Scholar]
- Laing NG. Congenital myopathies. Curr. Opin. Neurol. 2007;20:583–589. doi: 10.1097/WCO.0b013e3282ef6e69. [DOI] [PubMed] [Google Scholar]
- Littlefield R, Almenar-Queralt A, Fowler VM. Actin dynamics at pointed ends regulates thin filament length in striated muscle. Nat. Cell Biol. 2001;3:544–551. doi: 10.1038/35078517. [DOI] [PubMed] [Google Scholar]
- Littlefield R, Fowler VM. Defining actin filament length in striated muscle: rulers and caps or dynamic stability? Annu. Rev. Cell Dev. Biol. 1998;14:487–525. doi: 10.1146/annurev.cellbio.14.1.487. [DOI] [PubMed] [Google Scholar]
- Maciver SK, Pope BJ, Whytock S, Weeds AG. The effect of two actin depolymerizing factors (ADF/cofilins) on actin filament turnover: pH sensitivity of F-actin binding by human ADF, but not of Acanthamoeba actophorin. Eur. J. Biochem. 1998;256:388–397. doi: 10.1046/j.1432-1327.1998.2560388.x. [DOI] [PubMed] [Google Scholar]
- Mardahl-Dumesnil M, Fowler VM. Thin filaments elongate from their pointed ends during myofibril assembly in Drosophila indirect flight muscle. J. Cell Biol. 2001;155:1043–1053. doi: 10.1083/jcb.200108026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElhinny AS, Kolmerer B, Fowler VM, Labeit S, Gregorio CC. The N-terminal end of nebulin interacts with tropomodulin at the pointed ends of the thin filaments. J. Biol. Chem. 2001;276:583–592. doi: 10.1074/jbc.M005693200. [DOI] [PubMed] [Google Scholar]
- McElhinny AS, Schwach C, Valichnac M, Mount-Patrick S, Gregorio CC. Nebulin regulates the assembly and lengths of the thin filaments in striated muscle. J. Cell Biol. 2005;170:947–957. doi: 10.1083/jcb.200502158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKim KS, Heschl MF, Rosenbluth RE, Baillie DL. Genetic organization of the unc-60 region in Caenorhabditis elegans. Genetics. 1988;118:49–59. doi: 10.1093/genetics/118.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DM, Ortiz I, Berliner GC, Epstein HF. Differential localization of two myosins within nematode thick filaments. Cell. 1983;34:477–490. doi: 10.1016/0092-8674(83)90381-1. [DOI] [PubMed] [Google Scholar]
- Mohri K, Ono K, Yu R, Yamashiro S, Ono S. Enhancement of actin-depolymerizing factor/cofilin-dependent actin disassembly by actin-interacting protein 1 is required for organized actin filament assembly in the Caenorhabditis elegans body wall muscle. Mol. Biol. Cell. 2006;17:2190–2199. doi: 10.1091/mbc.E05-11-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohri K, Ono S. Actin filament disassembling activity of Caenorhabditis elegans actin-interacting protein 1 (UNC-78) is dependent on filament binding by a specific ADF/cofilin isoform. J. Cell Sci. 2003;116:4107–4118. doi: 10.1242/jcs.00717. [DOI] [PubMed] [Google Scholar]
- Mudry RE, Perry CN, Richards M, Fowler VM, Gregorio CC. The interaction of tropomodulin with tropomyosin stabilizes thin filaments in cardiac myocytes. J. Cell Biol. 2003;162:1057–1068. doi: 10.1083/jcb.200305031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano K, Mabuchi I. Actin-depolymerizing protein Adf1 is required for formation and maintenance of the contractile ring during cytokinesis in fission yeast. Mol. Biol. Cell. 2006;17:1933–1945. doi: 10.1091/mbc.E05-09-0900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obinata T. Contractile proteins and myofibrillogenesis. Int. Rev. Cytol. 1993;143:153–189. doi: 10.1016/s0074-7696(08)61875-6. [DOI] [PubMed] [Google Scholar]
- Ono K, Parast M, Alberico C, Benian GM, Ono S. Specific requirement for two ADF/cofilin isoforms in distinct actin-dependent processes in Caenorhabditis elegans. J. Cell Sci. 2003;116:2073–2085. doi: 10.1242/jcs.00421. [DOI] [PubMed] [Google Scholar]
- Ono K, Yu R, Mohri K, Ono S. Caenorhabditis elegans kettin, a large immunoglobulin-like repeat protein, binds to filamentous actin and provides mechanical stability to the contractile apparatuses in body wall muscle. Mol. Biol. Cell. 2006;17:2722–2734. doi: 10.1091/mbc.E06-02-0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono S. Purification and biochemical characterization of actin from Caenorhabditis elegans: its difference from rabbit muscle actin in the interaction with nematode ADF/cofilin. Cell Motil. Cytoskeleton. 1999;43:128–136. doi: 10.1002/(SICI)1097-0169(1999)43:2<128::AID-CM4>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Ono S. The Caenorhabditis elegans unc-78 gene encodes a homologue of actin-interacting protein 1 required for organized assembly of muscle actin filaments. J. Cell Biol. 2001;152:1313–1319. doi: 10.1083/jcb.152.6.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono S. Function of ADF/cofilin in muscle cells: an important regulator of actin cytoskeletal dynamics in myofibril assembly and muscle diseases. In: Fagan J, Davidson JN, Shimizu N, editors. Recent Developments in Cell Research. Vol. 1. Research Signpost; Kerala, India: 2003a. pp. 31–44. [Google Scholar]
- Ono S. Regulation of actin filament dynamics by actin depolymerizing factor/cofilin and actin-interacting protein 1: new blades for twisted filaments. Biochemistry. 2003b;42:13363–13370. doi: 10.1021/bi034600x. [DOI] [PubMed] [Google Scholar]
- Ono S. Mechanism of depolymerization and severing of actin filaments and its significance in cytoskeletal dynamics. Int. Rev. Cytol. 2007;258:1–82. doi: 10.1016/S0074-7696(07)58001-0. [DOI] [PubMed] [Google Scholar]
- Ono S, Baillie DL, Benian GM. UNC-60B, an ADF/cofilin family protein, is required for proper assembly of actin into myofibrils in Caenorhabditis elegans body wall muscle. J. Cell Biol. 1999;145:491–502. doi: 10.1083/jcb.145.3.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono S, Benian GM. Two Caenorhabditis elegans actin depolymerizing factor/cofilin proteins, encoded by the unc-60 gene, differentially regulate actin filament dynamics. J. Biol. Chem. 1998;273:3778–3783. doi: 10.1074/jbc.273.6.3778. [DOI] [PubMed] [Google Scholar]
- Ono S, Mohri K, Ono K. Microscopic evidence that actin-interacting protein 1 actively disassembles actin-depolymerizing factor/cofilin-bound actin filaments. J. Biol. Chem. 2004;279:14207–14212. doi: 10.1074/jbc.M313418200. [DOI] [PubMed] [Google Scholar]
- Ono S, Ono K. Tropomyosin inhibits ADF/cofilin-dependent actin filament dynamics. J. Cell Biol. 2002;156:1065–1076. doi: 10.1083/jcb.200110013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantaloni D, Carlier MF. How profilin promotes actin filament assembly in the presence of thymosin beta 4. Cell. 1993;75:1007–1014. doi: 10.1016/0092-8674(93)90544-z. [DOI] [PubMed] [Google Scholar]
- Pappas CT, Bhattacharya N, Cooper JA, Gregorio CC. Nebulin interacts with CapZ and regulates thin filament architecture within the Z-disc. Mol. Biol. Cell. 2008;19:1837–1847. doi: 10.1091/mbc.E07-07-0690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polet D, Lambrechts A, Ono K, Mah A, Peelman F, Vandekerckhove J, Baillie DL, Ampe C, Ono S. Caenorhabditis elegans expresses three functional profilins in a tissue-specific manner. Cell Motil. Cytoskeleton. 2006;63:14–28. doi: 10.1002/cm.20102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero S, Le Clainche C, Didry D, Egile C, Pantaloni D, Carlier MF. Formin is a processive motor that requires profilin to accelerate actin assembly and associated ATP hydrolysis. Cell. 2004;119:419–429. doi: 10.1016/j.cell.2004.09.039. [DOI] [PubMed] [Google Scholar]
- Schafer DA, Hug C, Cooper JA. Inhibition of CapZ during myofibrillogenesis alters assembly of actin filaments. J. Cell Biol. 1995;128:61–70. doi: 10.1083/jcb.128.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu N, Obinata T. Actin concentration and monomer-polymer ratio in developing chicken skeletal muscle. J. Biochem. (Tokyo) 1986;99:751–759. doi: 10.1093/oxfordjournals.jbchem.a135534. [DOI] [PubMed] [Google Scholar]
- Soeno Y, Abe H, Kimura S, Maruyama K, Obinata T. Generation of functional beta-actinin (CapZ) in an E. coli expression system. J. Muscle Res. Cell Motil. 1998;19:639–646. doi: 10.1023/a:1005329114263. [DOI] [PubMed] [Google Scholar]
- Stevenson TO, Mercer KB, Cox EA, Szewczyk NJ, Conley CA, Hardin JD, Benian GM. unc-94 encodes a tropomodulin in Caenorhabditis elegans. J. Mol. Biol. 2007;374:936–950. doi: 10.1016/j.jmb.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston RH. Muscle. In: Wood WB, editor. The Nematode C. elegans. Cold Spring Harbor Laboratory; 1988. pp. 281–335. [Google Scholar]
- Williams BD, Waterston RH. Genes critical for muscle development and function in Caenorhabditis elegans identified through lethal mutations. J. Cell Biol. 1994;124:475–490. doi: 10.1083/jcb.124.4.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt CC, Burkart C, Labeit D, McNabb M, Wu Y, Granzier H, Labeit S. Nebulin regulates thin filament length, contractility, and Z-disk structure in vivo. EMBO J. 2006;25:3843–3855. doi: 10.1038/sj.emboj.7601242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolven AK, Belmont LD, Mahoney NM, Almo SC, Drubin DG. In vivo importance of actin nucleotide exchange catalyzed by profilin. J. Cell Biol. 2000;150:895–904. doi: 10.1083/jcb.150.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashiro S, Gimona M, Ono S. UNC-87, a calponin-related protein in C. elegans, antagonizes ADF/cofilin-mediated actin filament dynamics. J. Cell Sci. 2007;120:3022–3033. doi: 10.1242/jcs.013516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashiro S, Mohri K, Ono S. The two Caenorhabditis elegans actin depolymerizing factor/cofilin proteins differently enhance actin filament severing and depolymerization. Biochemistry. 2005;44:14238–14247. doi: 10.1021/bi050933d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu R, Ono S. Dual roles of tropomyosin as an F-actin stabilizer and a regulator of muscle contraction in Caenorhabditis elegans body wall muscle. Cell Motil. Cytoskeleton. 2006;63:659–672. doi: 10.1002/cm.20152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengel JM, Epstein HF. Identification of genetic elements associated with muscle structure in the nematode Caenorhabditis elegans. Cell Motil. 1980;1:73–97. doi: 10.1002/cm.970010107. [DOI] [PubMed] [Google Scholar]