

Abstract
Seizures represent a common and serious complication of hypoglycemia. Here we studied mechanisms of control of hypoglycemic seizures induced by insulin injection in fasted and nonfasted rats. We demonstrate that fasting predisposes rats to more rapid and consistent development of hypoglycemic seizures. However, the fasting-induced decrease in baseline blood glucose concentration cannot account for the earlier onset of seizures in fasted versus nonfasted rats. Data obtained with c-Fos immunohistochemistry and [14C]2-deoxyglucose uptake implicate a prominent involvement of the substantia nigra reticulata (SNR) among other structures in the hypoglycemic seizure control. This is supported by data showing that fasting decreases the SNR expression of KATP channels, which link metabolism with activity, and is further confirmed with microinfusions of KATP channel agonist and antagonist. Data obtained with whole-cell and perforated patch recordings from SNR neurons in slices in vitro demonstrate that both presynaptic and postsynaptic KATP channels participate in the failure of the SNR to control hypoglycemic seizures. The results suggest that fasting and insulin-induced hypoglycemia can lead to impairment in the function of the SNR, leading thus to hypoglycemic seizures.
Keywords: hypoglycemia, fasting, Kir6.2, substantia nigra, intracellular recordings, 2-deoxyglucose
Introduction
Hypoglycemic seizures in humans (Malouf and Brust, 1985; Diabetes Control and Complications Trial Research Group, 1995; Davis et al., 1997) develop as a consequence of significantly decreased blood glucose, frequently in diabetic patients who mistakenly increase their insulin dose, miss a meal, or engage in excessive physical activity (Herbel and Boyle, 2000; Leckie et al., 2005). They also occur in patients with insulinoma or conditions such as the hyperammonemia-hypoglycemia syndrome (Hart and Frier, 1998). Although hypoglycemia can be resolved with prompt administration of glucose, there are still controversies about mechanisms involved in the expression and control of hypoglycemic seizures.
In vitro studies have shown that, in brain slices from forebrain structures, glucopenia invariably silences neuronal firing, shuts down preexisting epileptiform activity induced by removing Mg2+ from the perfusing solution (Kirchner et al., 2006), and suppresses synaptic transmission (Fan et al., 1988; Shoji, 1992; Akasu et al., 1996). However, in in vivo model systems, systemic hypoglycemia reliably induces seizures (Chapman et al., 1987; Panickar et al., 1998), consistent with the human data (Malouf and Brust, 1985; Diabetes Control and Complications Trial Research Group, 1995; Davis et al., 1997).
Extracellular glucose concentration regulates cell activity especially in cells containing specialized machinery translating the metabolic state into changes in excitability. Inward rectifying potassium channels represent an important link between the metabolism and excitability (Mourre et al., 1990a,b; Liss and Roeper, 2001; Yamada and Inagaki, 2005). These ATP-controlled (KATP) channels govern insulin release in pancreatic β-cells (Gloyn et al., 2004) and control excitability of neurons in response to metabolic changes in the brain (Allen and Brown, 2004) and stress adaptation in myocardial cells (Zingman et al., 2002). At normal glucose concentrations with abundant ATP present, these channels are actively closed. During metabolic stress (e.g., glucopenia or hypoxia), the channels open, allowing potassium leak out from the cell and to hyperpolarization, i.e., decreased excitability (Levin, 2001; Yamada et al., 2001; Yamada and Inagaki, 2005). The most metabolically sensitive version of these channels is a combination of four Kir6.2 pore-forming subunits and four sulfonylurea SUR1 binding sites peripherally found in pancreatic β-cells (Smith et al., 2007). In the brain, these SUR1/Kir6.2 channels are found in the highest concentration in the substantia nigra pars reticulata (SNR) (Mourre et al., 1989; Xia and Haddad, 1991; Xia et al., 1993; Hicks et al., 1994; Dunn-Meynell et al., 1998; Liss et al., 1999). This midbrain structure contains a reticulum of mostly GABAergic neurons significantly involved in control of seizures (Iadarola and Gale, 1981; Velíšková and Moshé, 2006). GABA-mediated hyperpolarization of GABAergic neurons especially in the anterior part of the SNR leads to silencing of these neurons and is associated with anticonvulsant effects. In contrast, increased neuronal firing in the anterior part of the SNR is associated with proconvulsant effects (Depaulis et al., 1994; Velíšková and Moshé, 2006).
In this report, we investigated control of hypoglycemic seizure by metabolic changes in the SNR using EEG recordings, c-Fos and [14C]2-deoxyglucose (2-DG) uptake, localized SNR microinfusions of KATP channel-affecting drugs, and whole-cell and gramicidin-perforated patch clamp in vitro.
Materials and Methods
Animals
All experiments have been approved by the Institutional Animal Care and Use Committee of the Albert Einstein College of Medicine and conform to the National Institutes of Health Revised Guide for the Care and Use of Laboratory Animals (1996). Adult [postnatal day 55 (P55) to P70] male Sprague Dawley rats (Taconic Farms) were used. For the whole-cell and perforated patch-clamp recordings in the SNR, male P21–P23 rats were used. Because our preliminary data indicated that there may be a difference in the occurrence of hypoglycemic seizures in rats with access to food ad libitum and food-restrained rats, two groups were randomly created: a group of nonfasted rats, which had access to food and tap water ad libitum until the experiments started and a group of fasted rats, which were limited in their access to food (but not water) for 14–16 h before the experiment (overnight before the experimental day). All efforts have been made to minimize pain and suffering, as well as the numbers of the animals.
Glucose measurements
Glucose was measured using One Touch glucose meters (Ultra and Basic models; LifeScan). These glucose meters were selected for the high measurement correlation with desktop glucose meters (Kirchner et al., 2006). Mixed peripheral blood (arterial, venous, and capillary) samples (1 or 5 μl) were obtained after snipping the tip of the tail. Mixed peripheral blood yields lower glucose readings than arterial blood (Suda et al., 1990). Because the volume of samples was minimal, there was no adverse effect to the rat's well being. Additionally, a single cut provided samples during the entire experiment, sometimes after a gentle tail massage. Statistical comparisons were performed using Student's t test (two groups) or ANOVA with post hoc Fisher's protected least square degree (PLSD) test (three or more group), with p always <0.05 (adjusted for multiple comparisons).
Hypoglycemia induction
On the experimental day, the rats were weighed and the baseline blood glucose was measured. Then the rats received 30 IU/kg insulin intraperitoneally (containing ∼25 IU/1 mg sodium salt crystalline; Sigma) (Vannucci and Vannucci, 1978; Kirchner et al., 2006; Velíšková et al., 2007) freshly dissolved in normal saline at 15 IU/ml. Blood glucose levels from mixed peripheral blood were determined every 30 min. We recorded the behavioral manifestations associated with hypoglycemia, including their incidence and latency to onset. Using logarithmic regression, we determined the best curve fit for the peripheral glucose levels after insulin injections. We also calculated glucose levels at the times when the behavioral manifestations were first observed by curvilinear extrapolation from the nearest points of measurement. Statistical comparisons between the fasted and nonfasted groups were performed using Student's t test, with p < 0.05.
EEG recordings
Adult male rats (n = 6) were anesthetized with a mixture of ketamine (50 mg/kg) and xylazine (7 mg/kg) intraperitoneally. Insulated stainless steel wire electrodes (Plastics One) were inserted stereotaxically (coordinates in mm from bregma): in the sensorimotor cortex [anteroposterior (AP), 0; lateral (L), 2; depth from the skull surface (D), 1], dorsal hippocampus (AP, 3.2; L, 4.5; D, 4.0), anterior part of the SNR (AP, 5.2; L, 2.2; D, 8.0), and pedunculopontine tegmental nucleus (PPTg) (AP, 7.5; L, 1.5; D, 7.5) The incisor bar was set at −3.5 mm. Additionally, one screw with an electrode contact was implanted in the nasal bone (to serve as a reference electrode for referential recordings) and one above the cerebellum to serve as ground. Electrodes were mounted in a six-pin connector (Plastics One), and the skull including the connector was covered with dental acrylic. After a week of recovery in the Association for Assessment and Accreditation of Laboratory Animal Care-approved animal facility with access to food and water ad libitum, rats were food restricted overnight (14–16 h) and connected to an EEG/video-monitoring system based on Harmonie Software (Stellate Systems). After the baseline EEG recordings, we injected 30 IU/kg insulin intraperitoneally and determined blood glucose levels every 30 min. We recorded behaviors and their EEG correlates. We continued to monitor the rats until the EEG became isoelectric. We then immediately rescued the rats with 5 ml of 20% glucose intraperitoneally and continued the EEG recordings and glucose measurements for 2 additional hours. If after 2 h, blood glucose level exceeded 50 mg/100 ml, we considered that the rat was successfully managing its glucose levels. If blood glucose was lower than 50 mg/100 ml, additional 3 ml of 20% glucose were injected intraperitoneally.
General immunohistochemistry procedure
For immunohistochemical procedures, we used standard 40-μm-thick sections (Velíšková and Velíšek, 2007). Thinner sections are fragile and can be easily damaged during the free-floating staining procedure. Free-floating sections were preincubated (room temperature) in 0.3% H2O2 in PBS, followed by three washes in PBS and incubated for 2 h in PBS with 0.25% Triton X-100 and 3% normal goat serum (NGS). Primary antibodies against either c-Fos [PC 38 (Calbiochem); 1:10,000 in PBS, 0.25% Triton X-100, and 3% NGS] or against Kir6.2 (1:200 in PBS; Santa Cruz Biotechnology) were added for 48 h at 4°C. After primary antibody incubation, sections were three times rinsed in PBS at room temperature, and then a secondary anti-rabbit antibody was applied (1:200, in PBS, 0.25% Triton X-100, and 3% NGS) for 2 h. After three additional rinses in PBS, sections were incubated in ABC reagent (in PBS with 0.5% Triton X-100), followed by PBS rinses and diaminobenzidine–nickel reaction. Sections were mounted on microscopy slides and coverslipped.
2-DG uptake study
The 2-DG technique is used for determination of brain energy metabolism using local cerebral metabolic rates of glucose (Pereira de Vasconcelos et al., 1992; Velíšková et al., 2005). These rates correlate well with the neuronal activation during pre-seizure and seizure states, reflecting altered synaptic activity (Ackermann et al., 1984; Nehlig et al., 1991). We assessed glucose uptake both before seizure occurrence and after the seizures developed using a modified 2-DG autoradiography technique with the subcutaneous administration of the tracer (Simmons et al., 1998; Velíšek et al., 2005; Velíšková et al., 2005). To determine the dynamics of preictal state (before seizures have developed), the experiment was designed to detect changes in 2-DG uptake during hypoglycemia (<40 mg/100 ml in fasted rats) but before seizure occurrence (mean time of onset at 100 min after insulin injection). Furthermore, we wanted to separate temporally early and late changes that occur within this time interval but also allow for a time period for hypoglycemia to emerge (usually 30 min), leaving an ∼70 min window to perform the study. Therefore, we decided to kill the rats 30 min after 2-DG injection with one 2-DG injection administered at 30 min after insulin and another one at 60 min after insulin. With this approach, we were able to determine changes at two time points before the seizures developed. However, postictally, there were no time limitations, so we killed the rats at 45 min after the seizures had occurred. This 45 min period is the period most often used in 2-DG experiments (Sokoloff et al., 1977; Brown et al., 2002).
Blood glucose measurements were taken before insulin/vehicle injection and then every 30 min until the animals were killed in deep urethane anesthesia. The brains were removed immediately, chilled in cooled methylbutane at −35°C, and stored at −70°C for additional processing. Sagittal sections (30 μm thick) were cut using a microtome, collected directly on microscopy slides, and exposed to Eastman Kodak Biomax MR film for 10 d along with [14C] standards (American Radiolabeled Chemicals). In autoradiography, thinner slices (Brown et al., 2002) provide better signal because C14 is a low-energy β-emitor. In thicker sections, the signal disperses. Developed films were scanned on the light box and analyzed using NIH Image 1.62 and ImageJ software (version 1.34a; Wayne Rasband, National Institutes of Health, Bethesda, MD). Measurements were focused especially on those structures in which inspection of the films indicated differences between controls, hypoglycemia, or hypoglycemia associated with seizures. Normalized optical density [optical density of the regions of interest divided by the optical density of the entire brain section; structure-to-section ratio (Velíšek et al., 2005)] was compared among the groups. Thus, we compared relative changes in the brain 2-DG uptake, allowing us to avoid hypoglycemia-associated changes in lumped sum constant used for calculation of absolute 2-DG (Sokoloff et al., 1977). The lumped sum constant is stable only at physiologic blood glucose levels, although it greatly increases during hypoglycemia (Suda et al., 1990).
Statistical comparisons of two groups were performed using Student's t test, with p preset to <0.05; three groups were compared using ANOVA with post hoc Fisher's protected least square degree test, with p < 0.05 adjusted for multiple comparisons.
Experiments were performed to determine the following.
Metabolic activation at early stages of significant hypoglycemia.
Two groups of fasted rats were used: one group injected with vehicle (“fasted + saline”) served as euglycemic controls (n = 6), and the other was injected with 30 IU/kg insulin intraperitoneally to induce hypoglycemia (“fasted + insulin”; n = 5). At 30 min after injection (fasted + saline, 98.8 ± 4.5 mg/100 ml blood glucose; fasted + insulin, 26.5 ± 4.9 mg/100 ml blood glucose), we administered 50 μCi/kg [14C]2-DG in saline subcutaneously and killed the rats 30 min later, i.e., 60 min after insulin/vehicle injection (fasted + saline, 111.6 ± 4.2 mg/100 ml blood glucose; fasted + insulin, 20.0 ± 5.1 mg/100 ml blood glucose). If any of the fasted + insulin rats developed seizures before death, they were not included. Thus, none of the hypoglycemic rats entering statistical evaluation experienced seizures at time of death. All comparisons were performed between controls and hypoglycemic rats within this group.
Metabolic activation at pre-seizure stages of hypoglycemia.
Two groups of fasted rats were used: one group was injected with vehicle (fasted + saline; controls, n = 6), and the other was injected with 30 IU/kg insulin intraperitoneally (fasted + insulin; hypoglycemic, n = 4). At 60 min after injection (fasted + saline, 100.4 ± 5.8 mg/100 ml blood glucose; fasted + insulin, 18.4 ± 0.4 mg/100 ml blood glucose), we administered 50 μCi/kg [14C]2-DG in saline subcutaneously and killed the rats 30 min later, i.e., 90 min after insulin/vehicle injection, a time point very close to mean seizure onset in fasted rats (fasted + saline, 106.4 ± 4.7 mg/100 ml blood glucose; fasted + insulin, 16.4 ± 0.8 mg/100 ml blood glucose). If any of the fasted + insulin rats developed seizures before death, they were not included. Thus, none of the hypoglycemic rats entering statistical evaluation experienced seizures at time of death. All comparisons were performed between controls and hypoglycemic rats within this group.
Metabolic activation after hypoglycemic seizures have occurred.
Three groups were used: six fasted rats with seizures (“fasted + insulin + seizure” group), four nonfasted rats (hypoglycemic “nonfasted + insulin” group) were injected with insulin (30 IU/kg, i.p.), and seven fasted rats injected with vehicle (“fasted + saline” group). All rats were injected with 50 μCi/kg [14C]2-DG at time of seizure onset (as per the fasted + insulin + seizure group) and had the following blood glucose levels (fasted + insulin + seizure, 19.1 ± 0.8 mg/100 ml; nonfasted + insulin, 29.8 ± 1.8 mg/100 ml; fasted + saline, 94.5 ± 3.7 mg/100 ml). The rats were killed 45 min after the 2-DG injection, and their blood glucose levels were tested (fasted + insulin + seizure, 16.0 ± 1.3 mg/100 ml; nonfasted + insulin, 29.5 ± 5.3 mg/100 ml; fasted + saline, 83.7 ± 4.1 mg/100 ml). All rats in the fasted + insulin + seizure group have developed seizures, and no seizures occurred in other groups. All comparisons were performed within this group.
c-Fos immunoexpression
Fasted and nonfasted rats were used to determine c-Fos expression as a marker of activation of brain nuclei (Morgan et al., 1987) during hypoglycemia and seizures associated with hypoglycemia. The rats were anesthetized with 8 ml/kg 20% urethane and perfused transcardially with 4% paraformaldehyde solution. Immediately after the animals were killed, the brains were removed and cryoprotected in 30% sucrose solution. Sagittal sections (40 μm thick) were cut and processed for c-Fos immunohistochemical detection. c-Fos product immunopositivity was visually inspected throughout the brain and semiquantitatively evaluated. We especially focused on those structures already prescreened with 2-DG during hypoglycemia or hypoglycemia associated with seizures.
We assigned score of 0 to brain regions containing no c-Fos immunopositivity, score 1 to nuclei containing scattered c-Fos-immunopositive cells (<10% of all cells), score 2 to nuclei with moderate c-Fos immunopositivity (10–50% of cells), and, finally, score 3 to nuclei with very high c-Fos immunopositivity (>50%) (Samoriski et al., 1997; Velíšek et al., 2007).
We focused the analysis on structures generally associated with seizure initiation or control. In addition, we analyzed the structures associated with food intake regulation and structures giving striking c-Fos immunopositivity after seizures. Therefore, we evaluated the following structures: orbital cortex, olfactory nuclei, lateral hypothalamus, hippocampus, anterior and posterior paraventricular nuclei of the thalamus, hypothalamic paraventricular nucleus, subthalamic nucleus (STN), SNR, intermediate layers of the superior colliculus, inferior colliculus, PPTg, pontine nuclei, vestibular nuclei, medial olive, and gigantocellular nucleus in the oblongata.
Statistical comparison between two groups was performed using Mann–Whitney U test, with p < 0.05. In experiments involving three groups, the scores were compared using nonparametric Kruskall–Wallis test, followed by post hoc pairwise Dunn's test. Level of significance was set to p < 0.05 adjusted for multiple comparisons using Holm–Sidak method. It should be emphasized that these tests return mean ranks of scores, which are plotted in the figures (not the actual scores).
Experiments were performed to determine the following.
Brain structures activated before hypoglycemic seizures.
In these experiments, only fasted rats were used because we demonstrated 100% incidence of hypoglycemic seizures in these rats (see Results), and, therefore, we could reasonably anticipate that all rats experiencing severe hypoglycemia would ultimately have developed seizures. Two groups were used: one injected with saline (fasted + saline, euglycemic; n = 4), and the other was injected with 30 IU/kg insulin intraperitoneally (fasted + insulin, hypoglycemic; n = 3). Glucose measurements were performed before insulin/vehicle injection, at 30, and 60, and 90 min after insulin, just before death. If any of the fasted + insulin rats developed seizures before death, it was discarded because of our interest was in pre-seizure state. Thus, none of the hypoglycemic rats entering statistical evaluation experienced seizures at time of death, i.e., 90 min after insulin injection.
Brain structures activated during hypoglycemic seizures.
Here we used our data showing that the incidence of hypoglycemic seizures in nonfasted rats is low (see Results), to distinguish between hypoglycemia-induced activation and hypoglycemic seizure-induced activation of brain structures at the same times after insulin injection. Thus, fasted and nonfasted rats were used. Fasted rats with seizures (fasted + insulin + seizure group; n = 3) and nonfasted rats (hypoglycemic nonfasted + insulin group; n = 3) were injected with insulin (30 IU/kg, i.p.), whereas additional fasted rats were injected with vehicle (fasted + saline group; n = 3). Thirty minutes after the seizure onset in the hypoglycemia + seizure group, 10 ml/kg 20% glucose solution was injected intraperitoneally to all hypoglycemic rats (or an equivalent volume of saline to normoglycemic rats) to ensure that the hypoglycemic rats survive for additional 90 min after the seizure, a time period needed for the expression of the c-Fos product (Silveira et al., 2002).
Kir6.2 immunoexpression
Kir6.2 immunoexpression was used to determine Kir6.2 expression after fasting. We compared chronic changes in relative numbers of Kir6.2-immunopositive cells in the SNR, STN, and pontine reticular nucleus, oral part (PnO) between fasted (n = 5) and nonfasted rats (overnight fasting; n = 5). Furthermore, we determined acute effects of blood glucose levels on Kir6.2 expression in fasted rats, 4 h after either 3 ml of saline (n = 5) or 3 ml of 20% glucose (n = 5) injection. We performed manual counts of immunopositive cells on matching sections, selecting three matching sections from both fasted and nonfasted rats, containing the structure of interest. Images of these structures were digitally captured on the computer, and the entire profile of the structure in the section has been recreated using Adobe Photoshop software (Adobe Systems). On these recreated images of the entire structure, the counts were performed. Three sections per structure per rat were averaged for the statistical comparisons. Results were compared using Student's t test with p < 0.05. Additionally, we used densitometry (Ravizza et al., 2002) to determine expression of the Kir6.2 protein in individual cells. On three sections per rat, immunointensity was measured in 10 immunopositive cells per section and averaged. Averages were compared between nonfasted (n = 6) and fasted (n = 6) rats.
SNR infusions
Under ketamine/xylazine anesthesia (50 and 7 mg/kg, i.p., respectively), we prepared the rats for stereotaxic implantation of guide cannulas (Plastics One) in the SNR (coordinates in mm: AP, 5.2; L, 4.0; D, 7.0; cannulas were declined 15° from sagittal plane). After 2 d recovery, we determined the baseline blood glucose level and then bilaterally infused in the anterior part of the SNR 0.5 μl of 5% 2-DG (n = 8; iso-osmotic) or 5% mannitol (n = 4; iso-osmotic) over 2 min period per site. After the infusion, the infusion cannula stayed inserted for an additional minute to prevent the backflow of the solution. Infusions of KATP channel drugs were performed similarly: For tolbutamide, a KATP channel blocker, two infusions of 4 mm solution in DMSO/β-cyclodextrine mixture (5:1 ratio, 0.25 μl/SNR) were delivered in nonfasted rats spaced by 24 h. Preliminary data indicated that a single infusion before insulin was inferior to long-term pretreatment. The second tolbutamide infusion immediately preceded insulin injection. Diazoxide, a KATP channel opener, was microinfused in a single infusion (0.25 μl of 4 mm solution in 0.1N NaOH–PBS mixture per site) into both SNRs in fasted rats just before insulin injection. Control rats received microinfusions of vehicle. Then, in all groups, we administered insulin (30 IU/kg, i.p.), followed the rats with serial blood glucose levels every 30 min, and recorded all seizure behaviors including latency to onset. Four hours after the insulin injection, the rats were killed in deep urethane anesthesia (7.5 ml/kg of 20% solution), and brains were removed and immediately frozen in chilled methylbutane (−35°C). The placement of cannulas in the SNR was verified with light microscopy in thionin-stained sections. Only the rats without brain lesions and with both cannulas placed symmetrically in the anterior part of the SNR were included in the analysis because the site specificity of the SNR effects on seizure has been shown (Thompson et al., 2000; Velíšková and Moshé, 2001).
In vitro recordings
For both whole-cell patch and gramicidin perforated patch-clamp recordings, P19–P22 Sprague Dawley male rats were deeply anesthetized with isoflurane and decapitated. The brain was quickly removed, and 300-μm-thick sagittal slices were cut using a vibratome (VT2000; Leica Microsystems) in ice-cold sucrose containing the following (in mm): 210 sucrose, 3.5 KCl, 1 CaCl2, 4 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, and 10 d-glucose. Slices were allowed to recover for at least 1 h at room temperature in artificial CSF (ACSF) containing the following (in mm): 124 NaCl, 2.5 KCl, 1 NaH2PO4, 26 NaHCO3, 2 CaCl2, 1.3 MgSO4, and 20 glucose. In those experiments in which we modified glucose concentrations (10, 6, or 2 mm), mannitol has been added to maintain osmolarity. All solutions were saturated with 95% O2/5% CO2 gas mixture. Slices were transferred to the recording chamber and perfused with oxygenated ASCF at a rate of 4 ml/min.
Whole-cell patch clamp.
The pipette filling solution contained the following (in mm): 140 KCl, 6 NaCl, 2 MgCl2, 10 HEPES, and 2 Mg-ATP, and pH adjusted to 7.3 with KOH.
Gramicidin perforated patch.
Gramicidin was used in the experiment to avoid the perturbation of the intracellular [ATP]i because it is capable of forming openings in the cell membrane, which allow to cross only monovalent cations and small uncharged molecules (Kyrozis and Reichling, 1995). The pipette filling solution contained the following (in mm): 140 KGln, 6 NaCl, 2 MgCl2, and 10 HEPES, and pH adjusted to 7.3 with KOH. Gramicidin at 1 mg/ml was added to pipette filling solution to obtain a final concentration of 0.1–0.3 μg/ml. After a tight seal was formed in voltage clamp, we waited until a decent perforation has been established (seal resistance, <100 MΩ without compensation) and then switched into current clamp. Recordings were made in current-clamp mode from electrophysiologically identified GABAergic neurons [i.e., presence of sag after negative current pulse, biphasic afterhyperpolarization, and fast spiking (Richards et al., 1997)] in the anterior part of the SNR using Axopatch 200B amplifier (Molecular Devices). Glutamate or GABA receptor antagonists were not used. All experiments were done at room temperature.
Recordings were started in the ACSF containing 20 mm glucose. After 5 min recording of baseline firing frequency, we switched to ACSF containing lower glucose concentrations (10, 6, and 2 mm glucose for the whole-cell patch; 6 and 2 mm glucose for the perforated patch). Slices were bathed 10 min in each glucose concentration. Action potentials were analyzed using MiniAnalysis Program (Synaptosoft). Firing frequency was measured in the last 10 s of each minute (i.e., we determined spiking frequency five times during baseline conditions and 10 times in each glucose concentration).
Results
Behavioral manifestations after insulin injection in fasted and nonfasted rats
The two groups (fasted and nonfasted) differed in basal glucose levels. Fasted rats had basal peripheral blood glucose of 86.8 ± 2.2 mg/100 ml (n = 26). Nonfasted rats had basal peripheral blood glucose of 123.2 ± 2.1 mg/100 ml (n = 23; p < 0.0001, Student's t test). Administration of insulin was associated with a progressive decrease in peripheral blood glucose levels in both fasted and nonfasted rats.
With decreasing blood glucose concentration, there was a decrease in motor activity and in reactivity to external stimuli in both groups. During severe hypoglycemia (<25 mg/100 ml), the following motor behaviors were observed: twitches, jumps, clonus of all four limbs, and barrel rotations. Because the twitches may have occurred as a response to an external stimulus and could not be easily verified by EEG as epileptic, we defined the syndrome of hypoglycemic seizures as consisting of jumps, clonus, and barrel rotations. EEG recordings showed discharges during clonic seizures and barrel rotations (see below), confirming that these are seizure behaviors. All 25 fasted rats responding to insulin administration with decreasing peripheral glucose displayed the hypoglycemic seizures (100% incidence). Although not every behavior was observed in every rat, all fasted rats developed barrel rotations within the 4 h observation period. Conversely, only 10 of 22 nonfasted rats with progressively decreasing peripheral glucose developed the hypoglycemic seizure syndrome (45.5% incidence) (Fig. 1 A, p < 0.0001 vs fasted group, Fisher's exact test). There were no statistically significant differences in blood glucose concentration between those nonfasted rats developing seizures and nonfasted rats without seizures over time (Fig. 1 B, p > 0.05, Student's t test) except for the 240 min after insulin injection. This is a time period long after the hypoglycemic seizure syndrome would have occurred, suggesting that peripheral glucose concentration cannot explain the differences in seizure occurrence in nonfasted rats. The latency to onset of all seizures was significantly shorter in fasted compared with nonfasted rats (Fig. 1 C, p < 0.001 in all cases, Student's t test).
Figure 1.

Hypoglycemic seizures induced by insulin in fasted and nonfasted rats. A, The incidence of insulin-induced (30 IU/kg, i.p.) seizures as well as barrel rotations was significantly higher in fasted (25 of 25) than in nonfasted (10 of 22) rats (*p < 0.0001, Fisher's exact test). B, Decrease in blood glucose over time in nonfasted rats experiencing seizures (n = 10) was not significantly different from nonfasted rats without seizures (n = 12), except for the last interval at 240 min after insulin (*p < 0.05, t test). Solid bar above the fitted curves indicates time of the first seizure onset (mean ± SEM) in the 10 rats. C, Hypoglycemic seizures occurred earlier in the fasted rats (n = 25) than in nonfasted (n = 10) rats (*p for all symptoms <0.001, t test). The first pair of bars indicates the latency to onset of any first seizure regardless of type (jumps, clonic seizures, and barrel rotations combined). The additional pairs of bars compare individual behaviors separately. D, Decrease in blood glucose over time in fasted and nonfasted rats experiencing seizures. There was a significant difference in initial blood glucose as well as in blood glucose at the time points up to 150 min after insulin (*p < 0.05, t test). This blood glucose decrease over time has a very precise logarithmic fit (see r 2 values). Dotted bar indicates time of the first seizure onset for the fasted group (n = 25; mean ± SEM), and the solid bar indicates time of the first seizure onset for the nonfasted group (n = 10; mean ± SEM).
Peripheral glucose levels in both fasted and nonfasted rats experiencing seizures fit well into logarithmic regression (for equations and r 2 values, see Fig. 1 D). Furthermore, the actual glucose levels were significantly different between nonfasted and fasted rats for up to the 150 min after insulin injections (p < 0.05, Student's t test).
To determine whether baseline peripheral blood glucose differences (lower blood glucose in fasting rats) are responsible for differential seizure onset in fasted and nonfasted rats, we used the same starting point for both groups: we calculated the latency to onset of the first seizure beginning from the time point, at which blood glucose in both groups decreased significantly to the level of 40 mg/100 ml. Still, there was a significant difference between the groups; seizures in the fasted group occurred significantly faster (after 78.0 ± 3.8 min) compared with the nonfasted group (106.0 ± 10.3 min; p < 0.01) (Table 1). These findings strongly indicate that the difference in peripheral blood glucose at the starting point cannot explain the differential latency to onset of hypoglycemic seizures.
Table 1.
Basic parameters of hypoglycemic seizures in fasted and nonfasted rats
| Parameter | Groups with progressively decreasing glucose |
p | |
|---|---|---|---|
| Fasted rats (n = 25) | Nonfasted rats (n = 22) | ||
| Incidence of any seizure symptom | 25 of 25 (100%) | 10 of 22 (45.5%) | <0.0001 |
| Peripheral glucose at the first seizure onset | 20.0 ± 0.5 mg/100 ml (n = 25) | 18.3 ± 1.4 mg/100 ml (n = 10) | NS |
| Latency to the onset of the first seizure | 100.1 ± 3.8 min (n = 25) | 172.4 ± 10.3 min (n = 10) | < 0.0001 |
| Latency to the onset of the first seizure from the beginning of significant hypoglycemia (defined as 40 mg/100 ml) | 78.0 ± 3.8 min (n = 25) | 106.0 ± 10.3 min (n = 10) | <0.01 |
EEG recordings
In fasted rats with implanted EEG electrodes, baseline glucose levels at 86.2 ± 6.1 mg/100 ml were not different from baseline glucose level in the fasted group in the experiment above (p > 0.05, Student's t test). The time of behavioral seizure onset (any behavioral manifestation) at 100.6 ± 6.4 min after insulin administration was not different from the fasted group described above. EEG seizures developed almost simultaneously in all recorded regions (Fig. 2 A,B) and later (at 128.0 ± 11.3 min) than motor seizures, indicating that our EEG electrode placements did not involve sites of seizure origin. We could not reliably evaluate short EEG discharges appearing synchronously with twitches and jumps attributable to the possible confounding effect of muscle activity artifacts. Clonic seizures and recurrent barrel rotations were associated with EEG discharges (Fig. 2 A,B).
Figure 2.

Electroencephalographic and behavioral events associated with hypoglycemia. A, EEG recordings obtained with barrel rotations during profound hypoglycemia. PPTg, Pedunculopontine tegmental nucleus, hippocampus, and (neo)cortex recordings. Seizure activity started as fast, low-amplitude spikes (arrow) almost simultaneously in all recorded areas. The barrel rotations occurred after the onset of the EEG discharge (the arrowhead marks the artifact induced by a barrel rotation). Calibration: 2 s, 500 μV. B, Individual frames from a video recording depicting a rat undergoing two barrel rotations. C, EEG recordings during a seizure developing during the recovery from hypoglycemia after intraperitoneal glucose administration. D, The behavioral correlate of the EEG seizure shown in C. The behavioral seizure consisted of rearing and face and forelimb clonus and was similar to forebrain seizures occurring after kindling, pilocarpine, or kainic acid (Racine, 1972; Ben-Ari, 1985; Cavalheiro et al., 1987).
After seizure occurrence, an isoelectric EEG line developed at 196.8 ± 3.4 min, and immediately the rats were rescued with an injection of 3 ml of 20% glucose. During the glucose rescue period, two of five rats with previous typical hypoglycemic seizures displayed a different seizure type consisting of clonic convulsions of face and forelimb muscles with fully preserved righting ability accompanied by chewing and salivation, motorically undistinguishable from stage 5 of kindling (Fig. 2 D) (Racine, 1972). In both rats, the clonic seizure type progressed into tonic-clonic seizures within 1 h after onset, with death occurring within an additional 20 min. The seizures were resistant to pentobarbital administration (50 mg/kg, i.p.). During the seizures, one of the rats had very high glucose levels (369 mg/100 ml = 20.5 mm), whereas the other rat had very low glucose levels (< 10 mg/100 ml; under 0.6 mm), suggesting impaired glucose control. This assumption was supported by irresponsiveness to additional intraperitoneal glucose injections. In both rats, EEG seizure discharges were generalized (Fig. 2 C).
Imaging studies: 2-DG uptake and c-Fos expression
Identification of structures activated before seizure development
These studies were performed in fasted rats that in our paradigm were likely to experience seizures at the appropriate time interval after the insulin injection. In the 2-DG uptake studies, we determined metabolic changes in brain regions involved before the onset of seizures. The change in 2-DG uptake is an equivalent to changes in inhibition or excitation within the affected structures and reflects synaptic activity. Therefore, we also examined c-Fos expression to identify specifically the structures with increased excitation during the preictal period.
2-DG uptake, temporal profile of structures involved in hypoglycemic seizures.
(1) At 60 min after insulin injection, [14C]2-DG was injected 30 min after insulin injection and the rats were killed 30 min later. We found decrease in 2-DG uptake in the insulin-injected hypoglycemic versus the control group in the SNR (Fig. 3 A, p < 0.01, t test) and also in the paraventricular nuclei of thalamus (Fig. 3 A, p < 0.05, t test). We also found increases in 2-DG uptake in the insulin-injected hypoglycemic group in the STN and intermediate layers of the superior colliculus (Fig. 3 A,D,E, p < 0.05, t test). The results in the SNR, subthalamic nucleus, and superior colliculus match the mutual connectivity of these structures (Bolam et al., 2000), which are part of the seizure controlling network (Velíšková et al., 2005): changes in the 2-DG uptake in these structures thus imply their involvement in the SNR controlling network during hypoglycemic seizures. (2) At 90 min after insulin injection, in this experiment, 2-DG was administered at 60 min after insulin and the rats were killed 30 min later. Significant decreases in 2-DG uptake were again found in the SNR and thalamic paraventricular nuclei and also in the orbital cortex and inferior colliculus (Fig. 3 B, p < 0.05, t test). Increases in 2-DG uptake were found in the hypothalamic paraventricular nucleus, PPTg, and PnO (Fig. 3 B, p < 0.05, t test). Results indicate that, with progressing hypoglycemia, the synaptic activity changes. Some structures keep the decreases found earlier, but increases in synaptic activation during hypoglycemia shift to different structures.
Figure 3.

Identification of structures activated before seizure development using c-Fos expression and 2-DG uptake. A, 2-DG uptake in fasted rats 30 min after the injection of either saline (normoglycemic, fasted + saline) or insulin (hypoglycemic, fasted + insulin). The relative uptake was determined in autoradiograms as the calibrated optical density value of the region of interest versus calibrated optical density of the entire section (structure-to-section ratio). Groups were compared using t test, *p < 0.05. There were significant decreases in 2-DG uptake during hypoglycemia in the paraventricular nuclei of the thalamus and SNR (both anterior and posterior part) and increases in the STN and intermediate layers of the superior colliculus. B, 2-DG uptake in fasted rats 60 min after the injection of either saline or insulin. The relative uptake was determined as above. With time, 2-DG uptake further decreased in the paraventricular thalamic nuclei, in the SNR, and in the inferior colliculus. Significant increases were observed in the paraventricular hypothalamic nucleus (PVN hypothalamus), PPTg, and pontine reticular nucleus, oral part (pontine nc.). C, Ranks of c-Fos scores in brain structures in the two groups of fasted rats (normoglycemic, fasted + saline; hypoglycemic, fasted + insulin) at 90 min after saline or insulin injection. The semiquantitative scores were assigned (see Materials and Methods) (Samoriski et al., 1997; Silveira et al., 2002). As the scores were evaluated using nonparametric Mann–Whitney test, mean ranks are shown with *p < 0.05. D, Example of brain 2-DG uptake in the fasted rat injected with saline at 30 min after injection; sagittal section at the level −1.9 mm according to Paxinos and Watson (1998). White arrowhead points to the intermediate layer of the superior colliculus, black arrowhead marks the STN, and the arrow points to the SNR. E, Example of brain 2-DG uptake in the fasted rat injected with insulin at 30 min of hypoglycemia; sagittal section at the level −1.9 mm according to Paxinos and Watson (1998). Please note the relative increase in the 2-DG uptake in the STN and the superior colliculus and a substantial decrease in the SNR, especially in the anterior part; compared with D.
c-Fos expression was studied at 90 min after insulin injection.
In the insulin-injected group, we found abundant c-Fos immunopositivity in the following structures: lateral hypothalamus, thalamic and hypothalamic paraventricular nuclei, subthalamic nucleus, pedunculopontine tegmental nucleus, pontine nuclei, and the medial olive (Fig. 3 C, p < 0.05, all Mann–Whitney U tests vs euglycemic controls). Hypoglycemic activation of thalamic and hypothalamic structures is most likely related to their function in food intake or vicinity to the ventricles and, therefore, suffering hypoglycemia firsthand. The prominent c-Fos expression in the midbrain/brainstem nuclei may suggest that these structures may be activated in the preictal state (Velíšková et al., 2005).
Activation during hypoglycemic seizures
Here, we used the low incidence of hypoglycemic seizures in nonfasted rats to distinguish between hypoglycemia-induced activation and hypoglycemic seizure-induced activation of brain structures to identify brain sites associated with hypoglycemic seizure propagation and control. Thus, the differences of both hypoglycemia + seizure and hypoglycemia groups from controls indicate effects of hypoglycemia, whereas differences between hypoglycemia + seizure group versus the hypoglycemia and control groups indicate effects of seizures.
2-DG uptake.
Decreases in 2-DG uptake were found in both hypoglycemia group and the group with hypoglycemic seizures (vs the normoglycemic group) in the olfactory nuclei and orbital cortex [Fig. 4 A, p < 0.05 vs normoglycemic group; p < 0.05 vs hypoglycemic (nonfasted) group; ANOVA with post hoc Fisher's PLSD test]. The increases in 2-DG uptake in both hypoglycemia and hypoglycemia + seizures appeared in the paraventricular hypothalamic nucleus. In the SNR (regardless of the subregion), there was a decrease in 2-DG uptake in the hypoglycemic group (in agreement with similar findings shown in Fig. 3 A,B compared with normoglycemic controls). However, as a result of hypoglycemia and seizures in the hypoglycemia + seizures group, there was a significant increase of 2-DG uptake in the SNR compared with the hypoglycemia group specifically in the SNR (Fig. 4 E–G). An additional decrease in hypoglycemia + seizures versus hypoglycemia group was found in the inferior colliculus and, versus the control normoglycemic group, in the anterior paraventricular thalamic nucleus. Images also indicate decreases in 2-DG uptake in the striatum, consistent with its role in the circuit (Velíšková and Moshé, 2006) and with the occurrence of barrel rotations during hypoglycemic seizures (Marrannes and Wauquier, 1988; Wurpel et al., 1988).
Figure 4.
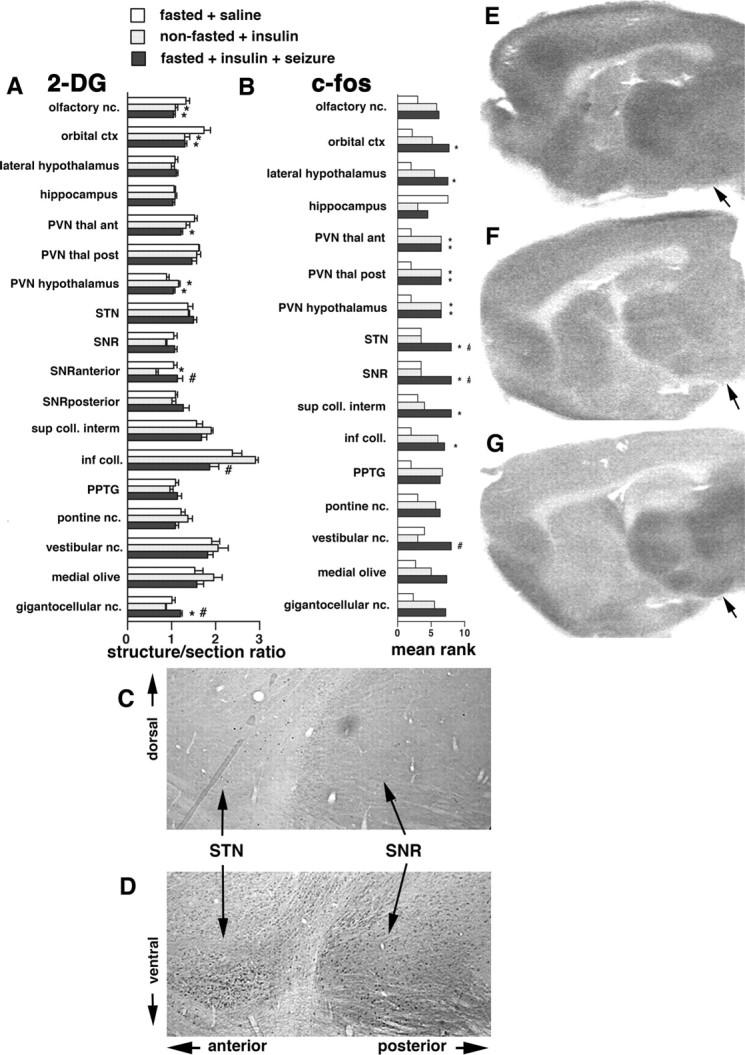
Identification of structures maintaining activation during and after hypoglycemic seizure. A, 2-DG uptake 45 min after the onset of hypoglycemic seizures in the fasted + insulin + seizure group or at matching times after the injection in the other two groups (fasted + saline and nonfasted + insulin). The relative uptake was determined in autoradiograms as described in Figure 3 (structure-to-section ratio). Statistical analysis was performed using ANOVA with post hoc Fisher's PLSD test; *p < 0.05 versus the fasted + saline group; # p < 0.05 versus the nonfasted + insulin group. Note that, in the nonfasted + insulin rats, the SNR metabolic activity is low compared with the fasted + saline and fasted + insulin + seizures rats. Accordingly, metabolic activity in superior colliculus is decreased in the fasted + insulin + seizure group as a complement to increased SNR metabolic activity of the same group. B, Ranks of c-Fos scores in brain structures in the three groups of rats (fasted + saline, nonfasted + insulin, and fasted + insulin + seizure) at 120 min after the seizure onset in the seizure group or matched times in the other two groups. Mean ranks are presented (*p < 0.05 vs fasted + saline; # p < 0.05 vs nonfasted + insulin group). From all the evaluated structures, we found significant increases in c-Fos expression in the rats with hypoglycemia and seizures compared with the rats with comparable hypoglycemia only in the SNR, STN, and the vestibular nuclei. C, Example of the c-Fos expression in the SNR and STN in a nonfasted rat with insulin-induced hypoglycemia at 120 min after the other group developed seizures. Note that there are only individual c-Fos-positive cells in both structures. D, Example of the c-Fos expression in the SNR and STN in a fasted rat with insulin-induced hypoglycemia and seizures at 120 min after the seizures occurred (matched time for the rat shown in E). Note that both the STN and SNR are packed with c-Fos-immunopositive cells, indicating strong activation of these structures. E, An example of 2-DG uptake in a sagittal brain section in a fasted rat with saline injection. The arrow points to the middle of the SNR. F, An example of 2-DG uptake in a sagittal brain section in a nonfasted rat with insulin injection. The 2-DG uptake in the SNR is decreased compared with E. G, An example of 2-DG uptake in a sagittal brain section of a fasted rat with insulin injection and seizures. The 2-DG uptake in the SNR is increased compared with F.
c-Fos.
The graph of the c-Fos expression (Fig. 4 B) based on mean ranks of c-Fos scores (Kruskal–Wallis analysis) shows that, during hypoglycemic seizures, there is a significant increase in c-Fos expression in the subthalamic nucleus (Kruskall–Wallis test, 6.171; p < 0.05) (Fig. 4 C,D) and the SNR (Kruskall–Wallis test, 7.714; p < 0.05) (Fig. 4 D,E) compared with both normoglycemic and hypoglycemic groups (post hoc Dunn's test, p < 0.05, corrected for multiple comparisons) (Fig. 4 A). Hypoglycemic activation was observed also in paraventricular nuclei of the thalamus and of the hypothalamus, which are located on the border of the lateral ventricles (Paxinos and Watson, 1998) and, thus, experience immediate changes in the CSF glucose concentration. The hypothalamic paraventricular nucleus is associated with food-seeking behavior and, as such, is activated when glucose is decreased (Suda et al., 1990; Ao et al., 2005). An additional control is provided by the activation of c-Fos in the vestibular nuclei in rats with seizures compared with the hypoglycemic group (Kruskall–Wallis test, 6.720; p < 0.05); the barrel rotations by their nature stimulate the vestibular apparatus.
Altered expression of Kir6.2 subunits in the SNR
Using immunohistochemistry, first we counted the numbers of cells expressing the Kir6.2 protein in the SNR, STN, and PnO in fasted (n = 5) and nonfasted (n = 5) rats. The SNR in fasted rats contained significantly fewer (almost one-third) Kir6.2-immunopositive cells than the SNR in nonfasted rats. (Fig. 5 A, p < 0.05, t test). The trend in the subthalamic nucleus was not significant. There was no difference in Kir6.2-immunopositive cell counts in the pontine reticular nucleus, oral part (Fig. 5 A, p > 0.80, t test) Second, using densitometry, we compared the relative immunodensity of the Kir6.2 staining in the cells in sections from fasted and nonfasted rats in the SNR region. There was significantly lower immunoexpression of Kir6.2 in the SNR in sections from fasted rats (n = 6) compared with nonfasted (n = 6) (Fig. 5 B, p < 0.05, t test). Although the results indicate that long-term glucose insufficiency (such as overnight fasting) is associated with decreases in the SNR Kir6.2 leading to the reduced capacity of the SNR neurons to hyperpolarize, this may also be the stress effect of overnight fasting. In the anterior part of the SNR, insufficient hyperpolarization (or depolarization) is associated with proconvulsant effects (Moshé and Sperber, 1990; Velíšková and Moshé, 2006).
Figure 5.
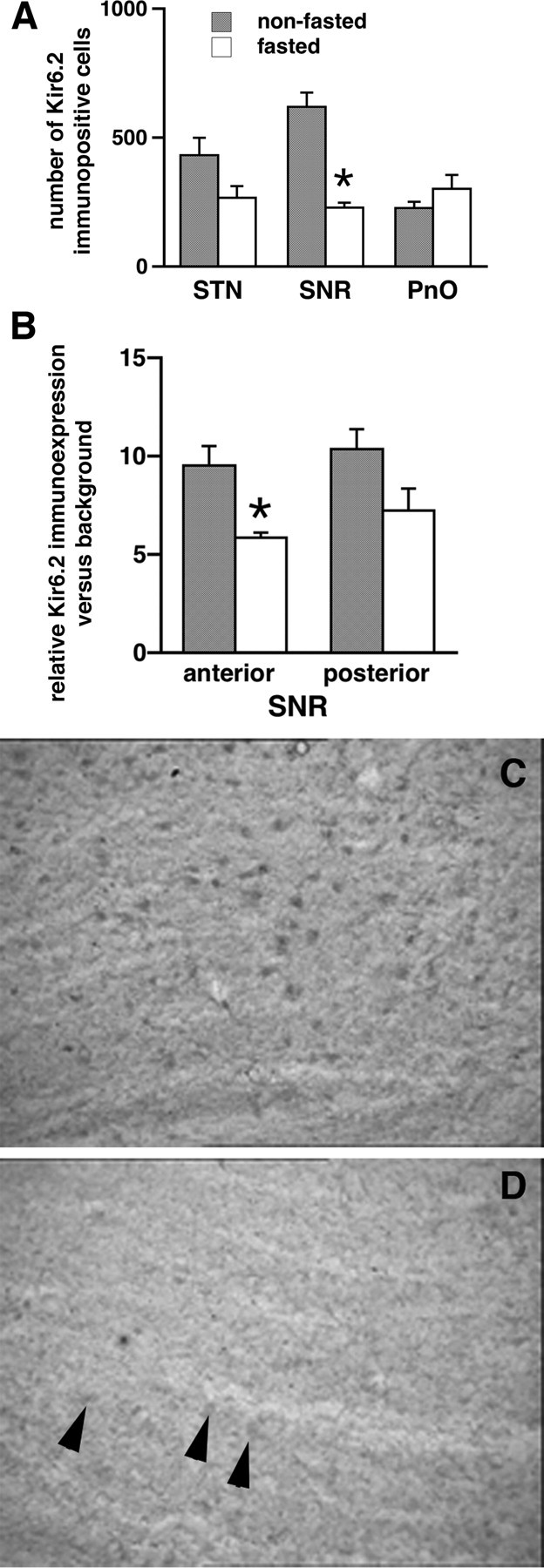
Kir6.2 expression in the SNR in fasted rats is decreased compared with nonfasted rats. A, Number of Kir6.2-immunopositive cells in the STN, SNR, and PnO of fasted (n = 5) and nonfasted (n = 5) rats. There was a significant decrease in the number of Kir6.2-immunopositive cells only in the SNR (*p < 0.05, t test). B, Densitometry (Ravizza et al., 2002) of the Kir6.2 immunoexpression in the SNR cells at the matching sections (10 cells at each of 3 sections per structure per rat) with the goal of comparison of the fasted (n = 6) and nonfasted (n = 6) rats. In the anterior part of the SNR of fasted rats, the Kir6.2 immunointensity was significantly lower than in nonfasted rats (*p < 0.05, t test). C, An example of a sagittal section through the SNR in a nonfasted rat demonstrating many Kir6.2-immunopositive cells. D, An example of a sagittal section through the SNR in a fasted rat demonstrating few Kir6.2-immunopositive cells.
Finally, we determined whether the decreases in the number of Kir6.2-immunopositive cells attributable to overnight fasting can be reversed by acute glucose administration. Two groups of overnight fasted rats with similar body weight (208.3 ± 4.8 vs 209.5 ± 2.5 g), and baseline blood glucose (76.0 ± 4.3 vs 72.8 ± 2.8 mg/100 ml) was injected with either saline (n = 4) or 3 ml of 20% glucose (n = 4), respectively. The rats were perfused after 4 h following the injection. Counting the Kir6.2-expressing cells did not reveal any differences between the glucose replenishment and saline groups. This result indicates that acute glucose substitution is incapable of altering Kir6.2 expression (data not shown), and, thus, the process of reinsertion of the Kir6.2 protein into the membrane leading to reoccurrence of immunoreactivity seems to be long term (Smith et al., 2007). Alternatively, stress related to the fasting may be responsible for internalization of the Kir6.2 protein, and, thus, the glucose refueling does not help.
Acute restriction of local SNR glucose has anticonvulsant effects
Previous experiments suggest an involvement of the anterior part of the SNR in hypoglycemic seizure control. In this experiment, we used to our advantage nonfasted rats with abundant Kir6.2 expression in the SNR (see above). Glucose availability in the anterior part of the SNR was decreased acutely by local bilateral microinfusions of 0.5 μl of 5% nonradioactive 2-DG (a glucose anti-metabolite). We hypothesized that this treatment would acutely limit ATP availability, thus opening the available Kir6.2 pores, resulting in hyperpolarization and, therefore, anticonvulsant effects. Control rats received equivalent volumes of 5% mannitol, a nonpermeable glucose analog. Baseline glucose concentration and glucose decrease with time were almost identical in both groups (Fig. 6 A). After insulin injection, there was a significant difference in the latency to onset of the first seizure (any of jumps, clonic seizures, or barrel rotations combined) of rats, which developed seizures as well as in the latency to onset of barrel rotations between the 2-DG (n = 8) and mannitol (n = 4) microinfused groups (Fig. 6 B, p < 0.05, Student's t test). The rats with 2-DG infusion in the SNR experienced longer latency to onset of both first seizure and barrel rotation, suggesting that, in the presence of hyperpolarizing Kir6.2 machinery, acute glucose insufficiency has anticonvulsant effects. The seizures in microinfused groups of nonfasted rats appeared earlier than in nonfasted rats in the initial experiment, consistent with the literature (Remler and Marcussen, 1984; Okada et al., 1986).
Figure 6.

Effects of drug microinfusions in the SNR on hypoglycemic seizures. A, Decrease of blood glucose with time after insulin injection in the two groups of nonfasted rats used for the experiment illustrated in B. There was no difference between the groups. There was a very good logarithmic fit, almost identical to the curve fit for the nonfasted rats in the Figure 1. B, Microinfusion of nonradioactive 2-DG, a glucose anti-metabolite, in the anterior part of the SNR (0.5 μl of the 5% solution per site), significantly delayed onset of first seizure and barrel rotation after insulin injection compared with rats microinjected with the same volume of 5% mannitol (mean ± SEM; *p < 0.05, t test). This finding is consistent with opening of postsynaptic KATP channels because of acute ATP deficiency resulting in hyperpolarization and decreased firing of the SNR neurons, and therefore, with anticonvulsant effects (Velíšková and Moshé, 2006). C, Double microinfusion of tolbutamide, a KATP channel blocker, in the anterior part of the SNR (0.25 μl of the 4 mm solution) of nonfasted rats at 24 h and immediately before insulin significantly accelerated onset of first seizure and barrel rotation compared with rats microinfused with solvent (DMSO/β-cyclodextrine mixture, 5:1 ratio; mean ± SEM; *p < 0.05, t test). This finding is consistent with closing of postsynaptic KATP channels resulting in depolarization and increased firing of the SNR neurons and, thus, proconvulsant effects (Velíšková and Moshé, 2006). D, Microinfusion of diazoxide, a KATP channel opener, in the anterior part of the SNR (0.25 μl of the 4 mm solution) of fasted rats did not change susceptibility to development of hypoglycemic seizures compared with rats microinfused with solvent (0.1N NaOH plus PBS), indicating that the effector system (postsynaptic KATP channels) may have been unavailable as suggested by decreased Kir6.2 subunit expression in fasted rats (Fig. 5).
SNR microinfusions of KATP channel blocker and opener
We tested whether, in nonfasted rats with abundant expression of postsynaptic KATP channels in the SNR, KATP channel blockade is associated with postsynaptic depolarization and, thus, would accelerate occurrence of hypoglycemic seizures. Tolbutamide infusion significantly decreased latency to onset of both the first seizure and barrel rotation compared with vehicle (Fig. 6 C, p < 0.05, Student's t test), consistent with closing KATP channels, postsynaptic membrane depolarization, and, therefore, proconvulsant effects (Velíšková and Moshé, 2006). In fasted rats with low SNR expression of postsynaptic KATP channels, we hypothesized that SNR microinfusion of KATP channel opener may be either anticonvulsant or will have no effect because of lack of the KATP channels. There was no effect of diazoxide compared with solvent (Fig. 6 D, p > 0.80, Student's t test). This is not surprising because not enough SNR neurons express KATP channels to mediate anticonvulsant effects of diazoxide (Fig. 5).
Recordings of SNR GABAergic cell activity in vitro
Rationale for the SNR recordings was to distinguish the role of presynaptic versus postsynaptic KATP channels in the SNR-mediated seizure control. Previous studies provide contradictory data about the respective roles of presynaptic and postsynaptic KATP channels in the control of seizures by the SNR. During et al. (1995) suggests a prominent role for presynaptic KATP channels on striatal terminals in the SNR associated with a decreased GABA release from those terminals. This results in increased firing of postsynaptic SNR neurons and proconvulsant effects. Yamada and Yuan (Yamada et al., 2001; Yuan et al., 2004), conversely, suggest a unique role of postsynaptic KATP channels completely discarding presynaptic participation. We used two different recording techniques to address this issue: whole cell and perforated patch clamp.
Whole-cell recordings with 2 mm ATP in the pipette
The purpose of this experiment was to determine contributions of presynaptic KATP channels to changes in the firing of SNR neurons during decreased extracellular glucose concentration. Thus, by the whole-cell approach, the ATP level in the SNR GABAergic cells has been clamped to 2 mm (n = 4 from 4 rats). No GABA or glutamate blockers were present in the ACSF. We found that, in correlation with decreasing extracellular glucose concentration, there was a significant increase in action potential firing rate (Fig. 7 A) associated with relevant increase in resting membrane potential (Fig. 7 B). The increase in firing frequency (occurring with a delay of 2–8 min after changes in glucose concentration) is consistent with the findings in mice (Yuan et al., 2004) and most likely follows either a decreased presynaptic GABA release from striatal terminals attributable to ATP deficiency associated with opening of Kir6.2 channels (During et al., 1995) or increased glutamate release from the subthalamic nucleus inputs. These data indicate a strong contribution of presynaptic inputs on the firing of SNR neurons.
Figure 7.

Single-cell recordings in the GABAergic cells of the SNR during decreases of extracellular glucose. A, Whole cell, 2 mm ATP in the electrode: frequency of GABAergic cell discharges (y-axis) is plotted versus extracellular glucose concentration (x-axis). The frequency of discharges significantly increased with decreasing extracellular glucose, reaching the highest firing rate at 2 mm extracellular glucose concentration (36 mg/100 ml). This indicates a significant contribution of presynaptic KATP channels in the increased postsynaptic firing, as postsynaptic ATP was clamped. B, Whole cell, 2 mm ATP in the electrode: resting membrane potential (y-axis) is plotted versus extracellular glucose concentration (x-axis). Resting membrane potential of the recorded cells significantly decreased in the decreasing glucose concentration. This finding is consistent with increased firing frequency. Furthermore, this illustrates that, in this case, the postsynaptic membrane cannot be affected by changes in intracellular ATP (clamped at 2 mm). Therefore, if postsynaptic intracellular ATP is kept constant, the effects of presynaptic ATP deficiency prevail, which result in a decreased release of GABA (During et al., 1995) from multiple GABAergic terminals predominantly of striatal origin (Bevan et al., 1996) and, thus, in increased postsynaptic firing. C, Gramicidin perforated patch clamp: frequency of GABAergic cell discharges (y-axis) is plotted versus extracellular glucose concentration (x-axis). This recording arrangement evaluated contribution of both presynaptic and postsynaptic KATP channels as postsynaptic ATP was allowed to fluctuate according to the metabolic state. The population of cells divided into two subpopulations: responders, which significantly decreased their firing rate with the decrease of extracellular glucose compared with nonresponders, in which the firing rate remained almost constant even after 10 min in 2 mm glucose concentration (p < 0.05, t test). In the hippocampus, this concentration of extracellular glucose may completely shut down synaptic transmission (Kirchner et al., 2006). D, Gramicidin perforated patch clamp: recording from electrophysiologically identified GABAergic neurons. The first neuron was spontaneously active with action potential firing at ∼19 Hz in 20 mm extracellular glucose concentration (top left). After decrease of extracellular glucose concentration to 2 mm, the frequency of action potentials decreased to 2 Hz (top right). This neuron is referred to as responder. Another neuron was spontaneously firing action potentials at ∼18 Hz in 20 mm extracellular glucose (bottom left). After extracellular glucose decreased to 2 mm, the firing frequency was maintained (bottom right). This neuron is referred to as a nonresponder.
Gramicidin perforated patch
In this arrangement, intracellular ATP was not clamped, and, therefore, the effects of both presynaptic and postsynaptic ATP channels were recorded. Here we investigated firing properties of SNR neurons with relatively intact intracellular environment because gramicidin pores permit passage of small Na+ and K+ ions. Our baseline recording in the attached patched mode with gramicidin indicated that baseline firing frequency of the SNR cells was slightly higher (16.7 ± 2.0 Hz; n = 8 from 6 rats) than in those cells recorded using whole-cell patch (11.0 ± 1.4 Hz, n = 4). This difference is consistent with different techniques used because patch dialysis in the whole-cell access alters firing frequency of the neurons. A decrease in glucose concentration from 20 to 6 mm did not produce any significant alteration in firing frequency of the SNR neurons; only a modest decrease occurred (Fig. 7 C). However, additional glucose concentration decrease to 2 mm split the SNR neurons into two groups. One group of neurons (n = 4; further referred to as responders) (Fig. 7 D, top) reacted by a profound decrease in firing to the decrease in glucose concentration, suggesting a loss of intracellular ATP, opening KATP channels, and hyperpolarization. The other group maintained the high firing rate, not significantly different from the firing rates at 20 or 6 mm extracellular glucose (referred to as nonresponders; n = 4) (Fig. 7 D, bottom), suggesting an impaired KATP channels control, e.g., because of insufficient of KATP channel presence in the membrane. The gramicidin perforated patch data indicate that there is also a significant postsynaptic component to the KATP channel control of neuronal firing in the SNR because some neurons do not shut down with hypoglycemia but continue firing even in very low glucose concentrations. These neurons may contribute to the SNR failure to control seizures in low glucose conditions.
Discussion
We demonstrate here that fasting predisposes rats to development of hypoglycemic seizures after insulin injection. However, fasting-induced decrease in baseline blood glucose concentration cannot account for the earlier onset of seizures in fasted versus nonfasted rats. Imaging data strongly implicate involvement of the SNR, PPTg, and superior colliculus in the hypoglycemic seizure control, with the SNR likely playing a central role. In addition, results suggest that metabolic stress (such as fasting) associated with impairment of hyperpolarizing machinery of KATP channels in the SNR may become a predisposing factor for seizure development as a result of failing SNR-mediated seizure control.
The 2-DG uptake changes in the SNR and superior colliculus suggest an important role of the SNR in seizure-controlling network in the hypoglycemic seizures. The SNR contains a high density of KATP channels and expresses exclusively the most sensitive type of KATP channel consisting of Kir6.2 and SUR1 subunits (Liss et al., 1999; Yamada and Inagaki, 2005). The presence of these channels determines seizure susceptibility, because the mice overexpressing SUR1 resist the induction of kainic acid seizures (Hernández-Sánchez et al., 2001). Indeed, it appears that both biosynthesis of these channels (Acosta-Martinez and Levine, 2007) as well as their clathrin-mediated endocytosis (Hu et al., 2003; Sivaprasadarao et al., 2007) is controlled by metabolic conditions. Unlike biosynthesis, endocytotic sorting mechanisms have the ability to rapidly adjust density of protein at the cell surface (Smith et al., 2007). Consistent with a previous report, we found that metabolic stress (in our experiments, overnight fasting) was associated with a decrease in the number of SNR neurons expressing Kir6.2 channels (Hu et al., 2003). Besides the direct effect of metabolic stress, stress associated with food restriction might also change the Kir6.2 expression because available reports indicate bidirectional changes in Kir6.2 expression after dexamethasone treatment (Hernández-Sánchez et al., 1999; Gu et al., 2007). The Kir6.2 decrease in fasted animals impairs hyperpolarization of the SNR neurons (Seino and Miki, 2003) during insulin-induced hypoglycemia, and this effect leads to faster development of hypoglycemic seizures (proconvulsant effect) compared with nonfasted rats with abundance of Kir6.2-expresing cells in the SNR. This process seems to happen faster and more reliably in vitro than in the brain in vivo, in which the glucostatic mechanisms are capable of long-term compensations. Therefore, it is possible that slow decrease in extracellular glucose over >15 min during in vitro recordings may have induced KATP channel endocytosis in ∼50% of recorded neurons (Hu et al., 2003) (approximately similar to the decrease in Kir6.2-immunopositive SNR cells in fasted rats compared with nonfasted), and those neurons maintained their firing rates even during profound extracellular glucopenia. This is similar to finding of others on the effects of glucose concentration on firing rates in the SNR neurons in mice, which requires a significant delay of 6–10 min (Yuan et al., 2004). These findings indicate that long-term global metabolic stress (global in terms of the whole brain) or focal events involving the SNR may become precipitating factors for the occurrence of different seizure types (not only hypoglycemic) controlled by the SNR system. This corresponds to our previous findings demonstrating decreased threshold for flurothyl seizures in rats rendered significantly hypoglycemic after insulin (Kirchner et al., 2006). This situation is also known in human conditions in which fasting is a precipitating factor for hypoglycemic seizures (Malouf and Brust, 1985). Interestingly, fasting is related to production of ketone bodies (Morris, 2005), and ketogenic diet is often used as an effective anticonvulsant treatment (Sankar and Sotero de Menezes, 1999). Studies indicate that presence of ketone bodies is associated with anticonvulsant effects in several seizure models (Stafstrom and Spencer, 2000). However, hypoglycemic seizures probably represent a special case of altered metabolism. Fasting before hypoglycemic seizures is not associated with anticonvulsant effects but rather with proconvulsant effects. This seems to be related to decreased expression of SNR KATP channels, which prevents effects of ketone bodies (Ma et al., 2007).
It should be emphasized that treatments that decreased activity of GABAergic neurons in the anterior part of the SNR lead to attenuation of seizures (Velíšková and Moshé, 2006). Original studies (Iadarola and Gale, 1981) showed that precisely localized bilateral SNR microinfusions of the GABAA receptor agonist muscimol produce anticonvulsant effects. In contrast, localized SNR microinfusions of the GABAA receptor antagonist bicuculline have proconvulsant effects (Sperber et al., 1989). In addition, suppression of the SNR glutamatergic input from the STN by the SNR microinfusions of NMDA receptor antagonist AP-7 decreases firing rate of SNR neurons and, thus, induces anticonvulsant effects (Turski et al., 1986; Velíšková et al., 2001). The fact that seizure activity is sensitive to manipulations of both GABA and glutamate receptor systems within the SNR suggests that both major nigral inputs (glutamatergic from the subthalamic nucleus and GABAergic from the striatum) are actively involved during seizures. These results indicate that anticonvulsant effects are associated with decreased activity of the GABAergic neurons in the anterior part of the SNR, leading to disinhibition of the output structures (Deniau and Chevalier, 1985): the superior colliculus and the PPTg (Bolam et al., 2000). Accordingly, bicuculline infusions in the superior colliculus or PPTg have anticonvulsant effects, but localized muscimol infusions as well as lesions in these structures are proconvulsant (Dean and Gale, 1989; Okada et al., 1989; Depaulis et al., 1990b; Redgrave et al., 1992).
In case of an acute in vivo glucose anti-metabolite poisoning in the SNR in rats with abundant Kir6.2-expressing neurons (i.e., nonfasted rats), hypoglycemic seizures occur later than in controls infused with equiosmotic mannitol. This indicates that acute glucose deficit translates in immediate lack of ATP, opening of KATP channels, and hyperpolarization of the SNR neurons associated with decreases in their firing rates and results in anticonvulsant effects (Ma et al., 2007). This effect is analogous to the effects of the microinfused GABAA receptor agonist muscimol (Velíšková and Moshé, 2001), which is associated with decreased firing of the SNR neurons and with anticonvulsant effects (Deniau et al., 1982; Depaulis et al., 1990a). Focal depletion of glucose in the structure controlling seizure expression (SNR) seems to have strikingly different effects compared with depletion of glucose in a brain structure with low seizure threshold (hippocampus), which easily develops epileptic focus (Abdelmalik et al., 2007).
Experiments with glucose rescue indicated that fast recovery from severe hypoglycemia may be also dangerous in terms of seizure control. However, the seizures developing during this recovery were semiologically different from hypoglycemic seizures, involving rearing, forelimb clonus, and tonic-clonic seizures and always leading to death. The variable blood glucose levels in those rats indicate impaired peripheral glucose control: liver plays an important role in glucose homeostasis after fasting (Klover and Mooney, 2004), with possible involvement of NAD+-dependent deacetylase sirtuin 1 (SIRT1), because hepatic knockdown of SIRT1 impairs fasting induction of gluconeogenesis and reduces glycemia (Rodgers and Puigserver, 2007). Another option is an impaired pancreatic involvement because, during hypoglycemia, both insulin and glucagon release are under KATP channel control (Muñoz et al., 2005) as well as under control of hypothalamus–brain stem hypoglycemia-induced vagal signaling (Moore et al., 1998; Wu et al., 2004). A possible explanation for the occurrence of these seizures is that the acute brain refueling from the peripheral source without appropriate peripheral control recovers activity in previously hypoglycemia-silenced brain regions such as cortex, hippocampus, and amygdala. Because the endogenous mechanisms involved in seizure control have failed, the ensuing seizures rapidly evolve to bilaterally clonic and tonic-clonic and, eventually, to death.
In summary, (1) Kir6.2 expression in the SNR is regulated by a metabolic stress (here fasting). Decreased expression of KATP channels in the SNR limits hyperpolarization capacity of SNR neurons, and, as a result, GABAergic neurons fire more, consistent with proconvulsant effects (Velíšková and Moshé, 2006). Indeed, 100% of fasted rats express hypoglycemic seizures. (2) In the SNR with KATP machinery present (nonfasted rats), acute glucose depletion induces hyperpolarizations of SNR neurons, an effect similar to the effect of microinfused muscimol (Deransart et al., 1998, 2001), which is associated with anticonvulsant effects (Depaulis et al., 1989; Velíšková and Moshé, 2001), whereas blockade of KATP channels by tolbutamide is proconvulsant. (3) Because we were unable to affect seizure incidence by limiting glucose availability in the SNR, this indicates that the SNR is not a site of origin of hypoglycemic seizures (supported by EEG findings); rather, it is a powerful control site. We can only speculate that hypoglycemic seizures may arise from structures in medulla oblongata, which are responsible for rhythmic breathing and heart control and maintain their rhythmic discharges even under severe metabolic suppression (Barman and Gebber, 2000; Mironov et al., 2005). Our findings imply that the SNR plays an important role in translating metabolic conditions into proconvulsant or anticonvulsant states. Therefore, control of the unique KATP channels in the SNR may become a significant part of hypoglycemic seizure treatment as well as seizure control in general.
Footnotes
This work was supported by the Heffer Family Medical Foundation, National Institutes of Health Grants NS-20253, NS-059504, and NS-056093, and a CURE Foundation Research grant. S.L.M. is Martin A. and Emily L. Fisher fellow in Neurology and Pediatrics. K.R. was supported by the Albert Einstein College of Medicine Summer Undegraduate Research Program. K.-L.P. was supported by the Harlem Children Society and is currently a Posse Foundation Scholar at the Franklin and Marshall College (Lancaster, PA). We gratefully acknowledge the late Dr. Marvin Cornblath for his advice.
References
- Abdelmalik PA, Shannon P, Yiu A, Liang P, Adamchik Y, Weisspapir M, Samoilova M, Burnham WM, Carlen PL. Hypoglycemic seizures during transient hypoglycemia exacerbate hippocampal dysfunction. Neurobiol Dis. 2007;26:646–660. doi: 10.1016/j.nbd.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Ackermann RF, Finch DM, Babb TL, Engel J., Jr Increased glucose metabolism during long-duration recurrent inhibition of hippocampal pyramidal cells. J Neurosci. 1984;4:251–264. doi: 10.1523/JNEUROSCI.04-01-00251.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta-Martínez M, Levine JE. Regulation of KATP channel subunit gene expression by hyperglycemia in the mediobasal hypothalamus of female rats. Am J Physiol Endocrinol Metab. 2007;292:E1801–E1807. doi: 10.1152/ajpendo.00700.2006. [DOI] [PubMed] [Google Scholar]
- Akasu T, Tsurusaki M, Shoji S. Depletion of glucose causes presynaptic inhibition of neuronal transmission in the rat dorsolateral septal nucleus. Synapse. 1996;24:125–134. doi: 10.1002/(SICI)1098-2396(199610)24:2<125::AID-SYN4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Allen TG, Brown DA. Modulation of the excitability of cholinergic basal forebrain neurones by KATP channels. J Physiol. 2004;554:353–370. doi: 10.1113/jphysiol.2003.055889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ao Y, Wu S, Go VL, Toy N, Yang H. Maintaining euglycemia prevents insulin-induced Fos expression in brain autonomic regulatory circuits. Pancreas. 2005;31:142–147. doi: 10.1097/01.mpa.0000172562.96168.59. [DOI] [PubMed] [Google Scholar]
- Barman SM, Gebber GL. “Rapid” rhythmic discharges of sympathetic nerves: sources, mechanisms of generation, and physiological relevance. J Biol Rhythms. 2000;15:365–379. doi: 10.1177/074873000129001468. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. Limbic seizure and brain damage produced by kainic acid: Mechanisms and relevance to human temporal lobe epilepsy. Neuroscience. 1985;14:375–403. doi: 10.1016/0306-4522(85)90299-4. [DOI] [PubMed] [Google Scholar]
- Bevan MD, Smith AD, Bolam JP. The substantia nigra as a site of synaptic integration of functionally diverse information arising from the ventral pallidum and the globus pallidus in the rat. Neuroscience. 1996;75:5–12. doi: 10.1016/0306-4522(96)00377-6. [DOI] [PubMed] [Google Scholar]
- Bolam JP, Hanley JJ, Booth PA, Bevan MD. Synaptic organisation of the basal ganglia. J Anat. 2000;196:527–542. doi: 10.1046/j.1469-7580.2000.19640527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown LL, Feldman SM, Smith DM, Cavanaugh JR, Ackermann RF, Graybiel AM. Differential metabolic activity in the striosome and matrix compartments of the rat striatum during natural behaviors. J Neurosci. 2002;22:305–314. doi: 10.1523/JNEUROSCI.22-01-00305.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalheiro EA, Silva DF, Turski WA, Calderazzo-Filho LS, Bortolotto ZA, Turski L. The susceptibility of rats to pilocarpine-induced seizures is age-dependent. Brain Res. 1987;465:43–58. doi: 10.1016/0165-3806(87)90227-6. [DOI] [PubMed] [Google Scholar]
- Chapman AG, Engelsen B, Meldrum BS. 2-amino-7-phosphonoheptanoic acid inhibits insulin-induced convulsions and striatal aspartate accumulation in rats with frontal cortical ablation. J Neurochem. 1987;49:121–127. doi: 10.1111/j.1471-4159.1987.tb03403.x. [DOI] [PubMed] [Google Scholar]
- Davis EA, Keating B, Byrne GC, Russell M, Jones TW. Hypoglycemia: incidence and clinical predictors in a large population- based sample of children and adolescents with IDDM. Diabetes Care. 1997;20:22–25. doi: 10.2337/diacare.20.1.22. [DOI] [PubMed] [Google Scholar]
- Dean P, Gale K. Anticonvulsant action of GABA receptor blockade in the nigrotectal target region. Brain Res. 1989;477:391–395. doi: 10.1016/0006-8993(89)91434-0. [DOI] [PubMed] [Google Scholar]
- Deniau JM, Chevalier G. Disinhibition as a basic process in the expression of striatal functions. II. The striato-nigra influence on thalamocortical cells of the ventromedial thalamic nucleus. Brain Res. 1985;334:227–233. doi: 10.1016/0006-8993(85)90214-8. [DOI] [PubMed] [Google Scholar]
- Deniau JM, Kitai ST, Donoghue JP, Grofova I. Neuronal interactions in the substantia nigra pars reticulata through axon collaterals of the projection neurons. An electrophysiological and morphological study. Exp Brain Res. 1982;47:105–113. doi: 10.1007/BF00235891. [DOI] [PubMed] [Google Scholar]
- Depaulis A, Snead OC, 3rd, Marescaux C, Vergnes M. Suppressive effects of intranigral injection of muscimol in three models of generalized non-convulsive epilepsy induced by chemical agents. Brain Res. 1989;498:64–72. doi: 10.1016/0006-8993(89)90399-5. [DOI] [PubMed] [Google Scholar]
- Depaulis A, Vergnes M, Liu Z, Kempf E, Marescaux C. Involvement of the nigral output pathways in the inhibitory control of the substantia nigra over generalized non-convulsive seizures in the rat. Neuroscience. 1990a;39:339–349. doi: 10.1016/0306-4522(90)90272-6. [DOI] [PubMed] [Google Scholar]
- Depaulis A, Liu Z, Vergnes M, Marescaux C, Micheletti G, Warter JM. Suppression of spontaneous generalized non-convulsive seizures in the rat by microinjection of GABA antagonists into the superior colliculus. Epilepsy Res. 1990b;5:192–198. doi: 10.1016/0920-1211(90)90038-w. [DOI] [PubMed] [Google Scholar]
- Depaulis A, Vergnes M, Marescaux C. Endogenous control of epilepsy: the nigral inhibitory system. Prog Neurobiol. 1994;42:33–52. doi: 10.1016/0301-0082(94)90020-5. [DOI] [PubMed] [Google Scholar]
- Deransart C, Vercueil L, Marescaux C, Depaulis A. The role of basal ganglia in the control of generalized absence seizures. Epilepsy Res. 1998;32:213–223. doi: 10.1016/s0920-1211(98)00053-9. [DOI] [PubMed] [Google Scholar]
- Deransart C, Lê-Pham BT, Hirsch E, Marescaux C, Depaulis A. Inhibition of the substantia nigra suppresses absences and clonic seizures in audiogenic rats, but not tonic seizures: evidence for seizure specificity of the nigral control. Neuroscience. 2001;105:203–211. doi: 10.1016/s0306-4522(01)00165-8. [DOI] [PubMed] [Google Scholar]
- Diabetes Control and Complications Trial Research Group. Implementation of treatment protocols in the diabetes control and complications trial. Diabetes Care. 1995;18:361–376. doi: 10.2337/diacare.18.3.361. [DOI] [PubMed] [Google Scholar]
- Dunn-Meynell AA, Rawson NE, Levin BE. Distribution and phenotype of neurons containing the ATP-sensitive K+ channel in rat brain. Brain Res. 1998;814:41–54. doi: 10.1016/s0006-8993(98)00956-1. [DOI] [PubMed] [Google Scholar]
- During MJ, Leone P, Davis KE, Kerr D, Sherwin RS. Glucose modulates rat substantia nigra GABA release in vivo via ATP- sensitive potassium channels. J Clin Invest. 1995;95:2403–2408. doi: 10.1172/JCI117935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan P, O'Regan PA, Szerb JC. Effect of low glucose concentration on synaptic transmission in the rat hippocampal slice. Brain Res Bull. 1988;21:741–747. doi: 10.1016/0361-9230(88)90041-x. [DOI] [PubMed] [Google Scholar]
- Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining GJ, Slingerland AS, Howard N, Srinivasan S, Silva JM, Molnes J, Edghill EL, Frayling TM, Temple IK, Mackay D, Shield JP, Sumnik Z, van Rhijn A, Wales JK, Clark P, Gorman S. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med. 2004;350:1838–1849. doi: 10.1056/NEJMoa032922. [DOI] [PubMed] [Google Scholar]
- Gu YT, Zhang H, Xue YX. Dexamethasone enhances adenosine 5′-triphosphate-sensitive potassium channel expression in the blood-brain tumor barrier in a rat brain tumor model. Brain Res. 2007;1162:1–8. doi: 10.1016/j.brainres.2007.05.053. [DOI] [PubMed] [Google Scholar]
- Hart SP, Frier BM. Causes, management and morbidity of acute hypoglycaemia in adults requiring hospital admission. QJM. 1998;91:505–510. doi: 10.1093/qjmed/91.7.505. [DOI] [PubMed] [Google Scholar]
- Herbel G, Boyle PJ. Hypoglycemia. Pathophysiology and treatment. Endocrinol Metab Clin North Am. 2000;29:725–743. doi: 10.1016/s0889-8529(05)70161-4. [DOI] [PubMed] [Google Scholar]
- Hernández-Sánchez C, Ito Y, Ferrer J, Reitman M, LeRoith D. Characterization of the mouse sulfonylurea receptor 1 promoter and its regulation. J Biol Chem. 1999;274:18261–18270. doi: 10.1074/jbc.274.26.18261. [DOI] [PubMed] [Google Scholar]
- Hernández-Sánchez C, Basile AS, Fedorova I, Arima H, Stannard B, Fernandez AM, Ito Y, LeRoith D. Mice transgenically overexpressing sulfonylurea receptor 1 in forebrain resist seizure induction and excitotoxic neuron death. Proc Natl Acad Sci U S A. 2001;98:3549–3554. doi: 10.1073/pnas.051012898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks GA, Hudson AL, Henderson G. Localization of high affinity [3H]glibenclamide binding sites within the substantia nigra zona reticulata of the rat brain. Neuroscience. 1994;61:285–292. doi: 10.1016/0306-4522(94)90231-3. [DOI] [PubMed] [Google Scholar]
- Hu K, Huang CS, Jan YN, Jan LY. ATP-sensitive potassium channel traffic regulation by adenosine and protein kinase C. Neuron. 2003;38:417–432. doi: 10.1016/s0896-6273(03)00256-3. [DOI] [PubMed] [Google Scholar]
- Iadarola MJ, Gale K. Cellular compartments of GABA in brain and their relationship to anticonvulsant activity. Mol Cell Biochem. 1981;39:305–329. doi: 10.1007/BF00232582. [DOI] [PubMed] [Google Scholar]
- Kirchner A, Velíšková J, Velíšek L. Differential effects of low glucose concentrations on seizures and epileptiform activity in vivo and in vitro. Eur J Neurosci. 2006;23:1512–1522. doi: 10.1111/j.1460-9568.2006.04665.x. [DOI] [PubMed] [Google Scholar]
- Klover PJ, Mooney RA. Hepatocytes: critical for glucose homeostasis. Int J Biochem Cell Biol. 2004;36:753–758. doi: 10.1016/j.biocel.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Kyrozis A, Reichling DB. Perforated-patch recording with gramicidin avoids artifactual changes in intracellular chloride concentration. J Neurosci Methods. 1995;57:27–35. doi: 10.1016/0165-0270(94)00116-x. [DOI] [PubMed] [Google Scholar]
- Leckie AM, Graham MK, Grant JB, Ritchie PJ, Frier BM. Frequency, severity, and morbidity of hypoglycemia occurring in the workplace in people with insulin-treated diabetes. Diabetes Care. 2005;28:1333–1338. doi: 10.2337/diacare.28.6.1333. [DOI] [PubMed] [Google Scholar]
- Levin BE. Glucosensing neurons do more than just sense glucose. Int J Obes Relat Metab Disord. 2001;25(Suppl 5):S68–S72. doi: 10.1038/sj.ijo.0801916. [DOI] [PubMed] [Google Scholar]
- Liss B, Roeper J. Molecular physiology of neuronal K-ATP channels. Mol Membr Biol. 2001;18:117–127. [PubMed] [Google Scholar]
- Liss B, Bruns R, Roeper J. Alternative sulfonylurea receptor expression defines metabolic sensitivity of K-ATP channels in dopaminergic midbrain neurons. EMBO J. 1999;18:833–846. doi: 10.1093/emboj/18.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Berg J, Yellen G. Ketogenic diet metabolites reduce firing in central neurons by opening K(ATP) channels. J Neurosci. 2007;27:3618–3625. doi: 10.1523/JNEUROSCI.0132-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malouf R, Brust JCM. Hypoglycemia: causes, neurological manifestations, and outcome. Ann Neurol. 1985;17:421–430. doi: 10.1002/ana.410170502. [DOI] [PubMed] [Google Scholar]
- Marrannes R, Wauquier A. Episodic barrel rotations induced by intrastriatal injection of quinolinic acid in rats. Inhibition by anticonvulsants. Pharmacol Biochem Behav. 1988;31:153–162. doi: 10.1016/0091-3057(88)90327-9. [DOI] [PubMed] [Google Scholar]
- Mironov SL, Hartelt N, Ivannikov MV. Mitochondrial K(ATP) channels in respiratory neurons and their role in the hypoxic facilitation of rhythmic activity. Brain Res. 2005;1033:20–27. doi: 10.1016/j.brainres.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Moore MC, Connolly CC, Cherrington AD. Autoregulation of hepatic glucose production. Eur J Endocrinol. 1998;138:240–248. doi: 10.1530/eje.0.1380240. [DOI] [PubMed] [Google Scholar]
- Morgan JI, Cohen DR, Hempstead JL, Curran T. Mapping patterns of c-fos expression in the central nervous system after seizure. Science. 1987;237:192–197. doi: 10.1126/science.3037702. [DOI] [PubMed] [Google Scholar]
- Morris AA. Cerebral ketone body metabolism. J Inherit Metab Dis. 2005;28:109–121. doi: 10.1007/s10545-005-5518-0. [DOI] [PubMed] [Google Scholar]
- Moshé SL, Sperber EF. Substantia nigra-mediated control of generalized seizures. In: Gloor G, Kostopoulos R, Naquet M, Avoli P, editors. Generalized epilepsy: cellular, molecular and pharmacological approaches. Boston: Birkhauser; 1990. pp. 355–367. [Google Scholar]
- Mourre C, Ben Ari Y, Bernardi H, Fosset M, Lazdunski M. Antidiabetic sulfonylureas: localization of binding sites in the brain and effects on the hyperpolarization induced by anoxia in hippocampal slices. Brain Res. 1989;486:159–164. doi: 10.1016/0006-8993(89)91288-2. [DOI] [PubMed] [Google Scholar]
- Mourre C, Widmann C, Lazdunski M. Sulfonylurea binding sites associated with ATP-regulated K+ channels in the central nervous system: autoradiographic analysis of their distribution and ontogenesis, and of their localization in mutant mice cerebellum. Brain Res. 1990a;519:29–43. doi: 10.1016/0006-8993(90)90057-i. [DOI] [PubMed] [Google Scholar]
- Mourre C, Smith ML, Siesjö BK, Lazdunski M. Brain ischemia alters the density of binding sites for glibenclamide, a specific blocker of ATP-sensitive K+ channels. Brain Res. 1990b;526:147–152. doi: 10.1016/0006-8993(90)90262-a. [DOI] [PubMed] [Google Scholar]
- Muñoz A, Hu M, Hussain K, Bryan J, Aguilar-Bryan L, Rajan AS. Regulation of glucagon secretion at low glucose concentrations: evidence for adenosine triphosphate-sensitive potassium channel involvement. Endocrinology. 2005;146:5514–5521. doi: 10.1210/en.2005-0637. [DOI] [PubMed] [Google Scholar]
- Nehlig A, Vergnes M, Marescaux C, Boyet S, Lannes B. Local cerebral glucose utilization in rats with petit mal-like seizures. Ann Neurol. 1991;29:72–77. doi: 10.1002/ana.410290113. [DOI] [PubMed] [Google Scholar]
- Okada R, Moshe SL, Wong BY, Sperber EF, Zhao DY. Age-related substantia nigra-mediated seizure facilitation. Exp Neurol. 1986;93:180–187. doi: 10.1016/0014-4886(86)90157-3. [DOI] [PubMed] [Google Scholar]
- Okada R, Negishi N, Nagaya H. The role of the nigrotegmental GABAergic pathway in the propagation of pentylenetetrazol-induced seizures. Brain Res. 1989;480:383–387. doi: 10.1016/0006-8993(89)90212-6. [DOI] [PubMed] [Google Scholar]
- Panickar KS, Purushotham K, King MA, Rajakumar G, Simpkins JW. Hypoglycemia-induced seizures reduce cyclic AMP response element binding protein levels in the rat hippocampus. Neuroscience. 1998;83:1155–1160. doi: 10.1016/s0306-4522(97)00466-1. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 4. San Diego: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Pereira de Vasconcelos A, el Hamdi G, Vert P, Nehlig A. An experimental model of generalized seizures for the measurement of local cerebral glucose utilization in the immature rat. II. Mapping of the brain metabolism using quantitative [14C]-2-deoxyglucose technique. Brain Res. 1992;69:243–259. doi: 10.1016/0165-3806(92)90165-s. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizures. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Ravizza T, Galanopoulou AS, Velíšková J, Moshé SL. Sex differences in androgen and estrogen receptor expression in rat substantia nigra during development: an immunohistochemical study. Neuroscience. 2002;115:685–696. doi: 10.1016/s0306-4522(02)00491-8. [DOI] [PubMed] [Google Scholar]
- Redgrave P, Simkins M, Overton P, Dean P. Anticonvulsant role of nigrotectal projection in the maximal electroshock model of epilepsy. I. Mapping of dorsal midbrain with bicuculline. Neuroscience. 1992;46:379–390. doi: 10.1016/0306-4522(92)90059-b. [DOI] [PubMed] [Google Scholar]
- Remler MP, Marcussen WH. The blood brain barrier lesion and the systemic convulsant model of epilepsy. Epilepsia. 1984;25:574–577. doi: 10.1111/j.1528-1157.1984.tb03464.x. [DOI] [PubMed] [Google Scholar]
- Richards CD, Shiroyama T, Kitai ST. Electrophysiological and immunocytochemical characterization of GABA and dopamine neurons in the substantia nigra of the rat. Neuroscience. 1997;80:545–557. doi: 10.1016/s0306-4522(97)00093-6. [DOI] [PubMed] [Google Scholar]
- Rodgers JT, Puigserver P. Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc Natl Acad Sci U S A. 2007;104:12861–12866. doi: 10.1073/pnas.0702509104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samoriski GM, Piekut DT, Applegate CD. Differential spatial patterns of Fos induction following generalized clonic and generalized tonic seizures. Exp Neurol. 1997;143:255–268. doi: 10.1006/exnr.1996.6368. [DOI] [PubMed] [Google Scholar]
- Sankar R, Sotero de Menezes M. Metabolic and endocrine aspects of the ketogenic diet. Epilepsy Res. 1999;37:191–201. doi: 10.1016/s0920-1211(99)00071-6. [DOI] [PubMed] [Google Scholar]
- Seino S, Miki T. Physiological and pathophysiological roles of ATP-sensitive K+ channels. Prog Biophys Mol Biol. 2003;81:133–176. doi: 10.1016/s0079-6107(02)00053-6. [DOI] [PubMed] [Google Scholar]
- Shoji S. Glucose regulation of synaptic transmission in the dorsolateral septal nucleus of the rat. Synapse. 1992;12:322–332. doi: 10.1002/syn.890120409. [DOI] [PubMed] [Google Scholar]
- Silveira DC, Sogawa Y, Holmes GL. The expression of Fos following kainic acid-induced seizures is age-dependent. Eur J Neurosci. 2002;15:329–344. doi: 10.1046/j.0953-816x.2001.01849.x. [DOI] [PubMed] [Google Scholar]
- Simmons JM, Ackermann RF, Gallistel CR. Medial forebrain bundle lesions fail to structurally and functionally disconnect the ventral tegmental area from many ipsilateral forebrain nuclei: implications for the neural substrate of brain stimulation reward. J Neurosci. 1998;18:8515–8533. doi: 10.1523/JNEUROSCI.18-20-08515.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaprasadarao A, Taneja TK, Mankouri J, Smith AJ. Trafficking of ATP-sensitive potassium channels in health and disease. Biochem Soc Trans. 2007;35:1055–1059. doi: 10.1042/BST0351055. [DOI] [PubMed] [Google Scholar]
- Smith AJ, Taneja TK, Mankouri J, Sivaprasadarao A. Molecular cell biology of KATP channels: implications for neonatal diabetes. Expert Rev Mol Med. 2007;9:1–17. doi: 10.1017/S1462399407000403. [DOI] [PubMed] [Google Scholar]
- Sokoloff L, Reivich M, Kennedy C, Des Rosiers MH, Patlak CS, Pettigrew KD, Sakurada O, Shinohara M. The [14C] deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedures and normal values in the conscious and anesthetized albino rat. J Neurochem. 1977;28:897–916. doi: 10.1111/j.1471-4159.1977.tb10649.x. [DOI] [PubMed] [Google Scholar]
- Sperber EF, Wurpel JN, Zhao DY, Moshé SL. Evidence for the involvement of nigral GABAA receptors in seizures of adult rats. Brain Res. 1989;480:378–382. doi: 10.1016/0006-8993(89)90211-4. [DOI] [PubMed] [Google Scholar]
- Stafstrom CE, Spencer S. The ketogenic diet: a therapy in search of an explanation. Neurology. 2000;54:282–283. doi: 10.1212/wnl.54.2.282. [DOI] [PubMed] [Google Scholar]
- Suda S, Shinohara M, Miyaoka M, Lucignani G, Kennedy C, Sokoloff L. The lumped constant of the deoxyglucose method in hypoglycemia: effects of moderate hypoglycemia on local cerebral glucose utilization in the rat. J Cereb Blood Flow Metab. 1990;10:499–509. doi: 10.1038/jcbfm.1990.92. [DOI] [PubMed] [Google Scholar]
- Thompson K, Anantharam V, Behrstock S, Bongarzone E, Campagnoni A, Tobin AJ. Conditionally immortalized cell lines, engineered to produce and release GABA, modulate the development of behavioral seizures. Exp Neurol. 2000;161:481–489. doi: 10.1006/exnr.1999.7305. [DOI] [PubMed] [Google Scholar]
- Turski L, Cavalheiro EA, Turski WA, Meldrum BS. Excitatory neurotransmission within substantia nigra pars reticulata regulates threshold for seizures produced by pilocarpine in rats: effects of intranigral 2-amino-7-phosphonoheptanoate and N-methyl-d-aspartate. Neuroscience. 1986;18:61–77. doi: 10.1016/0306-4522(86)90179-x. [DOI] [PubMed] [Google Scholar]
- Vannucci RC, Vannucci SJ. Cerebral carbohydrate metabolism during hypoglycemia and anoxia in newborn rats. Ann Neurol. 1978;4:73–79. doi: 10.1002/ana.410040114. [DOI] [PubMed] [Google Scholar]
- Velíšek L, Velíšková J, Ravizza T, Giorgi FS, Moshé SL. Circling behavior and [14C]2-deoxyglucose mapping in rats: possible implications for autistic repetitive behaviors. Neurobiol Dis. 2005;18:346–355. doi: 10.1016/j.nbd.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Velíšek L, Jehle K, Asche S, Velíšková J. Model of infantile spasms induced by N-methyl-D-aspartic acid in prenatally impaired brain. Ann Neurol. 2007;61:109–119. doi: 10.1002/ana.21082. [DOI] [PubMed] [Google Scholar]
- Velíšková J, Moshé SL. Sexual dimorphism and developmental regulation of substantia nigra function. Ann Neurol. 2001;50:596–601. doi: 10.1002/ana.1248. [DOI] [PubMed] [Google Scholar]
- Velíšková J, Moshé SL. Update on the role of substantia nigra pars reticulata in the regulation of seizures. Epilepsy Curr. 2006;6:83–87. doi: 10.1111/j.1535-7511.2006.00106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velíšková J, Velíšek L. β-Estradiol increases dentate gyrus inhibition in female rats via augmentation of hilar neuropeptide Y. J Neurosci. 2007;27:6054–6063. doi: 10.1523/JNEUROSCI.0366-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velíšková J, Liptáková S, Hussain S. The effects of N-methyl-d-aspartate antagonist 2-amino-7-phosphonoheptanoic acid microinfusions into the adult male rat substantia nigra pars reticulata are site-specific. Neurosci Lett. 2001;316:108–110. doi: 10.1016/s0304-3940(01)02379-5. [DOI] [PubMed] [Google Scholar]
- Velíšková J, Miller AM, Nunes ML, Brown LL. Regional neural activity within the substantia nigra during peri-ictal flurothyl generalized seizure stages. Neurobiol Dis. 2005;20:752–759. doi: 10.1016/j.nbd.2005.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velíšková J, Chudomel O, Poon KL, Marshall B, Velíšek L. The involvement of the substantia nigra pars reticulata in hypoglycemic seizures. Epilepsia. 2007;48(Suppl 5):106–108. doi: 10.1111/j.1528-1167.2007.01296.x. [DOI] [PubMed] [Google Scholar]
- Wu X, Gao J, Yan J, Owyang C, Li Y. Hypothalamus-brain stem circuitry responsible for vagal efferent signaling to the pancreas evoked by hypoglycemia in rat. J Neurophysiol. 2004;91:1734–1747. doi: 10.1152/jn.00791.2003. [DOI] [PubMed] [Google Scholar]
- Wurpel JN, Dundore RL, Bryan RM, Jr, Keil LC, Severs WB. Regional cerebral glucose utilization during vasopressin-induced barrel rotations or bicuculline-induced seizures in rats. Pharmacology. 1988;36:1–8. doi: 10.1159/000138340. [DOI] [PubMed] [Google Scholar]
- Xia Y, Haddad GG. Major differences in CNS sulfonylurea receptor distribution between the rat (newborn, adult) and turtle. J Comp Neurol. 1991;314:278–289. doi: 10.1002/cne.903140206. [DOI] [PubMed] [Google Scholar]
- Xia Y, Eisenman D, Haddad GG. Sulfonylurea receptor expression in rat brain: effect of chronic hypoxia during development. Pediatr Res. 1993;34:634–641. doi: 10.1203/00006450-199311000-00015. [DOI] [PubMed] [Google Scholar]
- Yamada K, Inagaki N. Neuroprotection by KATP channels. J Mol Cell Cardiol. 2005;38:945–949. doi: 10.1016/j.yjmcc.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Yamada K, Ji JJ, Yuan H, Miki T, Sato S, Horimoto N, Shimizu T, Seino S, Inagaki N. Protective role of ATP-sensitive potassium channels in hypoxia-induced generalized seizure. Science. 2001;292:1543–1546. doi: 10.1126/science.1059829. [DOI] [PubMed] [Google Scholar]
- Yuan H, Yamada K, Inagaki N. Glucose sensitivity in mouse substantia nigra pars reticulata neurons in vitro. Neurosci Lett. 2004;355:173–176. doi: 10.1016/j.neulet.2003.10.068. [DOI] [PubMed] [Google Scholar]
- Zingman LV, Hodgson DM, Bast PH, Kane GC, Perez-Terzic C, Gumina RJ, Pucar D, Bienengraeber M, Dzeja PP, Miki T, Seino S, Alekseev AE, Terzic A. Kir6.2 is required for adaptation to stress. Proc Natl Acad Sci U S A. 2002;99:13278–13283. doi: 10.1073/pnas.212315199. [DOI] [PMC free article] [PubMed] [Google Scholar]