

Abstract
ASAP1 is an Arf GTPase-activating protein (GAP) that functions on membrane surfaces to catalyze the hydrolysis of GTP bound to Arf. ASAP1 contains a tandem of BAR, pleckstrin homology (PH), and Arf GAP domains and contributes to the formation of invadopodia and podosomes. The PH domain interacts with the catalytic domain influencing both the catalytic and Michaelis constants. Tandem BAR-PH domains have been found to fold into a functional unit. The results of sedimentation velocity studies were consistent with predictions from homology models in which the BAR and PH domains of ASAP1 fold together. We set out to test the hypothesis that the BAR domain of ASAP1 affects GAP activity by interacting with the PH and/or Arf GAP domains. Recombinant proteins composed of the BAR, PH, Arf GAP, and Ankyrin repeat domains (called BAR-PZA) and the PH, Arf GAP, and Ankyrin repeat domains (PZA) were compared. Catalytic power for the two proteins was determined using large unilamellar vesicles as a reaction surface. The catalytic power of PZA was greater than that of BAR-PZA. The effect of the BAR domain was dependent on the N-terminal loop of the BAR domain and was not the consequence of differential membrane association or changes in large unilamellar vesicle curvature. The Km for BAR-PZA was greater and the kcat was smaller than for PZA determined by saturation kinetics. Analysis of single turnover kinetics revealed a transition state intermediate that was affected by the BAR domain. We conclude that BAR domains can affect enzymatic activity through intraprotein interactions.
The Bin, amphiphysin, RSV161/167 (BAR)2 domain is a recently identified structural element in proteins that regulate membrane trafficking (1–7). The BAR superfamily comprises three subfamilies: F-BAR, I-BAR, and BAR. The BAR group can be further subdivided into BAR, N-BAR, PX-BAR, and BAR-pleckstrin homology (PH). The BAR group domains consist of three bundled α-helices that homodimerize to form a banana-shaped structure. The inner curved face can bind preferentially to surfaces with similar curvatures. As a consequence, BAR domains can function as membrane curvature sensors or as inducers of membrane curvature. BAR domains also bind to proteins (8, 9). Several proteins contain a BAR domain immediately N-terminal to a PH domain, which also mediates regulated membrane association (10–13). In the protein APPL1 (9), the BAR-PH domains fold together forming a binding site for the small GTP-binding protein Rab5. Arf GTPase-activating proteins (GAPs) are regulators of Arf family GTP-binding proteins (14–18). Two subtypes of Arf GAPs have N-terminal BAR and PH domains similar to that found in APPL1.
Thirty-one genes encode Arf GAPs in humans (16–18). Each member of the family has an Arf GAP domain that catalyzes the hydrolysis of GTP bound to Arf family GTP-binding proteins. The Arf GAPs are otherwise structurally diverse. ASAP1 is an Arf GAP that affects membrane traffic and actin remodeling involved in cell movement and has been implicated in oncogenesis (19–22). ASAP1 contains, from the N terminus, BAR, PH, Arf GAP, Ankyrin repeat, proline-rich, and SH3 domains.
ASAP1 contains a BAR domain immediately N-terminal to a PH domain. The PH domain of ASAP1 is functionally integrated with the Arf GAP domain and may form part of the substrate binding pocket (23, 24). The PH domain binds specifically to phosphatidylinositol 4,5-bisphosphate (PIP2), a constituent of the membrane, leading to stimulation of GAP activity by a mechanism that is, in part, independent of recruitment to membranes (23, 25). The BAR domain of ASAP1 is critical for in vivo function of ASAP1, but the molecular functions of the BAR domain of ASAP1 have not been extensively characterized. Hypotheses related to membrane curvature have been examined. Recombinant ASAP1 can induce the formation of tubules from large unilamellar vesicles, which may be related to a function of ASAP1 in membrane traffic. The BAR domain might also regulate GAP activity of ASAP1. We have considered two mechanisms based on the known properties of BAR domains. First the BAR domain could regulate association of ASAP1 with membrane surfaces containing the substrate Arf1·GTP. The BAR domain could also affect GAP activity through an intramolecular association. In one BAR-PH protein that has been crystallized (APPL1), the two domains fold together to form a protein binding site (9). In ASAP1, the PH domain is functionally integrated with the GAP domain, raising the possibility that the BAR domain affects GAP activity by folding with the PH domain.
Here we compared the kinetics of recombinant proteins composed of the PH, Arf GAP, and Ankyrin repeat (PZA)3 or BAR, PH, Arf GAP, and Ankyrin repeat (BAR-PZA) domains of ASAP1 to test the hypothesis that the BAR domain affects enzymatic activity. We found kinetic differences between the proteins that could not be explained by membrane association properties. The results were consistent with a model in which the BAR domain affects transition of ASAP1 through its catalytic cycle.
EXPERIMENTAL PROCEDURES
Proteins—Bacterial expression vectors for His10-ASAP1-(325–724) (PZA) (26), ASAP1-(1–724)-His6 (BAR-PZA) (27) and myristoylated Arf1 (myrArf1) (28) have been described before. PZA and myrArf1 were expressed and purified from bacteria as described previously (24, 26, 28). Recombinant BAR-PZA was purified using a procedure modified from a previous report (27). After separating proteins with a HisTrap column (GE Healthcare), the fractions containing BAR-PZA were pooled and diluted 5-fold with 20 mm Tris to reduce the NaCl concentration to 100 mm. The diluted pool was then applied to a hydroxylapatite column that had been equilibrated with 50 mm potassium Pi, pH 7.0, 100 mm NaCl. The column was washed with 100 mm potassium Pi, pH 7.0, 100 mm NaCl. The protein was eluted with 500 mm potassium Pi, 100 mm NaCl. Protein concentrations were estimated using the Bio-Rad assay.
Preparation of Large Unilamellar Vesicles (LUVs)—All lipids were obtained from Avanti Polar Lipids. LUVs were prepared by extrusion (27). In lipid binding assays, we used LUVs containing molar ratios of either 40% phosphatidylcholine (PC), 25% phosphatidylethanolamine (PE), 15% phosphatidylserine (PS), 9.5% phosphatidylinositol (PI), 0.5% PIP2, and 10% cholesterol (+PS LUV) or 55% PC, 25% PE, 9.5% PI, 0.5% PIP2, and 10% cholesterol (-PS LUV). In fixed time point (3-min) GAP assays, we used LUVs containing 55% egg PC or 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), 20% PE, 15% PS, 7.5% PI, and 2.5% PIP2. The lipid mixtures were extruded through 1- or 0.1-μm pore filters. Single turnover and steady state kinetics were performed using LUVs containing 55% DSPC, 20% PE, 15% PS, 7.5% PI, and 2.5% PIP2. The lipid mixtures were extruded through 0.1-μm pore filters.
GAP Assays and Kinetic Analysis—The conversion of Arf1·GTP to Arf1·GDP was followed in one of two ways (26). In fixed time point assays and in single turnover kinetics, [α-32P]GTP bound to Arf1 was used as a substrate, and the conversion of protein bound [α-32P]GTP to [α-32P]GDP was measured. To determine initial rates for saturation kinetics, tryptophan fluorescence of Arf1 was monitored. Arf1·GTP has a greater quantum yield than does Arf1·GDP.
Fixed time point assays were used to determine the amount of ASAP1 required to hydrolyze 50% of the Arf bound GTP in 3 min, which we refer to as the C50 (26, 29). Either PZA or BAR-PZA was titrated into the reaction containing myrArf1·GTP as the substrate. Reactions were stopped after 3 min by adding 2 ml of ice-cold 20 mm Tris, pH 8.0, 100 mm NaCl, 10 mm MgCl2, and 1 mm dithiothreitol. Protein-bound nucleotide was trapped on nitrocellulose, eluted with formic acid, and separated by thin layer chromatography on polyethyleneimine-cellulose. In these experiments, only protein-bound GTP was analyzed.
Steady state kinetics was performed with a FluorMax3 spectrophotometer (Jobin Yvon Horiba, Edison, NJ) (24, 26) using 0.03 nm PZA or 0.5 nm BAR-PZA as enzyme and 0.25–15 μm myrArf1·GTP as substrate. The reaction contained 25 mm Hepes, pH 7.5, 100 mm NaCl, 1.5 mm MgCl2, 1 mm EDTA, 1 mm dithiothreitol, 100 μm GTP, and 500 μm LUVs. The excitation wavelength was 297 nm, and the emission wavelength was 340 nm. Fluorescence was recorded every 10 s. Initial rates were determined from the progress curves.
The single turnover assay and other rapid kinetic analyses were performed using an RQF-3 rapid chemical quench flow instrument (KinTek, Austin, TX) (24). MyrArf1 (3 μm) incubated with 25 mm Hepes, pH 7.5, 100 mm NaCl, 0.5 mm MgCl2, 1 mm EDTA, 1 mm ATP, 1 mm dithiothreitol, 0.125 nm [α-32P]GTP, and 500 μm LUVs for 60 min was used as the substrate. For experiments in which substrate was in excess of enzyme, 10 μm myrArf1 was incubated with 20 μm GTP to form Arf1·GTP. PZA (0.47–3.76 μm final concentration) or BAR-PZA (0.27–8.7 μm final concentration) with 500 μm LUVs was rapidly mixed with Arf1·GTP at 30 °C. The reaction was quenched with 3 m formic acid after 2–250 ms. GTP and GDP in the quenched reaction were separated by thin layer chromatography. In these experiments, unlike that used to determine C50, the protein-bound GTP was not separated from the free GTP.
Circular Dichroism (CD) Spectral Analysis—CD spectra were collected using a Jasco J720 CD spectropolarimeter for ASAP1 proteins. When collecting CD spectra of PZA, PH, ZA, and PH plus ZA, each protein was diluted to 2 μm in 20 mm sodium phosphate buffer, pH 7.0. When collecting CD spectra of BAR-PZA, BAR-PH, ZA, and BAR-PH plus ZA, each protein was diluted to 4 μm in PBS (137 mm NaCl, 2.7 mm KCl, 4.3 mm Na2HPO4, 1.47 mm KH2PO4, pH 7.4) to prevent the BAR-PZA from precipitating out of the solution.
Dynamic Light Scattering—Light scattering data were acquired at 90° at a wavelength of 633 nm and correlated with a BI-9000 AT autocorrelator (Brookhaven Instruments, Holtsville, NY). Data analysis was performed by modeling with particle size distributions with the maximum entropy method (30) as implemented in the software SEDFIT (31). The decay time is a measure of both diffusion (size-dependent) as well as rotation and other mechanical fluctuations. For particles larger than the wavelength of laser light used, multiple scattering events further complicate the quantitative interpretation toward Stokes radii. This is why we do not quantify the radii, but from the similarity of the autocorrelation functions we can conclude that the size-dependent translational diffusion and other mechanical movements are very similar.
Analytical Ultracentrifugation Sedimentation Velocity— Stock samples of BAR-PZA (6.7 μm), BAR-PH (14.1 μm), PZA (22.4 μm), and ZA (31.8 μm), purified by size exclusion with Sephacryl columns (GE Healthcare) in 200 mm K2PO4, pH 7.0, were used to prepare experimental samples at several different loading concentrations by serial dilution using the respective size exclusion chromatography running column buffer. The density (1.0238 g/ml) and viscosity (1.0759 centipoises) of the buffer were determined using a DMA5000 Density Meter (Anton Paar, Graz, Austria) and an AMVn Automated Micro Viscometer (Anton Paar), respectively. The protein partial specific volumes were determined to be 0.7334 ml/g (BAR-PZA), 0.7330 ml/g (BAR-PH), 0.7331 ml/g (PZA), and 0.7338 ml/g (ZA) from the amino acid sequence using the software SEDNTERP. 400-μl samples were loaded into 12-mm path length double sector cells and were centrifuged at 50,000 rpm at 20 °C in a ProteomeLab-XL-I analytical ultracentrifuge (Beckman Coulter, Palo Alto, CA) following the standard protocol. The evolution of the resulting concentration gradient was monitored by both the absorbance optics (280 nm) and the interference detection system.
Sedimentation coefficients for the protein constructs were obtained from the analysis of the sedimentation velocity data using the software SEDFIT. For this analysis, an average of 30 scans were loaded and modeled with a continuous c(s) distribution using a resolution of 200 s-values between 0.2 to 8.0 S and maximum entropy regularization (p = 0.68) (31). From the superposition of c(s) distributions obtained at different loading concentrations, the absence of concentration-dependent interactions was verified, and the ideal sedimentation was confirmed by obtaining molar mass estimates from c(s) close to the theoretical molar masses of the respective protein species. The s-value of the protein species was determined as the weight average sedimentation coefficient of the peak. The sequence molar mass and the measured sedimentation coefficient were utilized as input parameters in the program SEDNTERP to calculate values for the respective protein frictional ratio. Axial dimensions were calculated using a cylinder model assuming a hydration of 0.3 g/g. For calculating the hydrodynamic properties of the homology models, Protein Data Bank files were loaded into the modeling software Hydropro (version 7) (32).
Homology Modeling—The homology model of the ASAP1-(1–431) BAR-PH construct was generated using the APPL1 BAR/PH x-ray structure (Protein Data Bank code 2Q13) (9) with the Prime program (Schrödinger Inc., New York, NY) with N and C termini sequences (including the His tag sequence) beyond the x-ray structure generated in random coil conformations. The model was first generated with Prime as a monomer, superposed on the x-ray structure dimer, and minimized holding non-insert, non-termini backbone atoms fixed using the MacroModel program (Schrödinger Inc.).
The model for ASAP1-(1–724) BAR-PZA construct was generated by superposing PH domain residues 340–430 of the representative structure from the ensemble of PZA structures from Luo et al. (33) onto the PH domain of the ASAP1-(1–431) BAR-PH homology model monomer. Residues beyond position 310 for BAR-PH and before position 330 for PZA were removed, and the BAR-PH linker was regenerated using Prime as well as the C terminus His tag of the BAR-PZA construct. The result was superposed using the BAR-PH model as template to form the BAR-PZA dimer, and the structure was minimized using MacroModel with the non-linker, non-termini backbone atoms fixed. Secondary structure consensus predictions were performed using NPS@ (34).
To provide hypothetical sedimentation coefficients for the BAR-PH and BAR-PZA in conformations with no interdomain contacts, additional model structures were generated. The BAR, PH, and ZA linkers were first deleted, and the domains were displaced to a 10-Å minimum separation between domains and rotated to bring the domain termini to within 15-Å separation using Maestro (Schrödinger Inc.). The linker regions were then rebuilt using Prime.
Electron Microscopy—For negative stained images, a carbon-coated electron microscopy grid was placed on a 10-μl sample drop for 2 min, blotted with filter paper, chemically stained with 2% uranyl acetate for 2 min, blotted again, and air-dried.
Negative stained specimens were examined in a transmission electron microscope (Philips CM120, FEI Co.) operated at 100 kV. Images were recorded digitally on a 794 Gatan MultiScan charge-coupled device camera with the DigitalMicrograph software package (35).
RESULTS
Characterization of the Structural Properties of ASAP1
The experiments described here use recombinant proteins derived from ASAP1 that are represented schematically in Fig. 1A. The nomenclature we use for the recombinant proteins refers to the domain structure: the protein composed of the BAR and PH domains is called BAR-PH, the protein composed of the BAR, PH, Arf GAP, and Ank repeat domains is called BAR-PZA, the protein composed of the PH, Arf GAP, and Ank repeat domains is called PZA, and the protein composed of the Arf GAP and Ank repeat domains is called ZA.
FIGURE 1.

Structure of ASAP1. A, schematic of proteins used in the experiments. The recombinant proteins are schematized with amino acid coordinates indicated. PH, PZA, and ZA have N-terminal His tags. BAR-PZA and BAR-PH have a C-terminal His tag. B, homology models of BAR-PH and BAR-PZA. Shown are backbone ribbon diagrams of the BAR-PH model (top) and BAR-PZA (bottom) shaded from red at the N terminus and violet at the C terminus of each chain of the dimer. The red arrow in the BAR-PH model indicates for the chain on the right the putative region of contact between the BAR N-terminal extension and PH domain loop that forms part of the phosphoinositide binding site. C, analytical ultracentrifugation studies of BAR-PZA, BAR-PH, PZA, and ZA domains. Shown are the sedimentation coefficient distributions c(Pδ)(s) for the indicated protein domains that were obtained from the Bayesian analysis of sedimentation velocity data (42). The integrated weight average sedimentation coefficients, corrected for buffer density and viscosity (s20,w) and the sequence molar mass, were used to calculate the respective protein frictional ratio utilized in hydrodynamic shape modeling (Table 1).
Homology Modeling of ASAP1—The model of BAR-PH constructed using the x-ray structure of the homologous APPL1 (Fig. 1B) shows contacts between the BAR and PH domains in three regions. The loop between helices 2 and 3 of the BAR domain (ASAP1 residues 182–206) from one chain of the dimer packs against the PH domain of the other chain between β strands 5 and 6 (around residue 396) and the N-terminal end of the C-terminal helix (around residue 415). The linker region between the BAR and PH domains (residues 300–330) is 21 residues longer than the corresponding linker in the APPL1 structure. Although homology modeling does provide for insertions, the predicted structure for the additional linker residues is necessarily more speculative. A consensus of secondary structure prediction algorithms (34) predicts the additional linker residues to be in random coil conformation consistent with the homology model.
The third contact between the BAR and PH domains occurs between the BAR N-terminal residues of one chain and the loop between strands 3 and 4 of the PH domain (around residue 375) of the other chain. This loop forms part of the phosphoinositide binding site of the PH domain. ASAP1 contains 26 additional N-terminal residues compared with APPL1. As with the insertion in the BAR-PH linker, the additional residues of the N terminus are represented by random coil in the model. Secondary structure prediction algorithms predict an α helix not present in APPL1 that includes part of the additional N-terminal residues (ASAP1 residues 15–32). This putative helix would be located such that it could pack against part of the BAR domain, or it could interact with the PH domain in or near its phosphoinositide binding site.
The Arf GAP and Ank repeat (ZA) domain portion of the ASAP1-(1–724) BAR-PZA construct was modeled by taking the member nearest the average of the ensemble of PZA structures previously generated based on NMR, analytical ultracentrifugation, chemical protection, and mutagenesis data (33) and superposing the PH domain onto the PH domain of the BAR-PH homology model. The resulting model shows no contact between residues of the BAR domain analogous to APPL1 and the Arf GAP/Ank repeat domains. However, contact between the Arf GAP and Ank repeat domains and the additional N-terminal extension of the BAR domain could possibly occur if the additional residues adopt a relatively extended conformation. The linker between BAR and PH domains lies near the PH-ZA linker, and contacts between the additional BAR-PH linker residues and PH-ZA linker could also occur. The NMR data on the PZA construct indicated that the PH and ZA domains interact dynamically in an ensemble of contacts. If a similar dynamic interaction occurs in the BAR-PZA construct, then for some members of the ensemble, then possible N terminus to ZA domain or BAR-PH to PH-ZA linker contacts could be more likely.
Limited Proteolysis—We examined the structural relationship between the BAR, PH, and Arf GAP domains using limited proteolysis of BAR-PH, BAR-PZA, and PZA. Trypsin was titrated into a fixed time reaction, and the proteolytic products were examined by gel electrophoresis (Fig. 2A). The most sensitive cleavage site, affected by 1 ng of trypsin in a 25-μl reaction under these conditions, was between the BAR and PH domains consistent with a flexible linker between the two domains that was predicted from the homology modeling. The BAR fragment had additional cleavage sites apparent with 2–10 ng of trypsin. The fragment containing the PH, Arf GAP, and Ank repeat domains was less sensitive. Cleavage of the fragment generated from BAR-PZA or of recombinant PZA was observed with 50 ng of trypsin and occurred between the PH and Arf GAP domains (Fig. 2A and Ref. 25) consistent with the linker between the domains interacting with the Arf GAP domain as described in NMR studies (33).
FIGURE 2.

Structural analysis of ASAP1. A, limited proteolysis of ASAP1 reveals flexible linker between the BAR and PH domains. Panel a, BAR-PZA. Panel b, BAR-PH. Panel c, PZA. 4 μm of the indicated proteins was incubated with the indicated mass of trypsin for 10 min at 30 °C in 20 mm Hepes, pH 7.4, 100 mm NaCl, 2 mm MgCl2, 1 mm GTP. The reaction was stopped with 200 ng of soybean trypsin inhibitor, and products were analyzed by electrophoresis using a 10–20% polyacrylamide gel. Panel d, PH. The experiment performed as in panels a, b, and c but an 18% polyacrylamide gel to visualize the smaller fragments generated from the PH domain. The cleavage sites are indicated by arrows in the schematics. B, CD spectra of ASAP1 recombinant proteins. The spectra of the indicated proteins alone and in combination are shown. The spectra of the larger recombinant proteins were, in each case, the sum of the spectra of the small recombinant proteins, indicating that the domains independently folded. Panel a, PZA, PH, and ZA. Panel b, BAR-PZA, BAR-PH, and ZA. mdeg, millidegrees.
The protease sensitivities of the isolated PH domain and BAR-PH were also compared. We found that the isolated PH domain was relatively insensitive compared with the linker between the PH and BAR domain (Fig. 2A).
Hydrodynamics of ASAP1—To further examine the relationship among the BAR, PH, and Arf GAP domains, the sedimentation and frictional coefficients of BAR-PZA, BAR-PH, PZA, and ZA were determined in sedimentation velocity experiments (Fig. 1C and Table 1). Using the frictional coefficients, dimensions of the proteins, assuming a cylindrical shape, were determined. The experiments revealed that BAR-PH and BAR-PZA were exclusively dimers at concentrations of 1 μm. If the domains were a simple linear combination, the size of BAR-PZA should be a simple addition of BAR-PH (which is a dimer) and two ZAs (which are monomers) (see Fig. 1A). The dimensions were not additive whether modeled as a cylinder (Table 1) or an ellipse (not shown) consistent with a model in which the ZA domains are partially folded back onto the BAR domain.
TABLE 1.
Hydrodynamic properties of recombinant ASAP1 determined by sedimentation velocity experiments BAR-PZA and BAR-PH are the homology models shown in the Fig. 1A. BAR_PZA assumes a bead-on-a-string arrangement for the BAR and PZA segments of ASAP1. BAR_PH assumes a bead-on-string arrangement for the BAR and PH domains. BARP_ZA assumes that the BAR and PH domains form a bead and that ZA forms a second bead. BAR_P_ZA assumes that BAR, PH, and ZA are each beads on the string.
| Protein | swa | s20,wb | shc | f/fod | Axial dimensionse |
|---|---|---|---|---|---|
| BAR-PZAf | 5.1 | 5.9 | 6.6 | 1.82 | 32.8 × 3.3 |
| BAR_PZA | 5.9 | ||||
| BARP_ZA | 5.9 | ||||
| BAR_P_ZA | 5.3 | ||||
| BAR-PHf | 3.9 | 4.5 | 4.8 | 1.73 | 24.9 × 3.0 |
| BAR_PH | 4.2 | ||||
| PZA | 2.8 | 3.2 | 3.6 | 1.44 | 11.9 × 2.9 |
| ZA | 2.4 | 2.7 | 2.9 | 1.36 | 8.8 × 2.9 |
Weight average sedimentation coefficient at experimental conditions
Corrected to standard conditions (water, 20 °C)
Sedimentation coefficient determined for homology model using Hydropro7
Frictional ratio obtained with s20,w and molar mass using SEDNTERP
Axial dimensions (nm) of cylinder model (length × diameter) assuming hydration level of 0.3 g of water/gram of protein
These domains were present exclusively as dimers in solution above 0.1 mg/ml (1 μm)
To test the homology models further, we compared the determined sedimentation coefficients with theoretical sedimentation coefficients for the models presented in Fig. 1B as well as models assuming bead-on-string configurations for the domains (32) (Table 1). For both BAR-PZA and BAR-PH, the determined sedimentation coefficients were greater than the coefficient calculated for the completely bead-on-string models, indicated as BAR_P_ZA and BAR_PH in Table 1. The determined sedimentation coefficients were smaller than those calculated for the homology model presented in Fig. 1B; however, the determined values were also smaller than those calculated for ZA whose structural model was based on the crystal of the highly related Arf GAP ASAP2, which is not extended. Based on these results, we conclude that the BAR, PH, and Arf GAP domains fold back on each other. The domains may be mobile relative to one another, as described for PZA (33), which could account for differences between calculated and determined sedimentation coefficients.
Spectral Analysis of BAR-PZA— As one means of determining the effect of deletion of domains on the structure of ASAP1, CD spectra of BAR-PZA, BAR-PH, PZA, PH, and ZA were determined (Fig. 2B). In Fig. 2B, panel a, the spectra for PZA, PH, and ZA are shown. PZA and ZA have a peak at 208, and the spectrum of PZA was similar to the spectrum of PH and ZA combined. The spectrum of BAR-PZA is similar to the combined spectra of BAR-PH and ZA (Fig. 2B, panel b). The results indicate that the secondary structure of each domain is independent of the presence of the other domains. We could not examine BAR as an isolated protein because it was not stable without the PH domain, which is also consistent with the conclusion that the domains interact (27).
Contribution of the BAR Domain to Membrane Association
The substrate for ASAP1 is Arf1·GTP, which is restricted to membrane surfaces. Recruitment of ASAP1, at least in part, controls the GAP reaction. BAR domains are membrane association domains. We determined the effect of the BAR domain in ASAP1 on membrane association by comparing PZA and BAR-PZA (Fig. 3A). In these experiments, BAR-PZA and PZA were incubated with sucrose-filled LUVs of two diameters formed by extrusion through membranes with pores 1 or 0.1 μm in diameter (Fig. 3A). The LUVs were of two phospholipid compositions: one containing 15% PS and one without PS, both containing 0.5% PIP2. The extent of association of BAR-PZA and PZA was greater with LUVs containing PS than those without PS and was independent of vesicle diameter. BAR-PZA associated to a greater extent than PZA under every condition. We also examined the association of BAR-PZA and PZA to LUVs containing the saturated lipid DSPC and 2.5% PIP2.76 ± 4% of the BAR-PZA in the assay bound to the LUVs, whereas 64 ± 2% of PZA bound. We conclude that the BAR domain is a membrane association domain that contributes to recruiting ASAP1 to lipid bilayers.
FIGURE 3.

Membrane binding of ASAP1. A, contribution of the BAR domain to membrane association. BAR-PZA (0.7 μm) and PZA (1.2 μm) were incubated with 500 μm sucrose-loaded LUVs composed of either 40% PC, 25% PE, 15% PS, 9.5% PI, 0.5% PIP2, and 10% cholesterol (+PS) or 55% PC, 25% PE, 9.5% PI, 0.5% PIP2, and 10% cholesterol (-PS) and formed by extrusion through either a 1- or 0.1-μm pore filter. Vesicles were precipitated by centrifugation, and associated proteins were separated by SDS-PAGE. The amount of precipitated protein was determined by densitometry of the Coomassie Blue-stained gels with standards on each gel. Panel a, representative experiment. The raw data are shown from one experiment representative of three. The first five lanes are standards added to the gel to quantify the amount of protein that was precipitated with the vesicles. The numbers are the percentage of the total protein used for the binding experiment. Panel b, summary of three experiments. Averages and S.E. are presented in the figure. B, contribution of the PH domain/phosphatidylinositol 4,5-bisphosphate association for binding to LUVs. Panel a, comparison of PIP2-dependent binding of BAR-PZA and PZA. The experiment was performed as described in A but with +PS LUVs containing the indicated concentrations of PIP2. Data are the average and S.E. from three experiments. Panel b, effect of mutation of the PH domain on PIP2-dependent binding to PZA to LUVs. The experiment was performed as described in A but using the indicated recombinant ASAP1. Data are the average and S.E. of three experiments. Panel c, effect of mutation of the PH domain on PIP2-dependent binding of the BAR-PZA to LUVs. The data are the average and S.E. of three experiments. WT, wild type.
Both BAR and PH domains contributed to binding to LUVs (25, 27). To separate the contribution of each, we compared PIP2-dependent vesicle association of BAR-PZA and PZA (Fig. 3B). In the absence of PIP2, BAR-PZA bound to vesicles to a greater extent than did PZA. Binding of both proteins was increased by PIP2. BAR-PZA was more sensitive with a half-maximal effect of PIP2 at 0.2% in the LUVs in contrast to 0.8% with PZA. Mutations in the PH domain that affected PIP2 binding of PZA (23, 25) (Fig. 3B, panels b and c) also affected binding of BAR-PZA.
Effect of the BAR Domain on Enzymatic Power
The BAR domain affected membrane association of ASAP1. If the regulatory function of the BAR domain is recruitment, BAR-PZA should have greater enzymatic power than PZA consequent to a smaller Km. To test this prediction, we determined the amount of BAR-PZA and PZA required to hydrolyze 50% of the GTP on myrArf1 in a fixed time. We call the concentration of GAP for 50% hydrolysis the C50. It is inversely proportional to enzymatic power. In these experiments, we used LUVs of two diameters. The phospholipid composition was chosen to maximize binding, including 2.5% PIP2. For some LUVs, we changed the content of saturated lipid using distearoyl-PC in place of the lipid from egg (Fig. 4A). Contrary to the expectation based on recruitment, BAR-PZA had a greater C50 and, therefore, less enzymatic power than PZA under all conditions that we examined (Table 2). BAR-PZA was relatively insensitive to the size and composition of the vesicles. PZA was about twice as active on LUVs containing DSPC than on LUVs containing egg PC. We determined whether another BAR domain containing Arf GAP, ASAP3, was similar (Fig. 4B). ASAP3 uses Arf5 as a preferred substrate. We found that [ΔBAR]ASAP3 had 30-fold more enzymatic power than full-length ASAP3.
FIGURE 4.

Effect of BAR domain on activity of ASAP-type Arf GAPs. A, ASAP1. Panel a, PZA of ASAP1 was titrated into a reaction containing myrArf1-GTP and LUVs of two compositions with either DSPC or egg PC (total phospholipid concentration of 500 μm) extruded through two pore sizes, 1 or 0.1 μm. Panel b, an experiment similar to that described for A but with BAR-PZA as enzyme. Experiments were performed three times, and representative results are shown. B, ASAP3. ASAP3 or [ΔBAR]ASAP3 were titrated into a reaction containing LUVs composed of 55% DSPC, 20% PE, 15% PS, 7.5% PI, and 2.5% PIP2 and extruded through 0.1-μm pores. myrArf5 was the substrate. The plotted data are from a representative experiment. The C50 value presented is the mean ± S.E. from four experiments.
TABLE 2.
Effect of vesicle size and composition on the C50 values of PZA and BAR-PZA C50 values were determined as described previously (26). DSPC indicates LUVs containing 55% DSPC, 20% PE, 15% PS, 7.5% PI, and 2.5% PIP2. Egg PC indicates LUVs containing 55% egg PC, 20% PE, 15% PS, 7.5% PI, and 2.5% PIP2. 1 μm indicates LUVs were extruded through a filter with a 1-μm pore; 0.1 μm indicates extrusion through filters with a 0.1-μm pore. The results are the means ± S.E. of three experiments.
|
Enzyme
|
C50
|
||||||
|---|---|---|---|---|---|---|---|
| DSPC, 1 μm | DSPC, 0.1 μm | Egg PC, 1 μm | Egg PC, 0.1 μm | ||||
| nm | |||||||
| PZA | 0.017 ± 0.005 | 0.031 ± 0.004 | 0.076 ± 0.014 | 0.075 ± 0.016 | |||
| BAR-PZA | 1.00 ± 0.37 | 1.24 ± 0.05 | 0.90 ± 0.17 | 0.89 ± 0.09 | |||
These results were opposite of the predictions based on the BAR domain functioning as a curvature sensor and for the model in which curvature induction by the BAR domain stimulated GAP activity. We determined whether either BAR-PZA or PZA affected LUVs formed by extrusion through 0.1-μm pores. LUVs were incubated with 0.5 μm BAR-PZA or PZA for 5 min at 30 °C and then examined by negative stained electron microscopy (Fig. 5A). In contrast to previous work using larger LUVs containing cholesterol and unsaturated lipids (27), there were no BAR-induced changes in the shape of the LUVs used in these kinetic studies that we could detect.
FIGURE 5.

Role of membrane deformation in the effect of the BAR domain on GAP activity. A, effect of BAR-PZA on the geometry of LUVs lacking cholesterol and containing saturated lipids. Negative stained electron microscopy was used to analyze the shape of LUVs containing 55% DSPC, 20% PE, 15% PS, 7.5% PI, and 2.5% PIP2 formed by extrusion through 0.1-μm pores. The LUVs were incubated with either 0.5 μm BAR-PZA or 0.5 μm PZA. B, determination of size of vesicles used in experiments. The autocorrelation function of dynamic light scattering of LUVs is shown. LUVs containing either egg PC or DSPC and prepared by extrusion through a membranes of either 0.1- or 1.0-μm pores were analyzed by dynamic light scattering as described under “Experimental Procedures.” C, effect of BAR-PH on the activity of PZA. BAR-PH was titrated into a reaction containing 0.1 nm PZA, LUVs with 0.5% PIP2, and myrArf1·GTP as a substrate. The relative GAP activity was estimated as the ln(GTP present at time 0/GTP) as described previously (29). BAR-PH would not be predicted to bind to PZA (33).
The size of the LUVs might be affected by their lipid composition, which could affect GAP activity of PZA or BAR-PZA. To examine this possibility, the size distribution of LUVs containing either egg PC or DSPC and extruded through either 0.1- or 1.0-μm pore membranes was determined by dynamic light scattering. As shown in Fig. 5B the autocorrelation functions measured by dynamic light scattering have the same characteristic decay time for LUVs prepared by extrusion through membranes with the same pore size independent of LUV composition. From these data, we concluded that the LUV size was dependent on extrusion pore, not lipid composition.
We also tested the effect of BAR-PH on the activity of PZA. Like BAR-PZA, BAR-PH affects the curvature of LUVs of specific compositions (27). If the BAR domain inhibited activity because it changed the LUVs, then BAR-PH may also be inhibitory. We titrated BAR-PH to a concentration of 100 nm into a reaction mixture containing 0.2 nm PZA and found no effect on activity (Fig. 5C). We were not able to perform similar experiments with the isolated BAR domain because it is not stable (27). We conclude that the differences in activity of PZA that we observed were a consequence of vesicle composition, not curvature, and that the effect of the BAR domain on enzymatic activity was not consequent to inducing changes in the curvature of the lipids.
Effect of BAR Domain on kcat and Km
The kcat and Km for PZA and BAR-PZA were determined using LUVs containing DSPC and extruded through filters with 0.1-μm pores. PZA was examined first (Fig. 6). MyrArf1·GTP was titrated into the reaction, and initial rates were determined (Fig. 6A and Table 3). The kcat was 142.3 ± 5.7/s (as calculated from the Vmax), and the Km was 0.76 ± 0.13 μm. Single turnover kinetics was used as a second means to determine the kcat (Fig. 6, B and C, and Table 3). In single turnover experiments, kcat is measured directly, and the estimate is not affected if PZA were partly inactive. In these experiments, enzyme is titrated into the reaction. Reactions are initiated and terminated using a quench flow instrument. We found that the maximum velocity was achieved with 1.4 μm PZA with no statistically significant difference in reaction rate found with 1.4, 2.35, or 3.29 μm PZA (Fig. 6, B and C). If PZA were partly inactive, as a consequence of improper folding, the kcat determined by single turnover experiments would be greater than the kcat determined by saturation kinetics. Instead the kcat determined in single turnover experiments, 45.4 ± 4.5/s, was less than that determined in saturation kinetics (Table 3). A kinetic scheme consistent with this observation is presented in Fig. 7A. In the kinetic model, the enzyme undergoes a transition state intermediate that slowly returns to the ground state and that is able to bind substrate. If the conversion of E*S to E*P is much faster than the conversion of ES to E*S, the transition from E* to E is relatively slow, and E* can directly bind to S, then with S > Km and S > E, the rate is (k3·k4)/(k3 + k4). Under single turnover conditions, under the same assumptions, the observed rate would approach k2.
FIGURE 6.

Kinetic analysis of ASAP1-catalyzed hydrolysis of GTP bound to myrArf1. A and D, saturation kinetics. GAP activity for PZA or BAR-PZA was determined under conditions satisfying the steady state assumption. The conversion of Arf1·GTP to Arf1·GDP was monitored by fluorescence (24, 26). Arf1·GTP has a greater emission than Arf1·GDP; therefore the conversion results in a decrease in fluorescent signal. Reactions contained 0.03 nm PZA and 0.25–6 μm myrArf1·GTP or 0.5 nm BAR-PZA and 1–15 μm myrArf1·GTP. Reactions were initiated by the addition of ASAP1. Initial slopes of the change in fluorescence were determined from the progress curve. The plot of initial rate versus Arf1 concentration was fit to the Michaelis-Menten equation to estimate Vmax and Km. B and E, single turnover kinetic analysis. MyrArf1·GTP was rapidly mixed with the indicated concentrations of PZA or BAR-PZA. Reactions were quenched by rapid mixing with formic acid at the indicated time points. The data were fit to first-order rate equations to determine the observed rate constant, kobs. C and F, replot to determine kcat. The rates obtained from fitting the data in B and E were plotted against the concentrations of PZA or BAR-PZA in the reactions. The kcat was determined by fitting these data to kobs = [PZA]·kcat/([PZA] + Km, app) or kobs = [BAR-PZA]·kcat/([BAR-PZA] + Km, app). Representative experiments of three for each protein are shown.
TABLE 3.
Enzymatic parameters of PZA and BAR-PZA The values of kcat and Km were determined by saturation kinetics (ASAP1 << Arf1·GTP) and by single turnover kinetics (ASAP1 >> Arf1·GTP). The assays contained LUVs composed of 55% DSPC, 20% PE, 15% PS, 7.5% PI, and 2.5% PIP2 and extruded through 0.1-μm pore filters. The results are the mean ± S.E. of three experiments
|
Enzyme
|
Steady state
|
Single turnover
|
||
|---|---|---|---|---|
| kcat | Km | kcat | Km | |
| s–1 | μm | s–1 | μm | |
| PZA | 142.3 ± 5.7 | 0.76 ± 0.13 | 45.4 ± 4.5 | 0.85 ± 0.22 |
| BAR-PZA | 41.8 ± 8.1 | 6.3 ± 0.19 | 53.8 ± 4.9 | 1.18 ± 0.19 |
FIGURE 7.

Presteady kinetic analysis. A, reaction scheme with transition state intermediate that can bind substrate. This reaction scheme was the basis for the equations described under “Appendix.” E = ASAP1; S = Arf1·GTP; P = Arf1·GDP. B, comparison of ASAP1-catalyzed GTP hydrolysis with different substrate/enzyme ratios. Panels a and b, enzyme in excess of substrate. Single turnover kinetic analysis when PZA (1 μm) (panel a) or BAR-PZA (1 μm) (panel b) was in excess of substrate (12 pm) is shown. Panels c and d, substrate in excess of enzyme. An experiment similar to that in panels a and b except Arf·GTP concentration (2 μm) was in excess of PZA (panel c) or BAR-PZA (panel d) (1 μm). The data shown for panels c and d are the mean and S.E. for three experiments. The data shown for panels a and b are representative of four or more experiments.
In the kinetic model, PZA can stay in an activated state (E*) and rebind substrate. The equation describing the kinetic model under steady state conditions has squared substrate terms in the numerator and denominator but is difficult to experimentally distinguish from a simple hyperbola. However, the kinetic model also predicts a lag phase in product formation in presteady state kinetics when substrate is in excess of the enzyme. As predicted, using myrArf1·GTP at a concentration of 2 μm and 1 μm PZA a greater lag in myrArf1·GDP formation was observed than when 12 pm myrArf1·GTP was included in the reaction (Fig. 7B).
Saturation and single turnover kinetics were also determined for BAR-PZA. In saturation experiments, the Km was greater and the kcat was smaller than the parameters for PZA (Fig. 6D and Table 3). The change in Km is the opposite of what would be expected if the BAR domain were functioning to recruit the protein to the surface containing the substrate (36, 37). The kcat measured in single turnover experiments was similar to the kcat in saturation experiments (calculated from the Vmax) and to the kcat for PZA measured in single turnover experiments (Fig. 6, E and F, and Table 3). The concentration of enzyme to reach half-maximal activity was less than the Km measured in saturation experiments. In presteady state kinetics, the lag observed when Arf1·GTP was in excess of PZA was not evident if BAR-PZA were used as the enzyme (Fig. 7).
We considered molecular mechanisms by which the BAR domain could affect the enzymatic properties of ASAP1. Based on the homology structure, the N-terminal extension of the BAR domain contacts the PH and Arf GAP domains and could, consequently, affect GAP activity. As a test of this possibility, three mutant BAR-PZA recombinant proteins with parts of the N-terminal extension deleted were prepared: ASAP1-(6–724), ASAP1-(16–724), and ASAP1-(31–724). The enzymatic efficiency of each was determined by measuring the C50 (Fig. 8). Each protein with part of the loop deleted was more active than BAR-PZA.
FIGURE 8.

Effect of deleting the N-terminal loop of the BAR domain on the enzymatic power of ASAP1. The indicated recombinant ASAP1 proteins were titrated into a reaction mixture containing myrArf1·GTP and LUVs extruded through membranes with 0.1-μm pores and containing DSPC.
DISCUSSION
We examined the effect of the BAR domain of ASAP1 on the catalytic activity of the protein. BAR domains mediate association with membranes (2–4). A plausible hypothesis is that the BAR domain recruits ASAP1 to membranes containing the substrate myrArf1·GTP. BAR domains also bind intramolecularly to PH domains (9). The PH domain of ASAP1 is functionally integral to GAP activity (23). An alternative hypothesis is that the BAR domain may affect GAP activity through an intramolecular association with the PH and/or Arf GAP domains. To test for affects of the BAR domain on catalytic activity and to begin to discriminate between these mechanisms, kinetics of GTP hydrolysis catalyzed by a recombinant protein containing the BAR domain and one without it were compared. The BAR domain affected the kinetic parameters of ASAP1. The BAR domain of another Arf GAP, ASAP3, also affected catalysis. The effect on ASAP1 was dependent on an N-terminal extension of the BAR domain that may associate with the PH and Arf GAP domains. Our results exclude the effect of the BAR domain on enzymatic activity being secondary to recruitment to membranes containing the substrate Arf1·GTP. We propose a model in which the BAR domain controls a transition state intermediate in the catalytic cycle related to an effector function of ASAP1 and consider that BAR domains may have similar function in other Arf GAPs and Rho GAPs.
The most extensively examined functions of BAR domains are as sensors and inducers of membrane curvature (1–7). The BAR domain of ACAP1, an Arf GAP that is structurally related to ASAP1, has been reported to function as a curvature sensor (4). The BAR domain of ASAP1 can bend membranes as well (27), although we did not detect an effect on the curvature of the LUVs of the composition we used for our kinetic studies. In either case, the BAR domain mediates association with membranes, and the hypothesis that the BAR domain could regulate GAP activity by recruiting ASAP1 to membranes containing the substrate Arf1·GTP was reasonable. If the BAR domain affected GAP activity by this mechanism, BAR-PZA would be predicted to have more enzymatic power than PZA, and the BAR domain would be predicted to increase the affinity of ASAP1 for the substrate Arf1·GTP. Instead we found that BAR-PZA had less enzymatic power and a lower affinity for Arf1·GTP than did PZA. ASAP3 was similar in that the deletion of the BAR domain increased enzymatic power. This effect of the BAR domain may extrapolate to other proteins that have BAR domains associated with enzymatic domains (4, 14), such as other Arf GAPs and Rho GAPs.
Although membrane association has been the focus of most work on BAR domains, other functions have been reported. Arfaptin is a BAR domain that binds to Rac1·GDP and to Arf6·GTP (2, 38). The BAR and PH domains of APPL1 fold as a single structure forming a binding site for Rab5·GTP (9). ASAP1 also has a BAR-PH tandem (39). Furthermore the PH domain of ASAP1 is structurally critical for Arf GAP activity (23, 25). These observations led us to consider that the BAR domain might influence GAP activity by binding to the PH domain and/or Arf GAP domain. Homology modeling indicated that there could be an interaction between the N-terminal extension of the BAR domain and the PH and Arf GAP domains. We found that deletion of the entire BAR domain or the N-terminal extension increased GAP activity. Based on these results, we propose a model in which the N-terminal extension of the BAR domain interacts with the PH and Arf GAP domains to affect enzymatic activity. In this model, molecules that bind to the BAR domain could influence GAP activity as we have observed. FIP3, a Rab11- and Arf5/6-binding protein, stimulates the GAP activity of ASAP1 (40).
Our kinetic analysis of recombinant ASAP1 without a BAR domain (PZA) revealed that the kcat measured under conditions satisfying steady state assumptions was greater than the kcat measured in single turnover experiments. These results can be explained by a long lived transition state intermediate (E* in the schematic in Fig. 7), which is stabilized under some conditions, that is able to rebind substrate and catalyze GTP hydrolysis. This model was previously proposed based on the examination of mutant recombinant ASAP1 proteins in which the difference between kcat measured in steady state and single turnover experiments approached 10-fold under conditions in which no difference was observed with wild type protein (24). A difference between kcat determined by saturation and single turnover kinetics was not evident when using BAR-PZA as the enzyme. The BAR domain could affect kinetic parameters by affecting lifetime of and/or substrate binding to the transition state intermediate.
The effect of the BAR domain on catalytic activity could be consistent with a regulatory role of the domain in which a molecule binding to the BAR domain could affect GAP activity by an allosteric mechanism. Alternatively the BAR domain may have an effector role in the function of ASAP1. ASAP1 may undergo a significant conformational change on switching from the ground state of substrate binding to the transition state (E* in Fig. 7) toward catalysis as we first proposed when examining the movement of the C and N termini of a protein composed of the PH, Arf GAP, and Ank repeats of ASAP2 (23). The kinetic data reported here as well as mutational analysis of ASAP1 are consistent with an intermediate that has a distinct conformation. In this model, the position or conformation of the BAR domain would change through the catalytic cycle; this could link the function of the BAR domain with the binding of Arf1·GTP and the hydrolysis of GTP. Five other Arf GAPs, including ASAPs 2 and 3 and ACAPs 1, 2, and 3, and two Rho GAPs have a similar domain structure with N-terminal BAR domains (4, 14). The BAR domain may have a common function among these proteins.
APPENDIX
Steady State Equation—We use the following abbreviations in our derivations: E = Arf GAP; E* = Arf GAP in an activated state; S = Arf1·GTP; P = Arf1·GDP; N1 = first numerator constant, a composite of microconstants; N2 = second numerator constant, a composite of microconstants; D1 = first denominator constant; and D2 = second denominator constant.
We considered the reaction scheme depicted in Fig. 7 and assumed that the enzyme-substrate complexes are at steady state concentrations and that no product is initially present to derive an equation relating initial velocity to substrate concentration. The equation was derived using the King-Altman graphical method (41) and independently using standard linear algebra methods. Each yielded the equation in the form
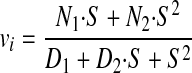 |
(Eq. 1) |
where N1 and N2 have the following values.
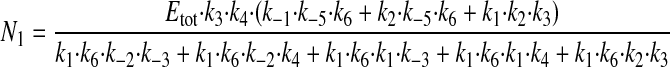 |
(Eq. 2) |
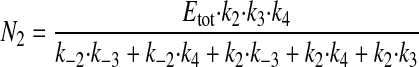 |
(Eq. 3) |
D1 has 16 numerator terms and five denominator terms, each composed of four microconstants. D2 has 21 numerator and five denominator terms, each composed of four microconstants. D1 and D2 incorporate all microconstants. D1 and D2 > 1, and D2 > D1. The predicted substrate dependence does not have significant or detectable sigmoidicity as we observed. At saturating substrate, vi → N2. If considering only the forward reactions, then vi → (k3·k4)/(k3 + k4).
Single Turnover—For single turnover kinetics, at saturating enzyme, the relationship of product to time has the following form.
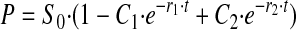 |
(Eq. 4) |
If k2 << k3, the equation approaches
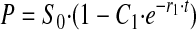 |
(Eq. 5) |
with r1 → k2 + k-2, which reduces to k2 if the back reaction is slow.
This work was supported, in whole or in part, by the intramural program at NCI and by the NIBIB, National Institutes of Health, Department of Health and Human Services. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: BAR, Bin, amphiphysin, RSV161/167; PH, pleckstrin homology; GAP, GTPase-activating protein; LUV, large unilamellar vesicle; Ank, Ankyrin; SH3, Src homology 3; PIP2, phosphatidylinositol 4,5-bisphosphate; myrArf1, myristoylated Arf1; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PS, phosphatidylserine; PI, phosphatidylinositol; DSPC, 1,2-distearoyl-sn-glycero-3-phosphocholine; CD, circular dichroism.
P stands for pleckstrin homology; Z is used to abbreviate Arf GAP domains because they contain a zinc binding motif; A is for ankyrin repeats.
References
- 1.Zhang, B., and Zelhof, A. C. (2002) Traffic 3 452-460 [DOI] [PubMed] [Google Scholar]
- 2.Habermann, B. (2004) EMBO Rep. 5 250-255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gallop, J. L., and McMahon, H. T. (2005) Nature 438 590-596 [DOI] [PubMed] [Google Scholar]
- 4.Peter, B. J., Kent, H. M., Mills, I. G., Vallis, Y., Butler, P. J. G., Evans, P. R., and McMahon, H. T. (2004) Science 303 495-499 [DOI] [PubMed] [Google Scholar]
- 5.Itoh, T., and De Camilli, P. (2006) Biochim. Biophys. Acta 1761 897-912 [DOI] [PubMed] [Google Scholar]
- 6.Lee, M. C. S., and Schekman, R. (2004) Science 303 479-480 [DOI] [PubMed] [Google Scholar]
- 7.Casal, E., Federici, L., Zhang, W., Fernandez-Recio, J., Priego, E. M., Miguel, R. N., DuHadaway, J. B., Prendergast, G. C., Luisi, B. F., and Laue, E. D. (2006) Biochemistry 45 12917-12928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hickson, G. R. X., Matheson, J., Riggs, B., Maier, V. H., Fielding, A. B., Prekeris, R., Sullivan, W., Barr, F. A., and Gould, G. W. (2003) Mol. Biol. Cell 14 2908-2920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu, G. Y., Chen, J., Liu, J., Brunzelle, J. S., Huang, B., Wakeham, N., Terzyan, S., Li, X. M., Rao, Z., Li, G. P., and Zhang, X. J. C. (2007) EMBO J. 26 3484-3493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Itoh, T., and Takenawa, T. (2002) Cell. Signal. 14 733-743 [DOI] [PubMed] [Google Scholar]
- 11.Lemmon, M. A. (2003) Traffic 4 201-213 [DOI] [PubMed] [Google Scholar]
- 12.Wenk, M. R., and De Camilli, P. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 8262-8269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lemmon, M. A., and Ferguson, K. M. (2000) Biochem. J. 350 1-18 [PMC free article] [PubMed] [Google Scholar]
- 14.Inoue, H., and Randazzo, P. A. (2007) Traffic 8 1465-1475 [DOI] [PubMed] [Google Scholar]
- 15.Randazzo, P. A., Inoue, H., and Bharti, S. (2007) Biol. Cell 99 583-600 [DOI] [PubMed] [Google Scholar]
- 16.Nie, Z. Z., and Randazzo, P. A. (2006) J. Cell Sci. 119 1203-1211 [DOI] [PubMed] [Google Scholar]
- 17.Randazzo, P. A., and Hirsch, D. S. (2004) Cell. Signal. 16 401-413 [DOI] [PubMed] [Google Scholar]
- 18.Gillingham, A. K., and Munro, S. (2007) Annu. Rev. Cell Dev. Biol. 23 579-611 [DOI] [PubMed] [Google Scholar]
- 19.Randazzo, P. A., Andrade, J., Miura, K., Brown, M. T., Long, Y. Q., Stauffer, S., Roller, P., and Cooper, J. A. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 4011-4016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bharti, S., Inoue, H., Bharti, K., Hirsch, D. S., Nie, Z., Yoon, H. Y., Artym, V., Yamada, K. M., Mueller, S. C., Barr, V. A., and Randazzo, P. A. (2007) Mol. Cell. Biol. 27 8271-8283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sabe, H., Onodera, Y., Mazaki, Y., and Hashimoto, S. (2006) Curr. Opin. Cell Biol. 18 558-564 [DOI] [PubMed] [Google Scholar]
- 22.Onodera, Y., Hashimoto, S., Hashimoto, A., Morishige, M., Yamada, A., Ogawa, E., Adachi, M., Sakurai, T., Manabe, T., Wada, H., Matsuura, N., and Sabe, H. (2005) EMBO J. 24 963-973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Che, M. M., Boja, E. S., Yoon, H.-Y., Gruschus, J., Jaffe, H., Stauffer, S., Schuck, P., Fales, H. M., and Randazzo, P. A. (2005) Cell. Signal. 17 1276-1288 [DOI] [PubMed] [Google Scholar]
- 24.Luo, R., Ahvazi, B., Amariei, D., Shroder, D., Burrola, B., Losert, W., and Randazzo, P. A. (2007) Biochem. J. 402 439-447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kam, J. L., Miura, K., Jackson, T. R., Gruschus, J., Roller, P., Stauffer, S., Clark, J., Aneja, R., and Randazzo, P. A. (2000) J. Biol. Chem. 275 9653-9663 [DOI] [PubMed] [Google Scholar]
- 26.Che, M. M., Nie, Z. Z., and Randazzo, P. A. (2005) Methods Enzymol. 404 147-163 [DOI] [PubMed] [Google Scholar]
- 27.Nie, Z., Hirsch, D. S., Luo, R., Jian, X., Stauffer, S., Cremesti, A., Andrade, J., Lebowitz, J., Marino, M., Ahvazi, B., Hinshaw, J. E., and Randazzo, P. A. (2006) Curr. Biol. 16 130-139 [DOI] [PubMed] [Google Scholar]
- 28.Ha, V. L., Thomas, G. M. H., Stauffer, S., and Randazzo, P. A. (2005) Method Enzymol. 404 164-174 [DOI] [PubMed] [Google Scholar]
- 29.Randazzo, P. A., Miura, K., and Jackson, T. R. (2001) Method Enzymol. 329 343-354 [DOI] [PubMed] [Google Scholar]
- 30.Livesay, A. K., Licinio, P., and Delaye, M. (1986) J. Chem. Phys. 84 5102-5107 [Google Scholar]
- 31.Schuck, P. (2000) Biophys. J. 78 1606-1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de la Torre, J. G., Huertas, M. L., and Carrasco, B. (2000) Biophys. J. 78 719-730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo, R., Miller Jenkins, L. M., Randazzo, P. A., and Gruschus, J. (2008) Cell. Signal. 20 1968-1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Combet, C., Blanchet, C., Geourjon, C., and Deleage, G. (2000) Trends Biochem. Sci. 25 147-150 [DOI] [PubMed] [Google Scholar]
- 35.Danino, D., Moon, K.-H., and Hinshaw, J. E. (2004) J. Struct. Biol. 147 259-267 [DOI] [PubMed] [Google Scholar]
- 36.Dennis, E. A. (1973) J. Lipid Res. 14 152-159 [PubMed] [Google Scholar]
- 37.Dennis, E. A. (1973) Arch Biochem. Biophys. 158 485-493 [DOI] [PubMed] [Google Scholar]
- 38.Tarricone, C., Xiao, B., Justin, N., Walker, P. A., Rittinger, K., Gamblin, S. J., and Smerdon, S. J. (2001) Nature 411 215-219 [DOI] [PubMed] [Google Scholar]
- 39.Brown, M. T., Andrade, J., Radhakrishna, H., Donaldson, J. G., Cooper, J. A., and Randazzo, P. A. (1998) Mol. Cell. Biol. 18 7038-7051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Inoue, H., Ha, V. L., Prekeris, R., and Randazzo, P. A. (2008) Mol. Biol. Cell 19 4224-4237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.King, E. L., and Altman, C. (1956) J. Phys. Chem. 60 1375-1378 [Google Scholar]
- 42.Brown, P. H., Balbo, A., and Schuck, P. (2007) Biomacromolecules 8 2011-2024 [DOI] [PMC free article] [PubMed] [Google Scholar]