

Abstract
The PI3K-Akt-FoxO1 pathway contributes to the actions of insulin and leptin in several cell types, including neurons in the CNS. However, identifying these actions in chemically identified neurons has proven difficult. To address this problem, we have developed a reporter mouse for monitoring PI3K-Akt signaling in specific populations of neurons, based on FoxO1 nucleocytoplasmic shuttling. The reporter, FoxO1 fused to green fluorescent protein (FoxO1GFP), is expressed under the control of a ubiquitous promoter that is silenced by a loxP flanked transcriptional blocker. Thus, the expression of the reporter in selected cells is dependent on the action of Cre recombinase. Using this model, we found that insulin treatment resulted in the nuclear exclusion of FoxO1GFP within POMC and AgRP neurons in a dose- and time-dependent manner. FoxO1GFP nuclear exclusion was also observed in POMC neurons following in vivo administration of insulin. In addition, leptin induced transient nuclear export of FoxO1GFP in POMC neurons in a dose dependent manner. Finally, insulin-induced nuclear export was impaired in POMC neurons by pretreatment with free fatty acids, a paradigm known to induce insulin resistance in peripheral insulin target tissues. Thus, our FoxO1GFP mouse provides a tool for monitoring the status of PI3K-Akt signaling in a cell-specific manner under physiological and pathophysiological conditions.
Keywords: FoxO1, POMC neurons, AgRP neurons, insulin, free fatty acids, insulin resistance
Introduction
The phosphoinositide 3-kinase (PI3K)-AKT signaling pathway is a key mediator of the metabolic actions of hormones such as insulin in peripheral tissues (Whiteman et al., 2002; Manning and Cantley, 2007). The PI3K system has also been reported to play a crucial role in energy homeostasis in the CNS (Morton et al., 2006; Plum et al., 2006a; Prodi and Obici, 2006). The PI3K-Akt signaling pathway is activated in the hypothalamus after administration of insulin and leptin (Niswender et al., 2001; Niswender et al., 2003). Both receptors are expressed in the CNS, and their deletion in the CNS results in metabolic alterations including obesity and glucose intolerance (Morton et al., 2006; Plum et al., 2006a; Prodi and Obici, 2006). Thus, it has been proposed that the PI3K-Akt pathway contributes to the metabolic actions of insulin and leptin within the CNS.
Two groups of neurons in the arcuate nucleus of the hypothalamus are thought to be key targets of insulin and leptin action. The first group coexpress the orexigenic peptides neuropeptide Y (NPY) and agouti-related peptide (AgRP). The other group express the anorexigenic neuropeptide α-MSH, which is a product of pro-opiomelanocortin (POMC) (Elmquist et al., 1999; Xu et al., 2005; Morton et al., 2006; Plum et al., 2006a; Prodi and Obici, 2006; Könner et al., 2007).
Recent evidence suggests that dysregulation of the PI3K-Akt pathway may directly contribute to the onset or development of diabetes (Elmquist and Marcus, 2003; Niswender et al., 2003; Prodi and Obici, 2006). Indeed, hypothalamic insulin resistance has been reported in animals fed with high fat diet (Wang et al., 2001; De Souza et al., 2005). However, the molecular mechanisms and the identity of the neurons underlying hypothalamic insulin resistance remain to be elucidated. Despite the hypothesized importance of the PI3K-Akt pathway in regulating coordinated neuronal responses, assessment of the activity of the PI3K pathway in chemically identified neurons has proven technically difficult. This is an issue as it is likely that signals such as leptin and insulin act differentially in neurons adjacent to each other such as the POMC and NPY/AgRP neurons that have opposite effects on food intake and body weight.
To circumvent these issues, we generated a “knock-in” reporter mouse in which FoxO1 fused with GFP (Nakamura et al., 2000) is used as a readout of PI3K-Akt signaling at the single cell level. FoxO1, a terminal component of the PI3K-Akt pathway, shuttles between the nucleus and the cytoplasm upon activation/inhibition of Akt (Kau et al., 2003; Accili and Arden, 2004; Van Der Heide et al., 2004) (see Fig. 1A). FoxO1 is the most extensively studied and best characterized of the FoxO family members with regards to the regulatory mechanisms of its nucleocytoplasmic transport. Direct phosphorylation of FoxO1 by Akt causes its nuclear exclusion by virtue of its nuclear export signal (Accili and Arden, 2004; Van Der Heide et al., 2004). This nucleocytoplasmic shuttling of FoxO1GFP has been successfully used as an indicator of the activation of the PI3K-Akt pathway in a cell culture system (Kau et al., 2003). Our novel FoxO1GFP reporter mouse model has enabled us to monitor in vitro and in vivo alterations in the PI3K-Akt signaling cascade within hypothalamic neurons responsible for energy homeostasis.
Figure 1.
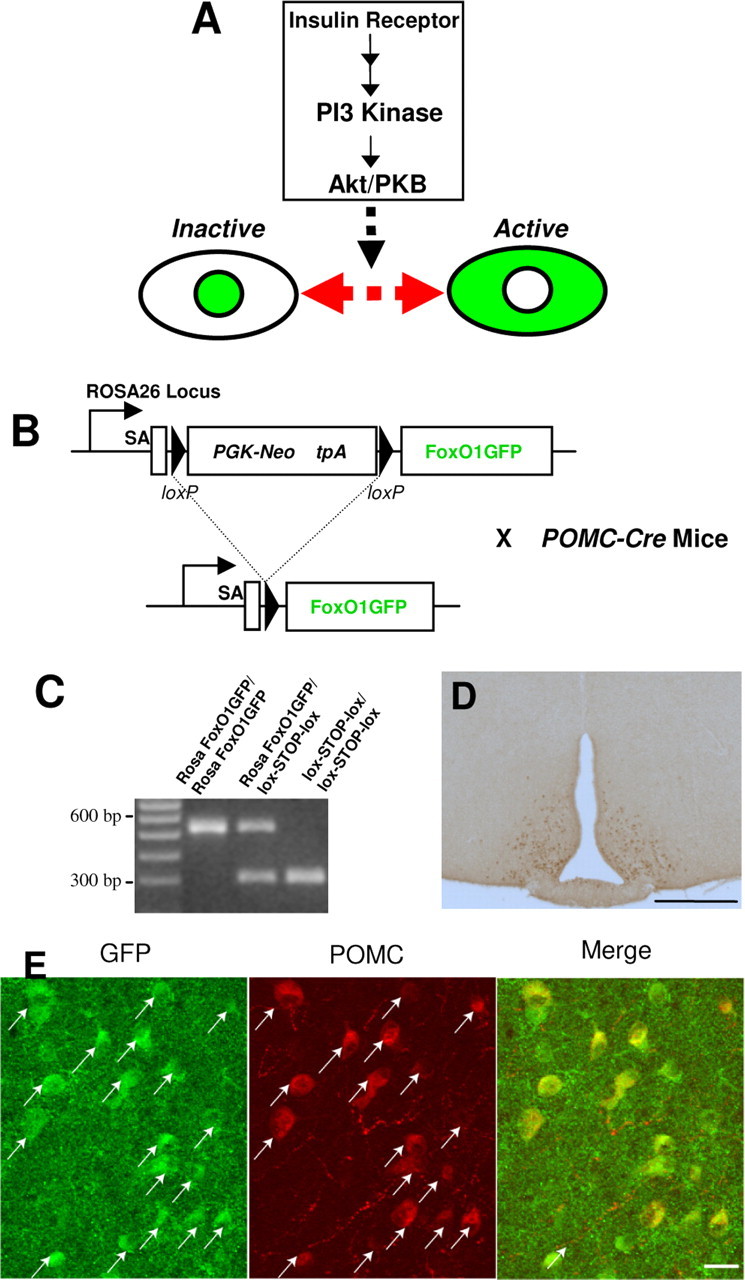
Generation and validation of FoxO1GFP mice. A, The PI3K-Akt pathway controls nucleocytoplasmic translocation of FoxO1GFP. Upon activation of the pathway, FoxO1GFP is rapidly exported from the nucleus. In contrast, inhibition of the pathway leads to nuclear import of FoxO1GFP. B, Mice expressing FoxO1GFP under the control of the Rosa26 promoter were generated by inserting FoxO1GFP into the Rosa26 locus. FoxO1GFP is only expressed in cells producing Cre recombinase because a STOP cassette (PGFk-Neo tpA) flanked by loxP sites was inserted between the Rosa26 promoter and FoxO1GFP. C, The genotypes of mice were shown by PCR analysis. 521 bp PCR product shows wild-type allele. The 298 bp band shows the presence of the FoxO1GFP gene. D, Immunohistochemical staining for GFP in a FoxO1GFP-POMC mouse (FoxO1GFP mouse × POMC-Cre mouse). Bregma −2.06 mm. Scale bar, 400 μm. FoxO1GFP was only observed in the hypothalamus. E, Double immunohistochemistry for β-endorphin (a marker for POMC neurons) and GFP was performed in FoxO1GFP-POMC mice. Arrows indicate the neurons that are doubly positive for both GFP and β-endorphin.
Materials and Methods
Generation of FoxO1GFP mice and AGRP-IRES-CRE mice.
FoxO1GFP was excised from pcDNA3-GFPFKHR (Addgene plasmid 9022) (Nakamura et al., 2000) by cutting with HindIII and XbaI, and then subcloned into pEGFP-C1 (Invitrogen) with HindIII and XbaI. The FoxO1GFP portion was excised from pEGFP-FoxO1GFP by digesting it with XhoI and BclI, and inserted into pBigT plasmid (Srinivas et al., 2001) digested with XhoI and BclI. This plasmid was then digested with PacI and AscI to release the entire floxed neo-tpA and FoxO1GFP assembly, and inserted into pROSA26PA (Soriano, 1999) digested with PacI and AscI. This plasmid was subsequently linearized with IsceI and used for electropo-ration. Using standard ES cell procedures, chimeric animals were obtained and mated with C57BL/6 mice to generate mice heterozygous for the Rosa26-FoxoGFP allele on a mixed C57BL6/J and 129Sv background. The targeting construct for AGRP-IRES-CRE mice was generated using ET cloning and related technologies within EL250 cells as described previously (Balthasar et al., 2004). A PCR was performed using the SIM-IRES-CRE BAC as a template using the following primers: CTTCTTCAATGCCTTTTGCTACTGCCGCAAGCTGGGTACGGCCACGAACCTCTGTAGTCGCACCTAGCCAAATTCCGCCCCTCTCCCT and ACCACAGCTTTAAGGTTAAACCGTCCCATCCTTTATTCTCATCCCCTGCCTTTGCCCAAACAACATCCATACAAAATATTAACGCTTACA. The IRES-Cre sequence flanked by AGRP homology arms was inserted into a mouse AGRP-containing bacterial artificial chromosome, at an insertion site located 3 base pairs after the AGRP stop codon (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). The final targeting construct, which consisted of the AGRP-IRES-Cre flanked by 4 kb and 1.3 kb AGRP homology arms, was electroporated into ES cells, and correct targeting was confirmed by Southern blot and PCR analyses. After germline transmission was established, the chimera carrying the recombinant allele was crossed onto a C57BL/6J background to yield N2F1 animals.
Hypothalamic organotypic slice culture.
The hypothalamic slices were made as essentially described (House et al., 1998). Briefly, FoxO1GFP mice pups, 8–11-d-old, were decapitated, and the brains were quickly removed. Hypothalamic tissues were blocked and sectioned at a thickness of 300 μm on a vibratome (VT1000 S, Leica) in chilled Gey's Balanced Salt Solution (Invitrogen) enriched with glucose (0.5%) and KCl (30 mm). The coronal slices containing the arcuate nucleus were then placed on Millicell-CM filters (Millipore; pore size 0.4 μm, diameter 30 mm), and then maintained at an air-media interface in MEM (Invitrogen) supplemented with heat-inactivated horse serum (25%, Invitrogen), Glucose (32 mm) and GlutaMAX (2 mm, Invitrogen). Cultures were typically maintained for 10 d in standard medium, which was replaced three times a week. After overnight incubation in low-serum (2.5%) MEM supplemented with GlutaMAX (2 mm), slices prepared from the FoxO1GFP-POMC mice were used for experiments. Palmitic acid containing media was prepared by conjugation of palmitic acid (Sigma) with fatty acid-free BSA (ICN), by a method modified from that described by Svedberg et al. (1990). Briefly, free fatty acids were dissolved in ethanol at 60°C and then was mixed with prewarmed BSA (5% in MEM supplemented with 12.5% HBSS) to yield a 10× stock concentration of 4 mm. This corresponds to 5:1 ratios of FFA/BSA, given 400 μm FFA in 0.5% BSA.
Immunohistochemistry for GFP.
Tissues were fixed with 4% formalin in PBS at 4°C overnight. After washing in PBS, the tissues were incubated overnight at room temperature in a rabbit anti-GFP primary antiserum (Invitrogen, 1:20,000) in PBT-azide (3% normal donkey serum (Jackson ImmunoResearch) with 0.25% Triton X-100 in PBS). After washing in PBS, the tissues were incubated in Alexa488 anti-rabbit IgG antibody (Invitrogen; 1:2000) for 3 h at room temperature. Tissues were mounted onto slides with Fluoromount-G (Southern Biotechnology Associates), and then visualized with Zeiss ApoTome. For in vivo studies, FoxO1GFP mice were perfused with 10% formalin, the brains were sectioned on a microtome, and GFP immunohistochemistry was performed as previously described (Balthasar et al., 2004).
Dual-label in situ hybridization histochemistry/immunohistochemistry for AgRP and GFP.
The protocol for in situ hybridization histochemistry (ISHH) was a modification of that previously reported (Liu et al., 2003; Kishi et al., 2005) Tissue was rinsed with DEPC-treated PBS, pH 7.0 for 1 h before being pretreated with 0.1% sodium borohydride (Sigma) in DEPC-PBS for 15 min. After washing in DEPC-PBS, tissue was briefly rinsed in 0.1 m TEA, pH 8.0 and incubated for 10 min in 0.25% acetic anhydride in 0.1 m TEA. The tissue was then rinsed in DEPC-treated 2× SSC before hybridization. Antisense mouse agouti-related protein (AgRP) 35S-labeled riboprobe was generated from cDNA template that includes positions 422–649 of GenBank accession number NM_007427.2. The plasmid was linearized with BamHI and subjected to in vitro transcription with T3 polymerase according to the manufacturer's protocols (Ambion). The 35S-labeled probe was diluted to 106 cpm/ml in 50% formamide, 10 mm Tris-HCl, pH 7.5 (Invitrogen), 5 mg of tRNA (Invitrogen), 100 mm dithiothreitol, 10% dextran sulfate, 0.1% SDS, 0.1% sodium thiosulphate, 0.6M NaCl, 0.5 mm EDTA, pH 8.0, 1× Denhardt's solution (Sigma), 1% sheared salmon sperm DNA (Sigma), and 2.5% total yeast RNA (Sigma) and applied to the free floating tissue. Sections were incubated overnight at 57°C. Tissue was rinsed four times in 4× sodium chloride/sodium citrate (SSC) before being incubated in 0.002% RNase A (Roche Applied Bioscience) diluted in 0.5 m NaCl, 10 mm Tris-HCl, pH 8.0, and 1 mm EDTA (RNase buffer) for 30 min at 37°C. After 30 min in RNase buffer, and two rinses at room temperature in 2× SSC, tissue was washed in increasing stringency with 2× SSC at 50°C, 0.2× SSC at 55°C, 0.2× SSC at 60°C for 1 h at each wash and then 0.1× SSC at 55°C for 30 min. After rinsing twice in PBS at room temperature, tissue sections were peroxide treated, blocked in normal donkey serum (Equitech), and incubated in GFP rabbit primary antiserum (Invitrogen; 1:20,000) overnight. The next day, tissues were incubated in biotinylated donkey antirabbit IgG (Jackson ImmunoResearch; 1:1000) for 2 h. Sections were then incubated in the avidin–biotin complex (ABC; Vector Elite Kit; 1:500) and incubated in 0.04% DAB and 0.01% hydrogen peroxide. Tissue was mounted on SuperFrost Plus slides (Fisher), dehydrated in 1 min incubations of increasing concentrations of ethanol (50, 70, 80, 95, and 100%), and delipidated for 5 min in chloroform. After washes in 100% and 95% ethanol, tissue was air-dried, and slides were placed in x-ray film cassettes with BMR-2 film (Kodak) for 1 d. Slides were then dipped in NTB photographic emulsion (Kodak), dried, and stored in desiccant-containing, foil-wrapped slide boxes at 4°C for 1 week. Slides were developed with Dektol developer (Kodak), dehydrated in increasing concentrations of ethanol, cleared in xylenes, and coverslipped with Permaslip.
Intracerebroventricular cannulation and injection.
FoxO1GFP–POMC males (8 weeks old) were implanted with indwelling intracerebroventricular cannulas described before (Zigman et al., 2005). Afterward, animals were housed in separate cages, and correct placement of the indwelling cannulas was validated 7 d later by observing the response to intracerebroventricular administration of 10 ng of angiotensin II. The mice deprived of food for 16 h with ad libitum access to water were injected with wortmannin (25 pmol, Calbiochem) or vehicle (1 μl) followed 1 h later by intracerebroventricular administration of insulin (100 pmol, human insulin from E. Lilly) or saline (1 μl). The mice were perfused with 10% formalin 20 min after intracerebroventricular injection, and brains were isolated for GFP immunohistochemical analysis.
Image analysis.
All fluorescence images were acquired as z-stacks comprising sequential x–y sections taken at 1.0 μm z-intervals by using an ApoTome imaging system (Imager Z1; Zeiss) with a ×20/1.4 objective (Zeiss), avoiding saturation of maximum pixel value. Image pixel intensity, as a measurement of fluorescence intensity, was measured within specific regions of the neuron [cytoplasmic (in the soma) and nuclear] as well as in regions outside the cell (background) with the AxioVision 4.1 software. After correction for background fluorescence, mean pixel intensity was used to determine the nuclear:cytoplasmic (N:C) ratio of fluorescence intensity. The mean N:C ratio was determined from 100 to 200 neurons for each condition. We defined neurons positive for cytoplasmic staining of GFP, for nucleus staining or for both staining as described in supplemental Figure 1, available at www.jneurosci.org as supplemental material. Neurons positive for cytoplasmic FoxO1GFP were defined as those with an N:C ratio of <1:2. Neurons with nuclear FoxO1GFP were ones with an N:C ratio of 2:1 or more. Neurons with an N:C ratio of between 1:2 and 2:1 were deemed as neurons showing both nuclear and cytoplasmic FoxO1GFP staining. The micrographs were generated by compiling Z-stacks of images taken at 1.0 μm z-intervals.
Results
Generation and characterization of FoxO1-GFP knock-in mice
We took advantage of the fact that PI3K-Akt dependent nuclear export of FoxO1GFP can be used as an indicator of the activity of the PI3K-Akt/PKB pathway (Kau et al., 2003) (Fig. 1A). FoxO1 fused with GFP, which has the same shuttling properties as endogenous FoxO1 (Frescas et al., 2005), was inserted into the endogenous Rosa26 locus using gene targeting, enabling ubiquitous expression of the reporter (Soriano, 1999). However, to achieve cell type specific expression of FoxO1GFP, a transcriptional blocking sequence (Srinivas et al., 2001) flanked by loxP sites was inserted between the Rosa26 promoter and the FoxO1GFP (Fig. 1B). Thus, the reporter is silent at baseline, but can be expressed in any cell type in a Cre-dependent manner. The targeting of FoxO1GFP into the Rosa26 locus was confirmed by Southern blot and PCR analysis (Fig. 1C).
To characterize the mice, we crossed FoxO1GFPmice with Pomc-Cre mice (FoxO1GFP-POMC mice) in which Cre is expressed in hypothalamic POMC neurons (Balthasar et al., 2004). POMC neurons are anorexigenic neurons that respond to insulin (Benoit et al., 2002; Plum et al., 2006a) and leptin (Elias et al., 1999; Elmquist et al., 1999; Cowley et al., 2001). Single label immunohistochemistry for GFP showed that FoxO1GFP then was expressed in a pattern similar to POMC (Fig. 1D). We then performed a double immunohistochemistry for β-endorphin (a POMC peptide product) and GFP (Fig. 1E). Quantification indicated that >90% of β-endorphin-positive neurons expressed FoxO1GFP, and that >90% of FoxO1GFP expressing neurons displayed β-endorphin immunoreactivity. Thus, the FoxO1GFP reporter was expressed eutopically and the vast majority of POMC neurons in the arcuate nucleus expressed FoxO1GFP.
FoxO1GFP dynamics in POMC neurons
To further validate our reporter system, we used hypothalamic organotypic slice cultures (House et al., 1998). Hypothalamic slices (300 μm thickness) of the FoxO1GFP-POMC mice were prepared, and maintained in culture for 10 d. We found that POMC neurons in the slices maintained for 2 weeks were morphologically and functionally intact. The slices were treated with different doses of insulin and then were subjected to immunohistochemistry for GFP to assess subcellular distribution of FoxO1GFP. For example, 30 min after the addition of 100 nm insulin, FoxO1GFP was excluded from the nuclei of >90% of POMC neurons (Fig. 2A,B). To evaluate whether the reporter provides measures for the PI3K-Akt activity in terms of quantity and dynamics, we quantified the ratios of nuclear to cytoplasmic fluorescent intensity of FoxO1GFP as described in Materials and Methods and supplemental Figure 1, available at www.jneurosci.org as supplemental material. A semiquantitative assessment revealed that insulin-induced nuclear exclusion of FoxO1 occurred in a concentration-dependent manner, reaching a plateau between 10 and 25 nm insulin (Fig. 2E). We found that insulin-induced cytoplasmic migration as early as 15 min that continued up to 120 min (Fig. 2F; supplemental Fig. 2A, available at www.jneurosci.org as supplemental material).
Figure 2.
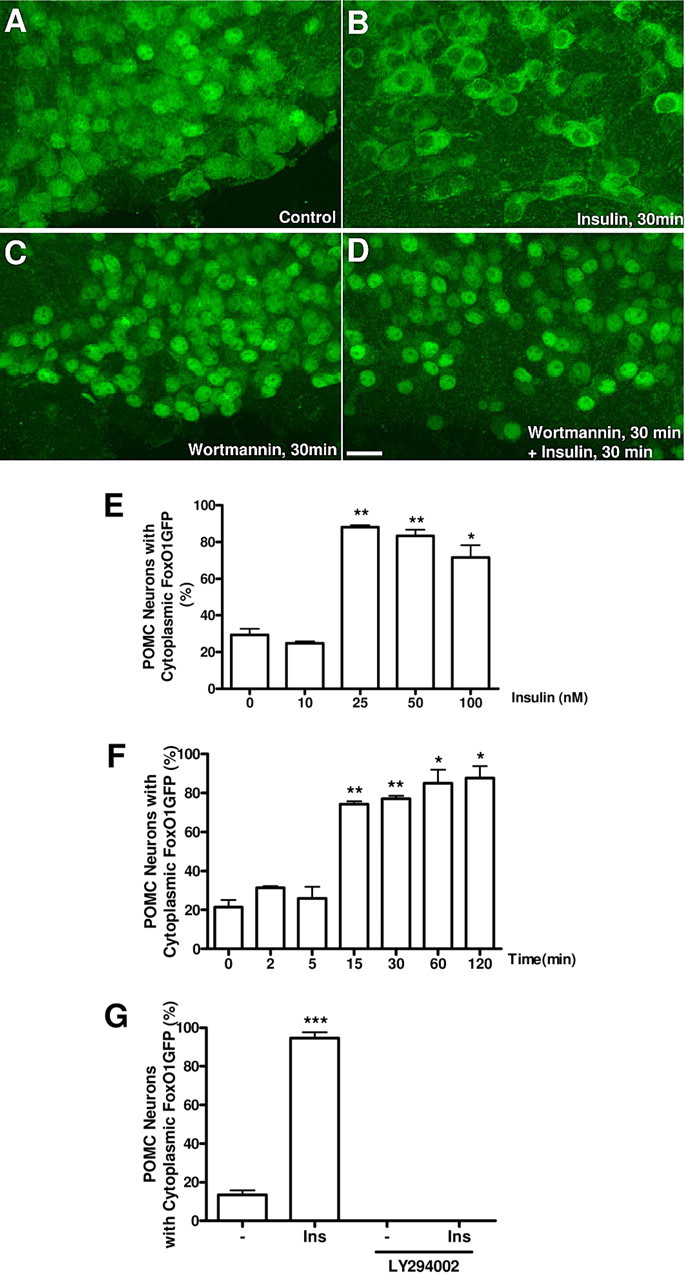
Dynamics and specificity of FoxO1GFP nuclear export within POMC neurons. A–D, Hypothalamic organotypic slices from FoxO1GFP-POMC mice were untreated (A) or treated with insulin (100 nm for 30 min) (B), wortmannin (100 nm for 30 min) (C) or wortmannin (100 nm for 30 min) followed by insulin (100 nm for 30 min) (D), and subjected to immunohistochemistry with an anti-GFP antibody. Scale bar, 20 μm. E, Dose-dependent nuclear exclusion of FoxO1GFP by insulin in POMC neurons. F, Time-dependent nuclear export of FoxO1GFP by insulin (100 nm) in POMC neurons. G, LY 294002 (100 nm for 30 min) blocked the nuclear export of FoxO1GFP by insulin (30 nm for 30 min). Values in E, F, and G represent the percentage of neurons with cytoplasmic FoxO1GFP. The criteria used here are described in Materials and Methods and supplemental Figure 1, available at www.jneurosci.org as supplemental material. ***p < 0.001, **p < 0.01 and *p < 0.05, compared with control by unpaired Student's t test.
We next tested whether insulin-induced nuclear export of FoxO1GFP is mediated by PI3K. Treatment with wortmannin, a PI3K inhibitor, led to prominent FoxO1GFP relocalization to the nucleus (Fig. 2C). In addition, wortmannin completely blocked the insulin-induced nuclear export of FoxO1GFP (Fig. 2D). We observed a similar inhibition following administration of another inhibitor of PI3K, LY 294002 (Fig. 2G). Treatment with LMB, an inhibitor for the nuclear export receptor (CRM1) (Fukuda et al., 1997), also resulted in FoxO1GFP nuclear sequestration. These data indicate that FoxO1 shuttles between the nucleus and cytoplasm in a PI3K- and CRM1-dependent manner in our novel model and that the nucleocytoplasmic shuttling of FoxO1GFP is an appropriate measure of activity of the PI3K/Akt pathway.
Effects of leptin on FoxO1 dynamics in POMC neurons
Leptin has been reported to increase phosphatidylinositol triphosphate (PIP3) production in POMC neurons (Xu et al., 2005; Plum et al., 2006b). Thus, we next examined the effect of leptin on FoxO1GFP dynamics in POMC neurons. We treated slices prepared from FoxO1GFP-POMC mice with increasing doses of leptin for 30 min. Leptin treatment induced nuclear export of FoxO1GFP in POMC neurons in a dose-dependent manner starting at 12 nm reaching a plateau at 120 nm (Fig. 3A, supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). Treatment of slices with leptin also resulted in transient nuclear exclusion of FoxO1GFP in POMC neurons in a time dependent manner (Fig. 3B; supplemental Fig. 2C–F, available at www.jneurosci.org as supplemental material). In particular, leptin-induced nuclear export was apparent 15 min after stimulation and persisted up to 30 min during constant exposure. Thereafter, the percentage of POMC neurons with cytoplasmic FoxO1GFP gradually returned close to control levels (Fig. 3B; supplemental Fig. 2C–F, available at www.jneurosci.org as supplemental material). Wortmannin completely inhibited the nuclear export of FoxO1GFP by leptin (Fig. 3C). We also tested whether or not FoxO1GFP is translocated to the cytoplasm from the nucleus of POMC neurons in response to insulin-like growth factor-1 (IGF-1) or IGF-2. We found that FoxO1GFP translocated to the cytoplasm in response to both IGF-1 and IGF-2 in a subset of POMC neurons (supplemental Fig. 3A–D, available at www.jneurosci.org as supplemental material).
Figure 3.
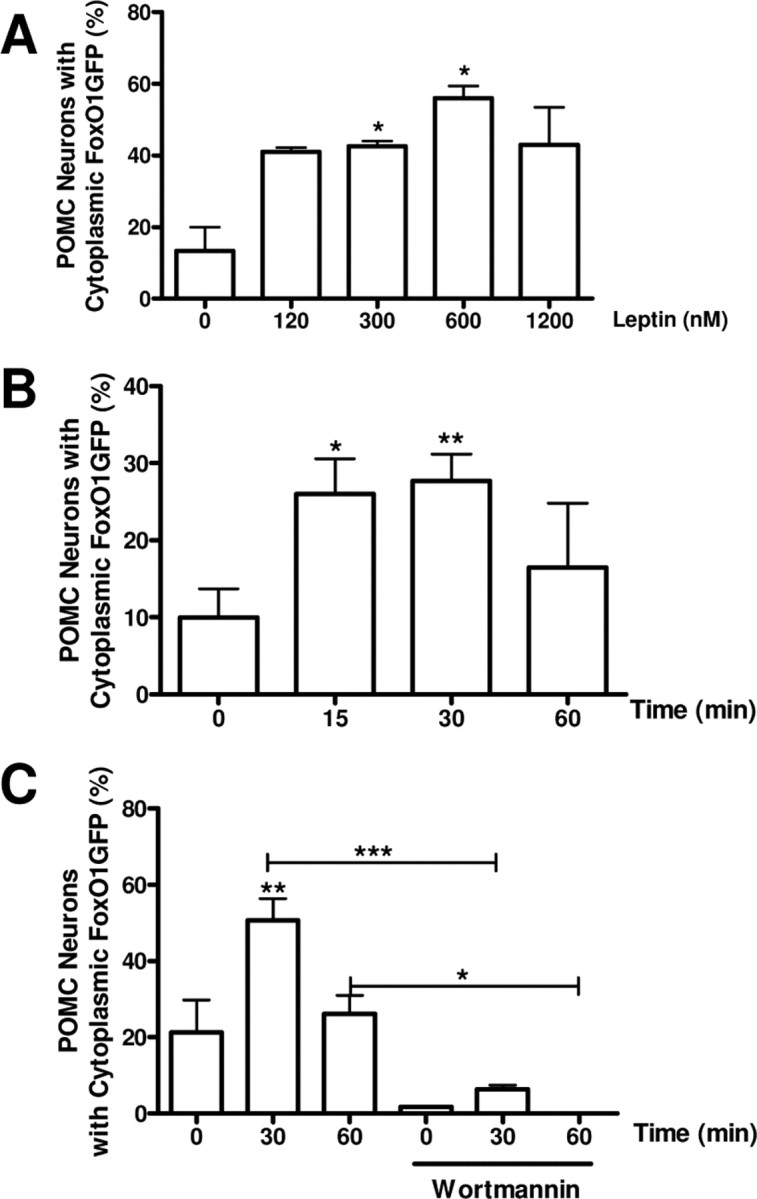
Effects of leptin on FoxO1GFP subcellular distribution. A, Slices expressing FoxO1GFP in POMC neurons were treated with increasing doses of leptin for 30 min. B, Time course of subcellular distribution of FoxO1GFP after exposure to 120 nm leptin. C, Inhibition of leptin-induced nuclear export of FoxO1GFP by wortmannin. Slices were pretreated with wortmannin (100 nm for 30 min), then followed by leptin stimulation (120 nm for indicated period). Subcellular localization of FoxO1GFP is plotted as the percentage of neurons with cytoplasmic FoxO1GFP as in Figure 2. **p < 0.01 and *p < 0.05, compared with control by unpaired Student's t test.
FoxO1GFP in AgRP/NPY neurons
We next evaluated FoxO1GFP dynamics within AgRP neurons, which are also responsive to insulin and leptin. However, unlike POMC neurons, NPY/AgRP neurons are orexigenic and are inhibited by leptin (Elmquist et al., 1999; Xu et al., 2005; Morton et al., 2006; Plum et al., 2006a; Prodi and Obici, 2006; Könner et al., 2007). For these studies, we generated AgRP-Cre mice in which a DNA cassette containing an internal ribosome entry site (IRES) followed by the coding sequence for Cre recombinase was inserted at a site located 3 base pairs after the AgRP stop codon (supplemental Fig. 4A, available at www.jneurosci.org as supplemental material) (Tong et al., 2008). To assess whether functional Cre protein was restricted to AgRP neurons, we crossed the AGRP-Cre mice with Z/EG Cre reporter mice in which GFP is expressed after Cre-mediated excision of a loxP-flanked lacZ gene (Novak et al., 2000). We assessed coexpression of Cre activity (immunohistochemical detection of GFP) and AgRP mRNA expression (in situ hybridization). We found that all Cre recombinase activity was restricted to AgRP neurons in the Arc (supplemental Fig. 4C,D, available at www.jneurosci.org as supplemental material), suggesting that GFP was expressed eutopically.
To examine FoxO1GFP dynamics in AgRP neurons, FoxO1GFP mice were crossed with AgRP-Cre mice, and hypothalamic organotypic slices of FoxO1GFP-AgRP mice were prepared. Insulin stimulation (100 nm) induced translocation of FoxO1GFP from the nucleus to the cytoplasm in AgRP neurons (Fig. 4A,B,D). As expected wortmannin treatment (100 nm) resulted in the strong nuclear accumulation of FoxO1GFP in AgRP (Fig. 4C,D). Thus, insulin activates PI3K activity in both POMC and NPY/AgRP neurons. We also assessed the responses of AgRP neurons to leptin treatment. FoxO1GFP was found in the cytoplasm ∼15 min after exposure to leptin (120 nm) and then shifted toward the nucleus at 60 min (Fig. 4E).
Figure 4.
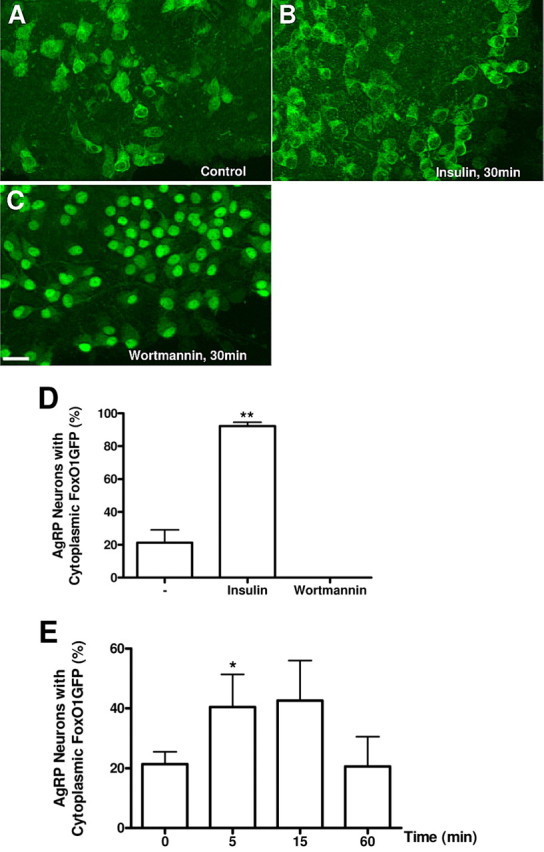
FoxO1GFP dynamics in AgRP neurons. A–C, Hypothalamic organotypic slices expressing FoxO1GFP in AgRP neurons were untreated (A) or treated with insulin (100 nm, 30 min) (B) or wortmannin (100 nm, 30 min) (C), and then fixed and stained with an anti-GFP antibody. Scale bar, 20 μm. D, Quantification of FoxO1GFP nuclear translocation in control, insulin or wortmannin treated organotypic slices of FoxO1GFP-AgRP mice. E, Effect of Leptin on FoxO1 subcellular localization in AgRP neurons. Slices expressing FoxO1GFP in AgRP neurons were treated with leptin (120 nm) for indicated time. Subcellular localization of FoxO1GFP is plotted as the percentage of neurons with cytoplasmic FoxO1GFP as in Figure 2. **p < 0.01, *p < 0.05 compared with control by unpaired Student's t test.
Dynamics of FoxO1GFP in POMC neurons in vivo
To assess whether FoxO1GFP mice could be used to effectively monitor PI3K-Akt signaling in identified neuronal populations in vivo, we performed intracerebroventricular (ICV) infusions of insulin alone (100 pmol) or wortmannin (25 pmol) followed by insulin (100 pmol) in 16 h fasted FoxO1GFP-POMC mice. Mice were perfused 20 min after ICV infusion and we then performed immunohistochemistry for GFP in the brains of treated mice. Similar to our in vitro data, a single ICV injection of insulin excluded FoxO1GFP from the nucleus of POMC neurons (Fig. 5B,D). In contrast, wortmannin completely relocalized FoxO1GFP to the nucleus even in the presence of insulin (Fig. 5C,D). This suggests that insulin-induced nuclear export of FoxO1GFP is mediated through the PI3K pathway. In control mice, FoxO1 was localized to both the nucleus and the cytoplasm in POMC neurons in fasted mice (Fig. 5A,D).
Figure 5.
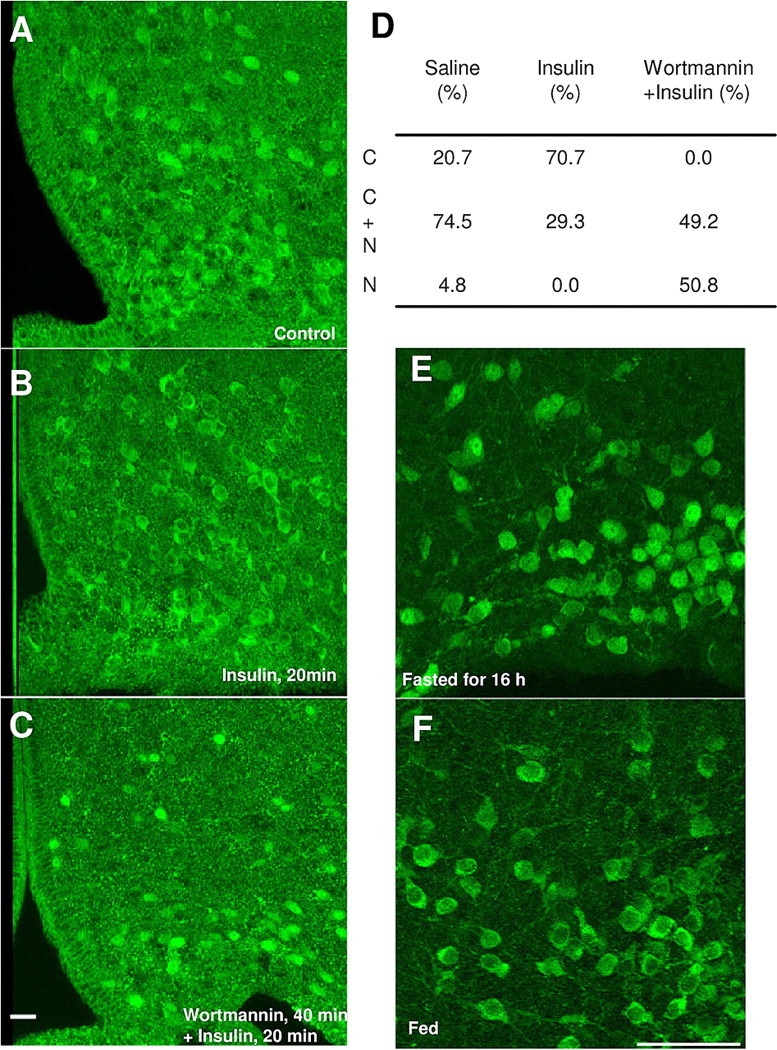
In vivo characterizations of FoxO1GFP mice. A–C, FoxO1GFP-POMC mice fasted for 16 h were administrated with saline (1 μl; i.c.v.) (A), insulin (100 pmol), or wortmannin (100 pmol) (B) 30 min before insulin (100 pmol) (C). Twenty minutes after injection, the mice were perfused and sliced (30 μm). Sections were immunohistochemically stained using an anti-GFP antibody. Scale bar, 20 μm. D, Quantification of FoxO1GFP nuclear translocation in control, insulin or wortmannin plus insulin treated FoxO1GFP-POMC mice. Subcellular localization of FoxO1GFP is presented as the percentage of POMC neurons with C, C+N or N. E, F, Subcellular localization of FoxO1GFP in POMC neurons in FoxO1GFP-POMC mice fasted for 16 h (E) or fed normally (F). Scale bar, 50 μm.
We further assessed whether FoxO1GFP may be able to detect physiological changes in PI3K-Akt signaling in POMC neurons by examining the reporter in fed and fasted mice. FoxO1GFP was observed in the cytoplasm of POMC neurons in fed mice (Fig. 5F; supplemental Fig. 5, available at www.jneurosci.org as supplemental material). In contrast, the fasted mice displayed significantly more localization of the reporter in the nuclear compartment (Fig. 5E; supplemental Fig. 5, available at www.jneurosci.org as supplemental material). These data suggest that FoxO1GFP can be used to monitor PI3K-Akt signaling in vivo under physiological conditions.
Free fatty acid-induced impairment of the insulin signaling in POMC neurons
High fat feeding or over nutrition has been reported to blunt hypothalamic insulin action, including induction of hypothalamic insulin resistance (Wang et al., 2001; De Souza et al., 2005). In addition, infusion of free fatty acids has been shown to induce insulin resistance in peripheral tissues by impairing the ability of insulin to suppress hepatic glucose production and to stimulate glucose uptake into skeletal muscle (Dresner et al., 1999; Lam et al., 2003; Boden et al., 2005). To determine whether POMC neurons lose their ability to respond to insulin after treatment of free fatty acids, we examined the responses of slices from the FoxO1GFP-POMC mice following exposure to FFAs in vitro. The organotypic slices of FoxO1GFP-POMC mice were pretreated with the saturated free fatty acid, palmitic acid (400 μm), for 6 h, and then stimulated at the threshold level of insulin concentration (20 nm insulin for 20 min) (Fig. 6A–C). A semiquantitative assessment revealed that insulin-induced nuclear export of FoxO1GFP was significantly impaired by pretreatment with palmitic acid at 400 μm (Fig. 6D); palmitic acid at lower dose (100 or 200 μm) also tended to blunt insulin-induced nuclear export of FoxO1GFP, although these responses did not reach statistical significance. Collectively, these data suggest that free fatty acids may cause insulin resistance by blocking the activity of the PI3K pathway within POMC neurons.
Figure 6.
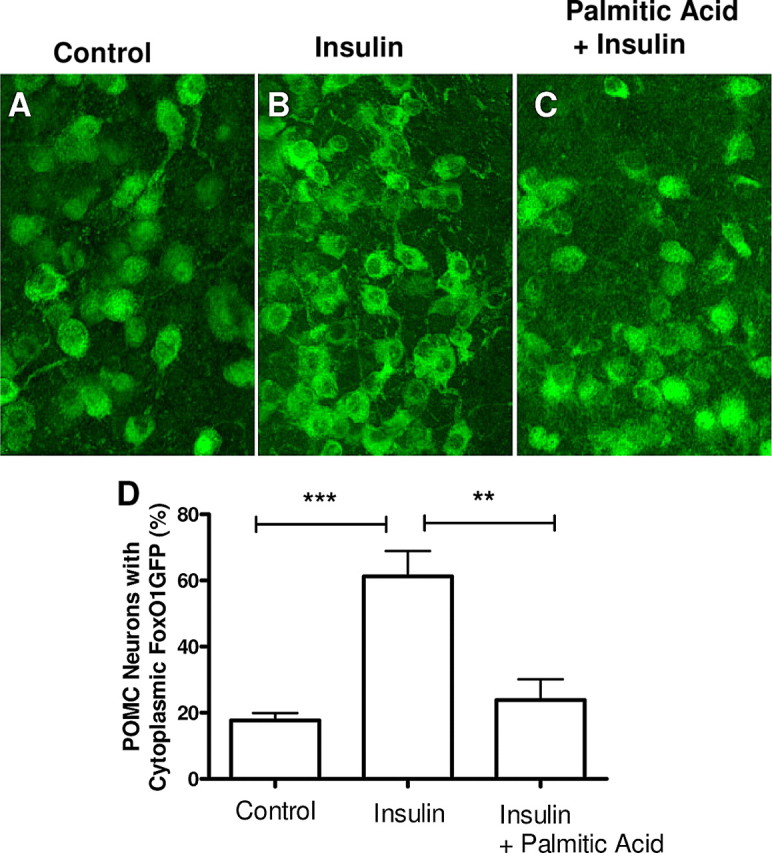
Impairment of POMC insulin signaling by palmitic acids. A–C, Hypothalamic organotypic slices expressing FoxO1GFP in POMC neurons were treated with BSA 6 h before either PBS (A) or insulin (B) (20 nm for 20 min) treatment or palmitic acid loaded on BSA (400 μm), 6 h followed by insulin treatment (20 nm for 20 min) (C). D, Quantification of FoxO1GFP nuclear translocation in control, insulin or palmitic acid followed by insulin treated organotypic slices of FoxO1GFP-POMC mice. Subcellular localization of FoxO1GFP is plotted as the percentage of neurons with cytoplasmic FoxO1GFP as in Figure 2. ***p < 0.001 compared with control; **p < 0.01 compared with insulin-treated slices, by unpaired Student's t test.
Discussion
Recent genetic, anatomic and pharmacological studies support the model that hypothalamic neurons are direct targets of several metabolic cues whose action is required to maintain energy balance. One key signal transduction cascade is the PI3K pathway which has been reported to be downstream of both leptin and insulin receptor signaling. However, the inherent molecular and cellular complexities of the CNS, has made it quite difficult to assess the CNS PI3K-Akt pathway with cellular resolution in chemically identified neurons. This relative lack of reagents has been the limiting factor in studying PI3K signaling in chemically identified neurons. We have developed a novel reporter mouse for monitoring nucleocytoplasmic shuttling of FoxO1 as a measure of activity of the PI3K-Akt pathway. The initial characterization of FoxO1GFP mice described indicates that FoxO1GFP translocation is useful as a specific and sensitive indicator of the activity of the PI3K-Akt pathway in vitro and in vivo.
Here, we found that insulin is able to exclude FoxO1 from the nucleus in POMC neurons and AgRP neurons. FoxO1 has been reported to directly control AgRP and POMC gene expression (Kim et al., 2006; Kitamura et al., 2006). FoxO1 increases AgRP mRNA and decreases POMC mRNA. Our data are consistent with this as insulin induces POMC expression and suppresses NPY/AgRP expression (Kim et al., 2006; Kitamura et al., 2006). Recent data has suggested that whole body glucose intolerance and insulin resistance may be due to defects in sensing and integrating multiple metabolic cues in the CNS (Elmquist and Marcus, 2003; Prodi and Obici, 2006). This includes hypothalamic insulin resistance, which may contribute to the onset or development of diabetes (Obici et al., 2002; Könner et al., 2007). The underlying mechanisms are still to be revealed. As a step toward this goal, we found that similar to peripheral tissues (Roden et al., 1996; Boden, 1997) free fatty acids induced cellular insulin resistance in hypothalamic POMC neurons using organotypic slice cultures from reporter mice.
POMC neurons play an essential role in controlling energy balance and glucose homeostasis, secreting α-MSH (a product of the POMC gene) that activates melanocortin receptors. Several studies have reported that the central melanocortin system can regulate peripheral insulin sensitivity independent of body weight regulation. For example, melanocortin-4 receptor (MC4r) knock-out mice showed insulin resistance before the onset of obesity (Huszar et al., 1997; Fan et al., 2000). Pharmacological activation of central melanocortin receptors enhances peripheral insulin sensitivity (Obici et al., 2001; Heijboer et al., 2005). Activation of serotonin 2C receptors acutely restores peripheral insulin sensitivity in an MC4r-dependent manner in diet-induced obese mice (Zhou et al., 2007). In addition, several reports have suggested that POMC neurons control whole body insulin sensitivity. For example, overexpression of α-MSH derived from POMC product attenuates insulin resistance in ob/ob mice (Mizuno et al., 2003). Also, mice lacking suppressor of cytokine signaling-3, which inhibits leptin and insulin signaling within POMC neurons, resulted in improvement of glucose homeostasis (Kievit et al., 2006). Thus, the dysfunction of POMC neurons may contribute to whole body insulin resistance.
POMC neurons are regulated by several humoral factors including insulin and leptin. Recent evidence suggests that both hormones activate PI3K signaling in hypothalamic neurons (Niswender et al., 2001, 2003; Plum et al., 2006b; Könner et al., 2007). Recently, we found that selective deletion of the PI3K regulatory subunits (p85α and β), abolished the ability of POMC neurons to acutely respond to leptin and insulin (Hill et al., 2008). This is consistent with observations that central insulin action was also blocked with pharmacological inhibition of the PI3K pathway (Niswender et al., 2003). Collectively, these findings support the model that when plasma free fatty acid concentration is elevated (over ∼1 mm) in diet-induced obesity, PI3K impairment in POMC neurons by free fatty acids may contribute to the pathophysiology of central insulin resistance and, subsequently, systemic insulin resistance.
FoxO1GFP reporter mice have several advantages for detecting PI3K-Akt signaling. First, the reporter mice can be used to detect inhibition of the PI3K-Akt pathway. This is contrast to the conventional methods, such as immunohistochemistry of anti Phospho-Akt or anti Phospho-FoxO1 antibodies, which assess PI3K pathway activation. Another advantage of the current model compared other approaches such as electrophysiological recordings is that the FoxO1GFP mouse expresses the reporter across the entire population of chemically identified neurons, enabling a broader view of the spatial and temporal patterns of activation or inhibition of the PI3K-Akt pathway. Previously, Xu et al. (2005) developed a system to monitor PIP3 production in POMC and AgRP neurons using GRP1-PH-GFP reporter that binds to PIP3 and then is translocated to the plasma membrane upon activation of PI3K. The adenovirus-mediated delivery successfully expressed the reporter in POMC and AgRP neurons, and the reporter was capable of monitoring PIP3 production in living neurons in response to insulin or leptin. Notably, our reporter model provides a reliable system to monitor the majority of chemically identified cells without the need for adenovirus delivery.
The requirement for cell-type specificity in investigating the CNS has become obvious from recent understanding of local neuro-circuits. For example, among the hypothalamic nuclei, the arcuate nucleus contains small, adjacent groups of neurons that are reciprocally regulated by several cues including leptin, glucose and neurotransmitters such as serotonin (Elmquist et al., 1999; Xu et al., 2005; Morton et al., 2006; Plum et al., 2006a; Prodi and Obici, 2006; Könner et al., 2007). Indeed, these two neuronal groups have opposite effects on food intake and body weight (Elmquist et al., 1999; Morton et al., 2006). Thus, it is important to ascertain the molecular mechanisms underlying the ability of these cues to regulate transcriptional and cellular activity of POMC and NPY/AgRP neurons. In addition, the Cre/loxP-mediated strategy used here makes it possible to selectively express the reporter and to illuminate the activity of the PI3K-Akt pathway in a specific population of neurons. Finally, although we have concentrated on neuronal cell types in the current study, it is important to note that this model would also be useful to study PI3K-dependent signaling in any cell type such as muscle, adipocytes, and hepatocytes. This is due to the ubiquitous expression of the reporter and the potential to activate its expression with specific Cre mice (e.g., fat and muscle specific lines).
One caveat of our approach is that it is possible that other signaling pathways could regulate the nucleocytoplasmic shuttling of FoxO1GFP affecting the interpretation of the reporter as an indicator of cellular activation or inhibition of the PI3K-Akt pathway. For example, it is possible that FoxO1 shuttling would be affected by other protein kinases, such as SGK, that are able to phosphorylate amino acid residues on FoxO1 responsible for determining its nucleocytoplasmic transport (Van Der Heide et al., 2004).
To assess this possibility, we used specific inhibitors for PI3K including wortmannin and LY294002. Around 10–20% of POMC neurons or AgRP neurons always showed cytoplasmic localization of FoxO1GFP even without leptin or insulin stimulation (Figs. 2E–G, 3A–C). This cytoplasmic staining seen in control completely disappeared when slices were treated with the inhibitors for PI3K, indicating that cytoplasmic localization of FoxO1GFP observed in controls would be attributed to activation of the PI3K-FoxO1 pathway. Despite this potential limitation, our approach adds a new tool to the list of reagents for investigating PI3K-Akt signaling. Furthermore, beyond regulation of energy homeostasis, FoxO1 is believed to be involved in a diverse set of physiological and pathological conditions ranging from cancer biology to aging. The reporter mouse presented here should also facilitate the investigation of FoxO1 functions in multiple tissues and in multiple physiological contexts.
Footnotes
This work was supported by National Institutes of Health Grants DK56116, MH61583, DK071320, and DK53301 to J.K.E., by an ADA Smith Family Foundation Pinnacle program award to J.K.E., and by Takeda Pharmaceutical Company, Japan. We thank Gui Lan Yao for help with organotypic slice culture. We thank Dr. William Sellers (Harvard University) for pcDNA3-GFP-FKHR plasmid.
The authors declare no competing financial interests.
References
- Accili D, Arden KC. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 2004;117:421–426. doi: 10.1016/s0092-8674(04)00452-0. [DOI] [PubMed] [Google Scholar]
- Balthasar N, Coppari R, McMinn J, Liu SM, Lee CE, Tang V, Kenny CD, McGovern RA, Chua SC, Jr, Elmquist JK, Lowell BB. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron. 2004;42:983–991. doi: 10.1016/j.neuron.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Benoit SC, Air EL, Coolen LM, Strauss R, Jackman A, Clegg DJ, Seeley RJ, Woods SC. The catabolic action of insulin in the brain is mediated by melanocortins. J Neurosci. 2002;22:9048–9052. doi: 10.1523/JNEUROSCI.22-20-09048.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boden G. Role of fatty acids in the pathogenesis of insulin resistance and NIDDM. Diabetes. 1997;46:3–10. [PubMed] [Google Scholar]
- Boden G, She P, Mozzoli M, Cheung P, Gumireddy K, Reddy P, Xiang X, Luo Z, Ruderman N. Free fatty acids produce insulin resistance and activate the proinflammatory nuclear factor-{kappa}B pathway in rat liver. Diabetes. 2005;54:3458–3465. doi: 10.2337/diabetes.54.12.3458. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdán MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, Saad MJ, Velloso LA. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology. 2005;146:4192–4199. doi: 10.1210/en.2004-1520. [DOI] [PubMed] [Google Scholar]
- Dresner A, Laurent D, Marcucci M, Griffin ME, Dufour S, Cline GW, Slezak LA, Andersen DK, Hundal RS, Rothman DL, Petersen KF, Shulman GI. Effects of free fatty acids on glucose transport and IRS-1–associated phosphatidylinositol 3-kinase activity. J Clin Invest. 1999;103:253–259. doi: 10.1172/JCI5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjorbaek C, Flier JS, Saper CB, Elmquist JK. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron. 1999;23:775–786. doi: 10.1016/s0896-6273(01)80035-0. [DOI] [PubMed] [Google Scholar]
- Elmquist JK, Marcus JN. Rethinking the central causes of diabetes. Nat Med. 2003;9:645–647. doi: 10.1038/nm0603-645. [DOI] [PubMed] [Google Scholar]
- Elmquist JK, Elias CF, Saper CB. From lesions to leptin: hypothalamic control of food intake and body weight. Neuron. 1999;22:221–232. doi: 10.1016/s0896-6273(00)81084-3. [DOI] [PubMed] [Google Scholar]
- Fan W, Dinulescu DM, Butler AA, Zhou J, Marks DL, Cone RD. The central melanocortin system can directly regulate serum insulin levels. Endocrinology. 2000;141:3072–3079. doi: 10.1210/endo.141.9.7665. [DOI] [PubMed] [Google Scholar]
- Frescas D, Valenti L, Accili D. Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J Biol Chem. 2005;280:20589–20595. doi: 10.1074/jbc.M412357200. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Asano S, Nakamura T, Adachi M, Yoshida M, Yanagida M, Nishida E. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature. 1997;390:308–311. doi: 10.1038/36894. [DOI] [PubMed] [Google Scholar]
- Heijboer AC, van den Hoek AM, Pijl H, Voshol PJ, Havekes LM, Romijn JA, Corssmit EP. Intracerebroventricular administration of melanotan II increases insulin sensitivity of glucose disposal in mice. Diabetologia. 2005;48:1621–1626. doi: 10.1007/s00125-005-1838-8. [DOI] [PubMed] [Google Scholar]
- Hill JW, Williams KW, Ye C, Luo J, Balthasar N, Coppari R, Cowley MA, Cantley LC, Lowell BB, Elmquist JK. Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J Clin Invest. 2008;118:1796–1805. doi: 10.1172/JCI32964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- House SB, Thomas A, Kusano K, Gainer H. Stationary organotypic cultures of oxytocin and vasopressin magnocellular neurones from rat and mouse hypothalamus. J Neuroendocrinol. 1998;10:849–861. doi: 10.1046/j.1365-2826.1998.00272.x. [DOI] [PubMed] [Google Scholar]
- Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- Kau TR, Schroeder F, Ramaswamy S, Wojciechowski CL, Zhao JJ, Roberts TM, Clardy J, Sellers WR, Silver PA. A chemical genetic screen identifies inhibitors of regulated nuclear export of a Forkhead transcription factor in PTEN-deficient tumor cells. Cancer Cell. 2003;4:463–476. doi: 10.1016/s1535-6108(03)00303-9. [DOI] [PubMed] [Google Scholar]
- Kievit P, Howard JK, Badman MK, Balthasar N, Coppari R, Mori H, Lee CE, Elmquist JK, Yoshimura A, Flier JS. Enhanced leptin sensitivity and improved glucose homeostasis in mice lacking suppressor of cytokine signaling-3 in POMC-expressing cells. Cell Metab. 2006;4:123–132. doi: 10.1016/j.cmet.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Kim MS, Pak YK, Jang PG, Namkoong C, Choi YS, Won JC, Kim KS, Kim SW, Kim HS, Park JY, Kim YB, Lee KU. Role of hypothalamic Foxo1 in the regulation of food intake and energy homeostasis. Nat Neurosci. 2006;9:901–906. doi: 10.1038/nn1731. [DOI] [PubMed] [Google Scholar]
- Kishi T, Aschkenasi CJ, Choi BJ, Lopez ME, Lee CE, Liu H, Hollenberg AN, Friedman JM, Elmquist JK. Neuropeptide Y Y1 receptor mRNA in rodent brain: distribution and colocalization with melanocortin-4 receptor. J Comp Neurol. 2005;482:217–243. doi: 10.1002/cne.20432. [DOI] [PubMed] [Google Scholar]
- Kitamura T, Feng Y, Kitamura YI, Chua SC, Jr, Xu AW, Barsh GS, Rossetti L, Accili D. Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nat Med. 2006;12:534–540. doi: 10.1038/nm1392. [DOI] [PubMed] [Google Scholar]
- Könner AC, Janoschek R, Plum L, Jordan SD, Rother E, Ma X, Xu C, Enriori P, Hampel B, Barsh GS, Kahn CR, Cowley MA, Ashcroft FM, Brüning JC. Insulin Action in AgRP-Expressing Neurons Is Required for Suppression of Hepatic Glucose Production. Cell Metab. 2007;5:438–449. doi: 10.1016/j.cmet.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Lam TK, van de Werve G, Giacca A. Free fatty acids increase basal hepatic glucose production and induce hepatic insulin resistance at different sites. Am J Physiol Endocrinol Metab. 2003;284:E281–E290. doi: 10.1152/ajpendo.00332.2002. [DOI] [PubMed] [Google Scholar]
- Liu H, Kishi T, Roseberry AG, Cai X, Lee CE, Montez JM, Friedman JM, Elmquist JK. Transgenic mice expressing green fluorescent protein under the control of the melanocortin-4 receptor promoter. J Neurosci. 2003;23:7143–7154. doi: 10.1523/JNEUROSCI.23-18-07143.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno TM, Kelley KA, Pasinetti GM, Roberts JL, Mobbs CV. Transgenic neuronal expression of proopiomelanocortin attenuates hyperphagic response to fasting and reverses metabolic impairments in leptin-deficient obese mice. Diabetes. 2003;52:2675–2683. doi: 10.2337/diabetes.52.11.2675. [DOI] [PubMed] [Google Scholar]
- Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443:289–295. doi: 10.1038/nature05026. [DOI] [PubMed] [Google Scholar]
- Nakamura N, Ramaswamy S, Vazquez F, Signoretti S, Loda M, Sellers WR. Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol Cell Biol. 2000;20:8969–8982. doi: 10.1128/mcb.20.23.8969-8982.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender KD, Morton GJ, Stearns WH, Rhodes CJ, Myers MG, Jr, Schwartz MW. Intracellular signalling. Key enzyme in leptin-induced anorexia. Nature. 2001;413:794–795. doi: 10.1038/35101657. [DOI] [PubMed] [Google Scholar]
- Niswender KD, Morrison CD, Clegg DJ, Olson R, Baskin DG, Myers MG, Jr, Seeley RJ, Schwartz MW. Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: a key mediator of insulin-induced anorexia. Diabetes. 2003;52:227–231. doi: 10.2337/diabetes.52.2.227. [DOI] [PubMed] [Google Scholar]
- Novak A, Guo C, Yang W, Nagy A, Lobe CG. Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis. 2000;28:147–155. [PubMed] [Google Scholar]
- Obici S, Feng Z, Tan J, Liu L, Karkanias G, Rossetti L. Central melanocortin receptors regulate insulin action. J Clin Invest. 2001;108:1079–1085. doi: 10.1172/JCI12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obici S, Feng Z, Karkanias G, Baskin DG, Rossetti L. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat Neurosci. 2002;5:566–572. doi: 10.1038/nn0602-861. [DOI] [PubMed] [Google Scholar]
- Plum L, Belgardt BF, Brüning JC. Central insulin action in energy and glucose homeostasis. J Clin Invest. 2006a;116:1761–1766. doi: 10.1172/JCI29063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plum L, Ma X, Hampel B, Balthasar N, Coppari R, Münzberg H, Shanabrough M, Burdakov D, Rother E, Janoschek R, Alber J, Belgardt BF, Koch L, Seibler J, Schwenk F, Fekete C, Suzuki A, Mak TW, Krone W, Horvath TL, Ashcroft FM, Brüning JC. Enhanced PIP3 signaling in POMC neurons causes KATP channel activation and leads to diet-sensitive obesity. J Clin Invest. 2006b;116:1886–1901. doi: 10.1172/JCI27123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodi E, Obici S. Minireview: the brain as a molecular target for diabetic therapy. Endocrinology. 2006;147:2664–2669. doi: 10.1210/en.2006-0143. [DOI] [PubMed] [Google Scholar]
- Roden M, Price TB, Perseghin G, Petersen KF, Rothman DL, Cline GW, Shulman GI. Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest. 1996;97:2859–2865. doi: 10.1172/JCI118742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svedberg J, Björntorp P, Smith U, Lonnroth P. Free-fatty acid inhibition of insulin binding, degradation, and action in isolated rat hepatocytes. Diabetes. 1990;39:570–574. doi: 10.2337/diab.39.5.570. [DOI] [PubMed] [Google Scholar]
- Tong Q, Ye CP, Jones JE, Elmquist JK, Lowell BB. Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci. 2008;11:998–1000. doi: 10.1038/nn.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Heide LP, Hoekman MF, Smidt MP. The ins and outs of FoxO shuttling: mechanisms of FoxO translocation and transcriptional regulation. Biochem J. 2004;380:297–309. doi: 10.1042/BJ20040167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Obici S, Morgan K, Barzilai N, Feng Z, Rossetti L. Overfeeding rapidly induces leptin and insulin resistance. Diabetes. 2001;50:2786–2791. doi: 10.2337/diabetes.50.12.2786. [DOI] [PubMed] [Google Scholar]
- Whiteman EL, Cho H, Birnbaum MJ. Role of Akt/protein kinase B in metabolism. Trends Endocrinol Metab. 2002;13:444–451. doi: 10.1016/s1043-2760(02)00662-8. [DOI] [PubMed] [Google Scholar]
- Xu AW, Kaelin CB, Takeda K, Akira S, Schwartz MW, Barsh GS. PI3K integrates the action of insulin and leptin on hypothalamic neurons. J Clin Invest. 2005;115:951–958. doi: 10.1172/JCI24301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Sutton GM, Rochford JJ, Semple RK, Lam DD, Oksanen LJ, Thornton-Jones ZD, Clifton PG, Yueh CY, Evans ML, McCrimmon RJ, Elmquist JK, Butler AA, Heisler LK. Serotonin 2C receptor agonists improve type 2 diabetes via melanocortin-4 receptor signaling pathways. Cell Metab. 2007;6:398–405. doi: 10.1016/j.cmet.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigman JM, Nakano Y, Coppari R, Balthasar N, Marcus JN, Lee CE, Jones JE, Deysher AE, Waxman AR, White RD, Williams TD, Lachey JL, Seeley RJ, Lowell BB, Elmquist JK. Mice lacking ghrelin receptors resist the development of diet-induced obesity. J Clin Invest. 2005;115:3564–3572. doi: 10.1172/JCI26002. [DOI] [PMC free article] [PubMed] [Google Scholar]