

Abstract
Diminished bioavailability of nitric oxide is a hallmark of endothelial dysfunction and is associated with a broad spectrum of vascular disorders such as impaired angiogenesis. Because Rac1, a Rho family member, mediates cellular motility and generation of reactive oxygen species, it could be involved in the regulation of endothelial nitric oxide production. However, the pathophysiological consequences of postnatal endothelial Rac1 deletion on endothelial function have not been determined. We generated endothelial-specific Rac1 haploinsufficient mice (EC-Rac1+/-) using Cre-loxP technology. The EC-Rac1+/- mice have decreased expression and activity of endothelial nitric oxide synthase (eNOS), impaired endothelium-dependent vasorelaxation, and mild hypertension compared with control (Rac1+/flox) mice. Hind limb ischemia model and aortic capillary sprouting assay showed that eNOS activity and angiogenesis was impaired in EC-Rac1+/- mice. Indeed, Rac1 promotes eNOS gene transcription through p21-activated kinase but not NADPH oxidase, increases eNOS mRNA stability, and enhances eNOS activity by promoting endothelial uptake of l-arginine. These findings indicate that endothelial Rac1 is essential for endothelium-dependent vasomotor response and ischemia-induced angiogenesis. These effects of Rac1 on endothelial function are largely due to the upregulation of eNOS through multiple mechanisms that are mediated, in part, by p21-activated kinase. Therapeutic strategies to enhance Rac1 function, therefore, may be important for preventing endothelial dysfunction.
Keywords: angiogenesis, endothelium, hypertension, nitric oxide synthase, signal transduction
Endothelium-derived nitric oxide (NO) is an important determinant of endothelium-dependent vasodilation and contributes to the anti-inflammatory and antithrombotic properties of the endothelium. Endothelial dysfunction is characterized by impaired synthesis, release, and activity of endothelial-derived NO and is thought to contribute to the development and progression of various vascular disorders such as vasoconstriction, hypertension, atherosclerosis, and peripheral artery occlusive disease.1 Therefore, understanding the pathophysiological mechanisms of endothelial dysfunction would provide important insights into therapeutic strategies for preventing cardiovascular disease.
Endothelial NO synthase (eNOS) is the primary source of endothelium-derived NO. The steady-state mRNA level of eNOS is one of the principal determinants of eNOS expression and activity and NO bioavailability.2 Numerous exogenous stimuli and conditions have been shown to alter eNOS mRNA level through both transcriptional and posttranscriptional mechanisms. For example, the eNOS mRNA is upregulated by laminar flow, vascular endothelial growth factor, transforming growth factor-beta and lysophosphatidylcholine and downregulated by tumor necrosis factor-α and oxidized low-density lipoprotein.2 The enzymatic activity of eNOS is further regulated by multiple posttranslational mechanisms involving protein-protein association, phosphorylation of eNOS, and l-arginine uptake through cationic amino acid transporter-1 (CAT-1).3,4
The small GTP-binding proteins belonging to the Rho GTPases are versatile signal transducers activated by extracellular humoral and mechanical stimuli. They mediate actin cytoskeletal rearrangements, gene expression, cell cycle progression, and many other important cellular functions.5 Two members of the Rho GTPase family, RhoA and Rac1, have been shown to regulate endothelial cellular events such as polarization, permeability, and adhesion to leukocytes in distinct and overlapping ways.6,7 Of note, inhibition of RhoA and its effector Rho-kinase leads to stabilization and upregulation of eNOS mRNA and improved endothelial function.8,9 However, the role of Rac1 in regulating eNOS expression and activity is not known.
Endothelial Rac1 is activated by fluid shear stress and vascular endothelial growth factor and mediates their effects on cytoskeletal rearrangements and motility, in part, through p21-activated kinase (PAK).7,10 Most importantly, these 2 stimuli upregulate and activate eNOS as well and require NO to exert their vasodilatory and angiogenic effects.2,11 Furthermore, both Rac1 and eNOS are required for endothelial motility, proliferation, survival, and permeability.7,10-12 These findings suggest a possible link between Rac1 and eNOS for the regulation of endothelial function. In contrast, Rac1 could also activate NADPH oxidase and promote the formation of reactive oxygen species (ROS), which, depending on the timing, location, and amount produced, may decrease NO bioavailability.12 Therefore, it is not known what the net effect of Rac1 is on endothelial function in vivo. Thus, to directly address this question, we developed mice with endothelial-specific knockout of Rac1 using a conditional gene targeting strategy. The aim of the study was to determine the role of Rac1 on postnatal endothelial function and angiogenesis.
Materials and Methods
Animals
All animal protocols were approved by the Harvard Medical School’s Standing Committee on Animal Welfare and Protection. Age-matched male litters (12 to 20 weeks) were used for experiments. Rac1 conditional allele knock-in mice were developed as described13 and backcrossed at least 8 times onto the C57BL/6 strain. Tie2-Cre transgenic mice (Jackson Laboratory, Bar Harbor, Me)14 and Tie2-CreERT2 transgenic mice were maintained on a C57BL/6 background.15
Cell Culture
Mice heart endothelial cells (MHECs) were isolated by affinity selection method using Dynabeads sheep antirat IgG (Invitrogen, Carlsbad, Calif) and anti-PECAM1 antibody (BD Biosciences, San Jose, Calif).16
GTP-Rac1 Pull Down Assay
GTP-Rac pull down assay was performed as described.17
Measurement of NADPH Oxidase Activity
NADPH oxidase activity was measured using lucigenin chemiluminescence.18
Assessment of Nitric Oxide Release, Endothelial Nitric Oxide Synthase Activity, l-Arginine Uptake, and cGMP Level
NO release in the culture media was measured by a chemiluminescence method using Sievers 280i nitric oxide analyzer NOA (Sievers Instruments, Inc, Boulder, Colo).19 Cellular eNOS activity and l-arginine incorporation was measured by pulsing cells with l-[3H] arginine (5 μCi/well) for 30 minutes. l-[3H] arginine and l-[3H] citrulline was separated by Dowex 50WX8-400 column.20 eNOS activity and cGMP level in aorta homogenate was measured with a NOS assay kit (Calbiochem, Gibbstown, NJ) and cGMP Enzyme immunoassay Biotrak system (GE Health Care, Bucks, UK), respectively.
RNA Isolation, Northern Blotting, and Quantitative Reverse Transcriptase-Polymerase Chain Reaction
Total RNA was extracted using Trizol reagent (Invitrogen, Carlsbad, Calif). Poly (A)+ RNA was purified from total RNA using oligo (dT) cellulose. Total RNA (20 to 30 μg) and poly (A)+ RNA (2 μg) was subjected to Northern blot analysis. One-step real-time reverse transcriptase-polymerase chain reaction was performed using Quan-tiTect SYBR Green RT-PCR kit (Qiagen, Valencia, Calif). The relative mRNA level was determined by normalization with GAPDH mRNA or 18S rRNA.
Immunoblotting and Immunoprecipitation
Cell and tissue lysates (20 to 50 μg protein) were used for immunoblotting. The blots were visualized using an enhanced chemiluminescence system (GE Health Care, Bucks, UK). For immunoprecipitation, cell lysates were incubated with CAT-1 antibody (Santa Cruz, Santa Cruz, Calif) and protein G Sepharose (GE Health Care). The immunoprecipitate was subjected to immunoblotting with PAK1/2/3 antibody (Cell Signaling, Danvers, Mass).
Reporter Assay
Transfection of reporter constructs was performed using Lipofectamine 2000 (Invitrogen). The luciferase activity was assessed using Dual-Luciferase Reporter Assay system (Promega, Madison, Wis).
Assessment of mRNA Stability
Confluent bEND.3 cells were treated with transcriptional inhibitor 5,6-dichlorobenzimidazole 1-β-d-ribofuranoside (50 μmol/L) in the absence or presence of 100 μmol/L Rac1 inhibitor for 6, 12, 18, 24, and 30 hours. eNOS mRNA expression was assessed by Northern blot analysis and quantified by densitometry.
Blood Pressure Measurement
Noninvasive measurement of blood pressure was conducted using the tail-cuff method (BP-2000 Blood Pressure Analysis System; Visitech, Apex, NC).
Vasomotor Tone Analysis
Measurement of aortic contractile tension was performed as described.21
Mice Hind Limb Ischemia
Hind limb ischemia was initiated by ligation and excision of the left femoral artery.22 Mice were anesthetized through intraperitoneal injection of ketamine (60 mg/kg) and xylazine (10 mg/kg). Some mice were administered Nω-Nitro-l-arginine methyl ester (l-NAME) hydrochloride in drinking water (0.2 g/L) throughout the whole observation period up to 28 days.22 Laser Doppler perfusion image was recorded with a laser Doppler perfusion image analyzer (Moor Instruments, Inc, Wilmington, Del).
Aortic Microvessel Sprouting Assay
Aortic segments were embedded in Matrigel (BD Biosciences, Bedford, Mass) and cultured until day 8.
Statistical Analysis
All statistical analysis was carried out by analysis of variance followed by Fisher test. P<0.05 was considered to be significant.
Results
Generation of Endothelial-Specific Rac1 Knockout Mice
To establish EC-specific Rac1 knockout mice, we crossed transgenic mice expressing Cre recombinase under the control of Tie2 promoter and enhancer (Tie2-Cre Tg)14 to mice harboring Rac1 conditional allele (designated as “c”) with exon 1 flanked by loxP sites (Figure 1A).13 This mating did not yield the birth of Tie2-Cre Rac1c/c mice, although other genotypes were present at mendelian ratios, indicating the demise of Tie2-Cre Rac1c/c embryos (Figure 1B). Accordingly, we used EC-Rac1 haploinsufficient mice (Tie2-Cre Rac1c/+) to study the role of Rac1 in postnatal vascular endothelium (Figure 1B-C). Both EC-Rac1+/- and control mice (Rac1c/+) grew up normally without difference in body and organ weight (see Supplementary Data, Table S1). The MHECs isolated from EC-Rac1+/- showed approximately 50% reduction in the levels of total Rac1 protein and GTP-bound active form of Rac1 (Figure 1D). Effective inhibition of Rac1 signaling in EC-Rac1+/- endothelium was further demonstrated by approximately 40% attenuation in NADPH oxidase activity (Figure 1D). Because Tie2-Cre Tg reportedly undergo Cre-mediated recombination in hematopoietic cells as well as in ECs,14 we also generated inducible EC-Rac1 knockout mice (Tie2-CreERT2 Rac1c/c; Figure 1B-C). In the Tie2-CreERT2 Tg system, administration of tamoxifen induces recombination in a vast majority of ECs and less than 0.2% of hematopoietic cells as reported previously.15
Figure 1.
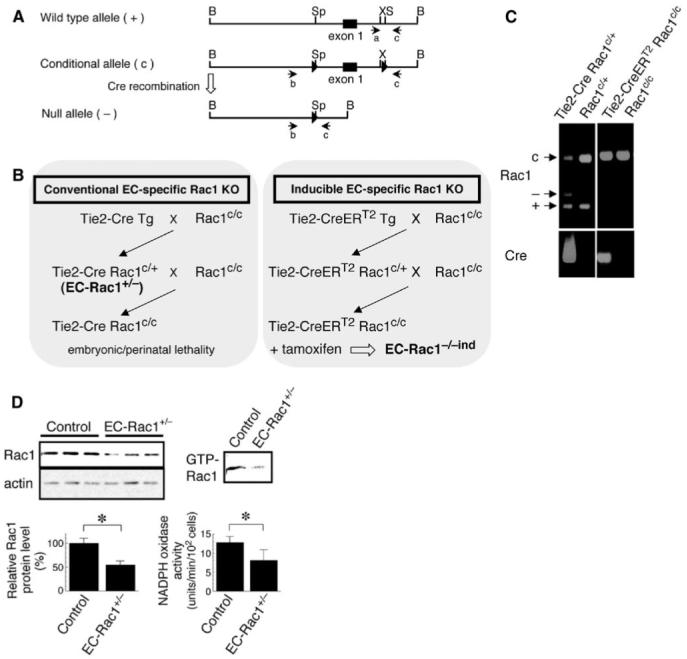
Generation of endothelial-specific Rac1 knockout mice. A, Schematic maps of the wild-type, conditional, and null alleles of the Rac1 gene. Expression of Cre induces homologous recombination of conditional allele into null allele. Polymerase chain reaction (PCR) primers for genotyping are shown under each allele. B, Strategies for generation of conventional (left) and inducible (right) endothelial-specific Rac1 knockout mice. C, Representative PCR genotyping. c: conditional (242 bp); -: null (140 bp); +: wild-type allele (115 bp) of Rac1 gene. Note the presence of null allele in EC-Rac1+/- (Tie2-Cre Rac1c/+) showing the occurrence of homologous recombination in the tail vascular endothelium, whereas no such recombination is observed in inducible EC-Rac1 knockout (Tie2-CreERT2 Rac1c/c) without tamoxifen treatment. D, Immunoblotting analysis of Rac1 protein expression in the MHECs isolated from EC-Rac1+/- and control (Rac1c/+) mice (left panels). Right upper panel, the level of GTP-bound Rac1 as assessed by PAK-CRIB domain pull down assay. Right lower panel, NADPH oxidase activity determined as lucigenin chemiluminescence inhibitable by DPI (10 μmol/L). *P<0.05. Data are mean±SEM.
Inhibition of Endothelial Rac1 Downregulates Endothelial Nitric Oxide Synthase
We first investigated the effect of endothelial Rac1 haploin-sufficiency on the expression and activity of eNOS in mice organs. Quantitative reverse transcriptase-polymerase chain reaction and Northern blot analysis revealed that the eNOS mRNA in the aorta and brain of EC-Rac1+/- mice was decreased by 40% to 60% as compared with controls (Figure 2A-B). Western blot analysis further demonstrated reduced expression of eNOS protein in EC-Rac1+/- heart (Figure 2C). Consistent with the reduced expression of eNOS, the eNOS activity in EC-Rac1+/- aortas was decreased by 25% (Figure 2D). Furthermore, the level of cGMP, the downstream effector of NO signaling, was attenuated by 40% in EC-Rac1+/- aortas.
Figure 2.
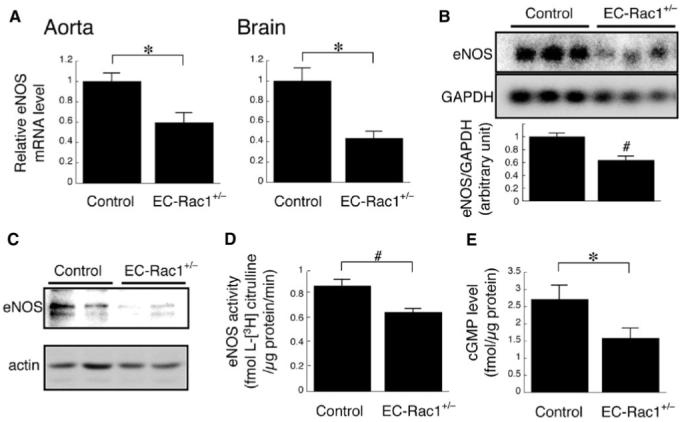
Reduced expression and activity of eNOS in EC-Rac1+/- mice. A, Quantitative reverse transcriptase-polymerase chain reaction analysis of the eNOS mRNA in the aorta and brain from EC-Rac1+/- (n=6) and control mice (n=5). B, Northern blot showing the eNOS mRNA expression in brain poly (A)+ RNA. The bar graph shows densitomentric quantification. C, Western blot showing the eNOS protein expression in the heart. D, eNOS activity in the aorta as determined by l-[3H] arginine to l-[3H] citrulline conversion (n=7 for each group). E, cGMP level in the aorta as determined by enzyme-linked immunosorbent assay (n=6 for each group). *P<0.05; #P<0.01. Data are mean±SEM.
Inhibition of Endothelial Rac1 Blunts Endothelial Regulation of Vascular Tone
In agreement with the downregulation of eNOS, Rac1+/- MHECs showed significantly diminished NO release after stimulation with Ca++ ionophore and acetylcholine (ACh; Figure 3A). To ascertain the physiological relevance of Rac1-mediated eNOS regulation, we investigated ex vivo the vasomotor response of mice aortic rings. EC-Rac1+/- aorta contracted more intensely to phenylephrine than control (Figure 3B). Blockade of NOS with l-NAME had little effect on the contractility of EC-Rac1+/- aorta but potentiated that of control to a similar extent as EC-Rac1+/-. In addition, l-NAME by itself evoked contraction in control mice aorta more profoundly than in EC-Rac1+/- (see Supplementary Data, Figure S1A). ACh-induced endothelium-dependent relaxation was markedly depressed in EC-Rac1+/- aorta compared to control (Figure 3C). Importantly, the diminished endothelium-dependent relaxation was recapitulated in tamoxifen-induced EC-Rac1 KO mice aorta, confirming the defective NO release from the EC-Rac1+/- endothelium (Figure 3D).
Figure 3.
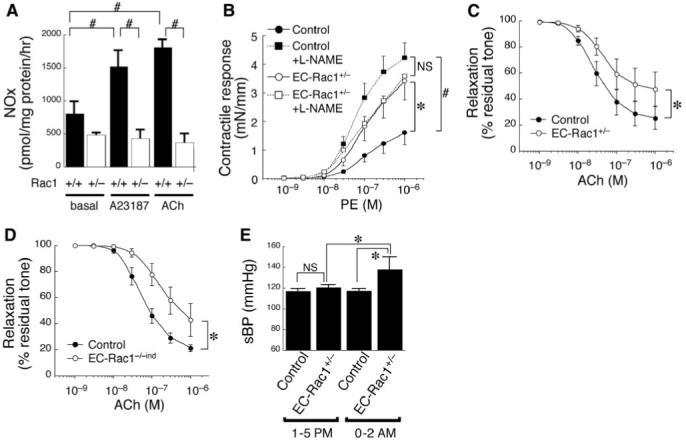
EC-Rac1+/- mice exhibit impaired endothelium-dependent vasomotor response and mild hypertension. A, NO production from Rac1+/+ and Rac1+/- MHECs as determined by the cumulative NOx level (nitrite and nitrate) in the culture supernatant without or with 30-minute stimulation with A23187 (10 μmol/L) or ACh (10 μmol/L). Data are mean±SEM from triplicate experiments. B-C, Vascular reactivity of EC-Rac1+/- and control mice aortic rings to phenylephrine without or with l-NAME (B, n=8 for each group) and ACh (C, n=8). D, Vascular reactivity to ACh of inducible EC-Rac1 knockout (EC-Rac1-/-ind, n=5) and control mice aortas (n=8) treated with tamoxifen. E, Systolic blood pressure (sBP) of EC-Rac1+/- (n=14) and control mice (n=12) was measured by the tail cuff method at diurnal resting period (1:00 to 5:00 pm) and nocturnal active period (12:00 to 2:00 am). *P<0.05; #P<0.01. Data are mean±SEM.
Inhibition of Endothelial Rac1 Causes Subclinical Hypertension
Vascular contractile response is an important peripheral component of the regulatory system of blood pressure. Given the deteriorated vasomotor regulation by EC-Rac1+/- endothelium, we characterized the hemodynamic profile of these mice. The systolic blood pressure, as assessed by tail-cuff method, showed tendency of elevation in EC-Rac1+/- mice at the resting, daytime phase (1:00 to 5:00 pm). The genotypic difference in systolic blood pressure became highly significant during the active, nocturnal phase (12:00 to 2:00 am, 137.8±12.4 versus 117.2±2.2 mm Hg, P<0.002; Figure 3E; Supplementary Data, Table S2). The hypertension in EC-Rac1+/- mice is compatible with the exaggerated vascular contractility caused by endothelial dysfunction; however, the blood pressure elevation is modest and subclinical likely because, in part, of the sensitization in smooth muscle relaxation to NO in EC-Rac1+/- mice (see Supplementary Data, Figure S1B).
Rac1 and Endothelial Nitric Oxide Synthase Constitute an Essential Axis for Angiogenesis
Previous studies demonstrated the essential roles of Rac1 and eNOS in the angiogenic process.10,11 We, therefore, explored the pathophysiological link between Rac1 and eNOS in mediating angiogenesis using a mice model of hind limb ischemia. EC-Rac1+/- and control mice underwent surgical resection of the left femoral artery followed by continuous administration of l-NAME or vehicle up to 4 weeks (Figure 4A-B; Supplementary Data, Figure S2). EC-Rac1+/- mice showed substantial retardation in blood flow recovery and exacerbated clinical symptoms compared with controls, suggesting the required role of endothelial Rac1 in ischemia-induced angiogenesis. The blockade of NOS with l-NAME in control mice similarly blunted blood flow recovery, although to a lesser extent than the genetic inhibition of Rac1 in EC-Rac1+/- mice. Importantly, l-NAME had no additive effect with endothelial Rac1 inhibition to further aggravate the blood flow recovery in EC-Rac1+/- mice. This suggests that inhibition of endothelial Rac1 in EC-Rac1+/- mice diminishes neovascularization, in part through suppression of NO synthesis.
Figure 4.
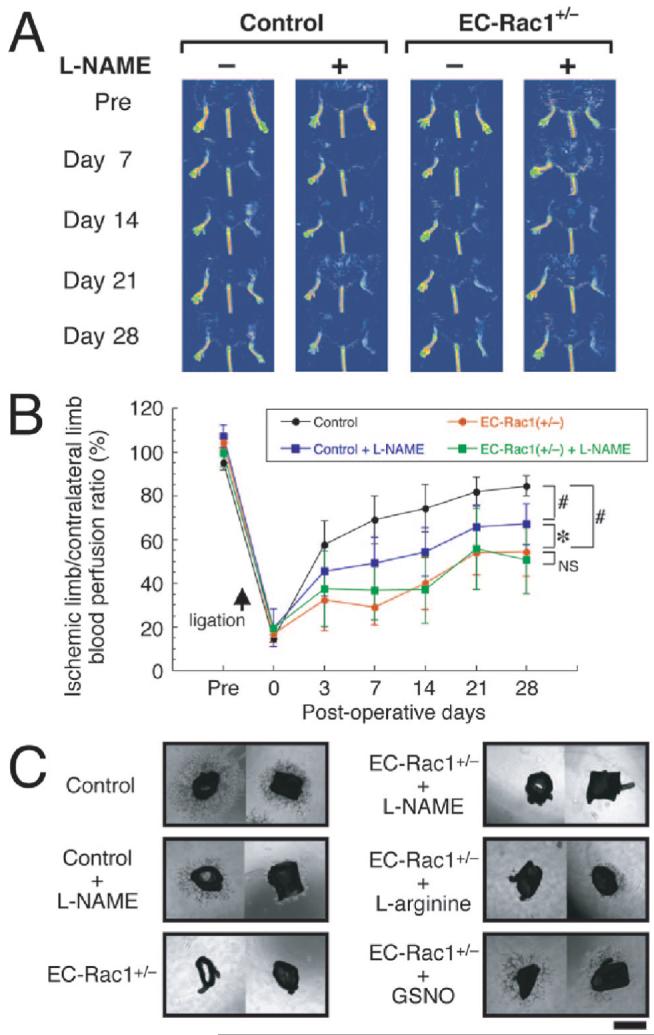
Essential role of Rac1-eNOS axis in neovascularization. A, Representative serial laser Doppler perfusion images (LDPI) after hind limb ischemia of the 4 experimental groups; control without l-NAME (n=8); control with l-NAME (n=7); EC-Rac1+/- without l-NAME (n=9); EC-Rac1+/- with l-NAME (n=9). B, Time course of ischemic limb/contralateral limb blood perfusion ratio in LDPI after hind limb ischemia. *P<0.05; #P<0.01. Data are mean±SEM. C, Microvessel sprouting was assessed using aortic explants from control (n=2) and EC-Rac1+/- mice (n=2) without or with 1 mmol/L l-NAME, 10 mmol/L l-arginine, or 100 μmol/L S-nitrosoglutathione. Scale bar=1 mm. Higher magnification images and quantitative analysis of the capillary area are found in Supplementary Data, Figure S3.
To further corroborate the role of eNOS in Rac1 signaling, we assessed the capillary-forming capacity of aortic explants in Matrigel as an ex vivo model of angiogenesis (Figure 4C; Supplementary Data, Figure S3). At day 8 after implantation, the aortic segments from control mice exhibited robust capillary formation. The vessel sprouting from control mice aortas was markedly inhibited by l-NAME, indicating the importance of nitric oxide synthase in this angiogenesis model. The sprouting from EC-Rac1+/- mice aorta was almost totally abolished, a finding consistent with the severely retarded blood flow recovery in hind limb ischemia. Importantly, supplementation with eNOS substrate l-arginine and S-nitrosoglutathione, an NO donor, partially but markedly restored the microvessel formation from EC-Rac1+/- aortas. Collectively, these results suggest that endothelial Rac1-eNOS axis constitutes an important pathway that mediates angiogenic response.
Rac1 Regulates Endothelial Nitric Oxide Synthase Expression at Transcriptional and Posttranscriptional Levels
To further ascertain the specificity of our finding that the genetic inhibition of endothelial Rac1 downregulates eNOS, we used different methods to inhibit Rac1 in cultured ECs (Figure 5A). Adenovirus-mediated transduction of dominant-negative Rac1 profoundly attenuated eNOS mRNA and protein levels in human aortic endothelial cells as compared with the control virus. In addition, treatment of murine microvascular endothelial cells (bEnd.3) with a specific Rac1 inhibitor NSC23766 (100 μmol/L) consistently suppressed the Rac1 mRNA expression as compared with vehicle up to 3 days.23 To study the mechanisms by which Rac1 regulates eNOS expression, we investigated the effects of NSC23766 on the eNOS promoter activity. Both dominant-negative Rac1 and NSC23766 repressed the eNOS promoter activity by 60% in bovine aortic ECs and by 30% to 35% in bEND.3 cells (Figure 5B). Importantly, transduction of dominant-negative PAK1 (K299R PAK1)24 in bEND.3 cells repressed the eNOS promoter activity with the similar potency as dominant-negative Rac1 and NSC23766. By contrast, an NADPH oxidase inhibitor apocynin and an antioxidant N-acetylcysteine did not decrease, but rather increased the eNOS promoter activity. These results indicate that Rac1 transactivates eNOS promoter most likely through PAK, and not a NADPH oxidase/ROS cascade. We next examined the effect of NSC23766 on the stability of eNOS mRNA in ECs treated with a transcriptional inhibitor 5,6-dichlorobenzimidazole 1-β-d-ribofuranoside (Figure 5C). The half-life of eNOS mRNA was 40 hours in bEND.3 cells untreated with NSC23766. Inhibition of Rac1 with NSC23766 destabilized eNOS mRNA and shortened its half-life to 17 hours.
Figure 5.
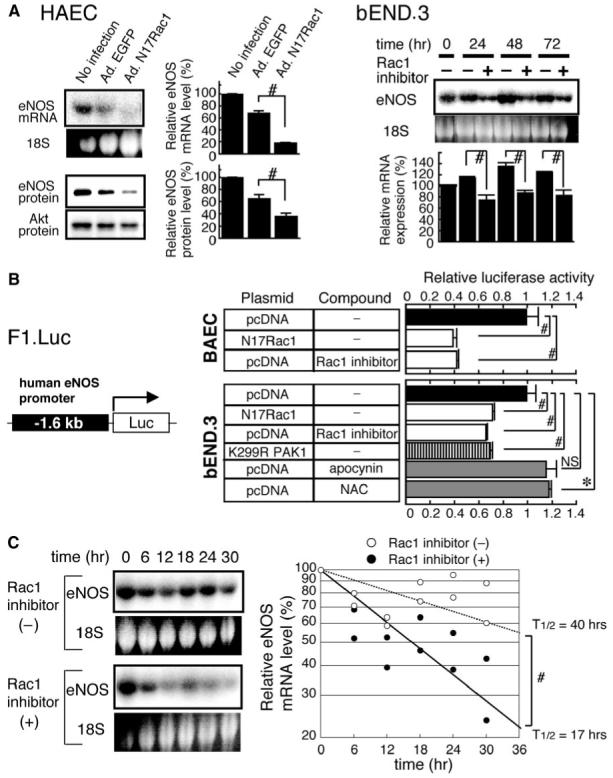
Rac1 mediates the expression of eNOS mRNA through transcriptional and posttranscriptional mechanisms. A, Effect of Rac1 inhibition on eNOS mRNA and protein levels in cultured ECs as assessed by Northern and immunoblot analyses. Left panel, human aortic endothelial cells (HAEC) were infected with adenoviruses encoding enhanced green fluorescent protein or dominant negative Rac1 (N17Rac1) at 50 MOI for 24 hours. Right panel, bEND.3 cells were treated without or with 100 μmol/L Rac1 inhibitor (NSC23766) for 24 to 72 hours. 18S: EtBr-stained bands corresponding to 18S rRNA. B, Effect of Rac1 inhibition on eNOS promoter activity. Left panel, the structure of luciferase reporter construct harboring -1.6 kb upstream sequence of human eNOS gene (F1. Luc). Right panel, the promoter activity of F1. Luc in BAEC and bEND.3 cells cotransfected with pcDNA3, N17Rac1, or dominant negative PAK1 (K299R PAK1) expression vector and subsequently treated with vehicle, Rac1 inhibitor, apocynin (30 μmol/L) or NAC (10 mmol/L). Data are mean±SEM of the relative luciferase activity from triplicate assays. C, Effect of Rac1 inhibition on eNOS mRNA stability. Confluent bEND.3 cells were treated with 5,6-dichlorobenzimidazole 1-β-d-ribofuranoside (50 μmol/L) in the absence or presence of 100 μmol/L Rac1 inhibitor for indicated periods. eNOS mRNA expression was assessed by Northern blot analysis (left) in duplicate and quantified by densitometry (right panel). *P<0.05; #P<0.01.
Pharmacological Blockade of Rac1 Acutely Worsens Endothelial Function
Although the decrease in eNOS expression explains the endothelial dysfunction caused by chronic inhibition of endothelial Rac1 in our genetic mice model, it does not exclude the possibility of Rac1 modulation of eNOS function at the posttranslational level. Indeed, 30-minute pretreatment with NSC23766 dose-dependently attenuated ACh-induced relaxation of wild-type mice aortic rings (Figure 6A). The inhibitory effect of NSC23766 on endothelium-dependent relaxation is not due to toxicity, because the endothelium-dependent relaxation was at least partially reversible by washout of NSC23766. The ability of Rac1 inhibitor to acutely suppress endothelium-dependent relaxation indicates that Rac1 mediates not only the expression of eNOS, but also the posttranslational maintenance of eNOS activity. Posttranslational regulation of eNOS involves multiple mechanisms, including the phosphorylation of eNOS by Akt at Ser1177. Previous studies demonstrated that Rac1 activates Akt in certain cell types.25 However, the phosphorylation level of Akt and eNOS was not decreased by 30-minute treatment of bEND.3 cells with NSC23766 as compared with the vehicle (Figure 6B).
Figure 6.
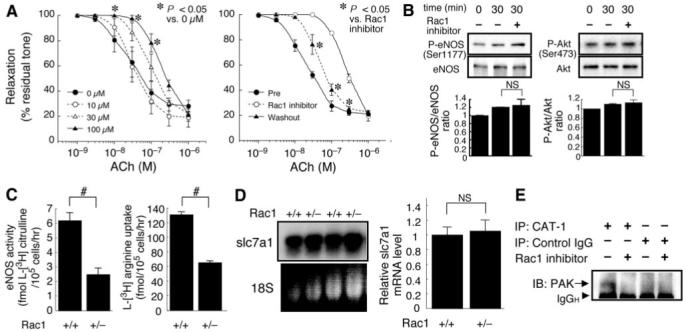
Rac1 promotes endothelial uptake of l-arginine and maintains eNOS function at the posttranslational level. A, Acute inhibition of Rac1 worsens endothelium-dependent vasorelaxation. Left panel, vascular reactivity to ACh was studied using wild-type mice aorta rings (n=3) pretreated with Rac1 inhibitor (0 to 100 μmol/L) for 30 minutes. Right panel, vasorelaxation to ACh was attenuated by Rac1 inhibitor (100 μmol/L) and partially restored following washout for 30 minutes. B, Effect of Rac1 inhibition on the phosphorylation of eNOS (Ser1177) and Akt (Ser473). bEND.3 cells were treated without or with 100 μmol/L Rac1 inhibitor for 30 minutes and harvested for immunoblotting analysis. C, Effect of Rac1 inhibition on endothelial uptake of l-arginine. Rac1+/+ and Rac1+/- MHECs were incubated with 5 μCi/mL (approximately 86.2 nM) l-[3H] arginine for 30 minutes. The eNOS activity and l-arginine uptake was determined as intracellular level of l-[3H] citrulline and l-[3H] arginine, respectively. D, The steady-state mRNA level of slc7a1 gene encoding CAT-1 in Rac1+/+ and Rac1+/- MHECs was determined by Northern blot (left) and real-time reverse transcriptase-polymerase chain reaction (right panel, n=6 for each group). E, Effect of Rac1 inhibition on the association of PAK with CAT-1. bEND.3 cells were treated without or with 100 μmol/L Rac1 inhibitor for 30 minutes. The cell lysates were immunoprecipitated with anti-CAT-1 antibody or IgG (negative control) and subjected to immunoblotting with anti-PAK antibody. *P<0.05; #P<0.01.
Inhibition of Rac1 Attenuates Endothelial Uptake of l-arginine
During the course of multiple measurements of eNOS activity in MHECs, we noticed that the decreased eNOS activity in Rac1+/- ECs was associated with reduced l-arginine uptake (Figure 6C). Because ECs import l-arginine through CAT-1, we examined the expression of slc7a1, the gene encoding CAT-1 (Figure 6D).26 There was no difference in slc7a1 mRNA expression between Rac1+/+ and Rac1+/- ECs, indicating that the reduced l-arginine uptake by Rac1+/- ECs was not caused by slc7a1 downregulation. To identify protein-protein interaction that may potentially mediate regulation of CAT-1 by Rac1, we performed coimmunoprecipitation studies with the bEND.3 cell lysates after 30-minute treatment with NSC23766 (Figure 6E). We found that PAK forms tertiary complex with CAT-1, and this association is dependent on Rac1 activity. Although the mechanistic relevance of Rac1-induced PAK binding to CAT-1 needs to be addressed by further investigation, it may constitute an underlying mechanism for the Rac1 mediation of endothelial CAT-1 activity and l-arginine uptake.
Discussion
Endothelial nitric oxide synthase is constitutively expressed in healthy vascular endothelium, and its downregulation is associated with various vascular diseases such as atherosclerosis.1,2 Therefore, understanding the mechanisms by which eNOS expression and activity is regulated is important for understanding the pathogenesis of vascular disease. Previous studies demonstrated that Rac1 GTPase mediates endothelial cell motility, proliferation, survival, and endothelial barrier formation7,10,12; all these processes are also known to involve eNOS.2,11 In addition, endothelial Rac1 is activated by and mediates cellular effects of shear stress and vascular endothelial growth factor,7,10,12 stimuli that increase the expression and activity of eNOS.2,11 These findings suggest a potential link between Rac1 and eNOS. Interestingly, inhibition of small GTP-binding proteins through isoprenoid depletion with statins has been shown to upregulate eNOS, possibly through inhibition of the Rho/Rho-kinase pathway.8 However, Rac1 is also isoprenylated and could also be affected by statin therapy. Therefore, it is not known whether inhibition of Rac1 is beneficial or detrimental for endothelial function in vivo.
Using a genetic approach, we showed that Rac1 is required for the maintenance of eNOS expression and activity and that eNOS is an important downstream mediator of Rac1 signaling in terms of angiogenesis. The EC-Rac1+/- mice have impaired endothelium-dependent vasodilation, mild hypertension, and retarded angiogenesis. Given that laminar shear stress is an important determinant of basal eNOS expression and activity, our finding that endothelial Rac1 is required for the maintenance of eNOS activity in vivo likely reflects the role of Rac1 in mediating shear regulation of eNOS.2,7 Indeed, the downregulation of eNOS in EC-Rac1+/- mice as compared with controls was more evident in organs than in isolated mice ECs cultured under static condition (NS and JKL, unpublished data). Another important function of Rac1 is the production of ROS through its effector NADPH oxidase, especially under pathological conditions such as chronically inflamed blood vessels. The ROS scavenges NO and diminishes NO bioavailability.12 Therefore, in the setting of such diseased states with high ROS production, reduction of ROS production through Rac1 inhibition could be beneficial for increasing overall NO bioavailability. However, our data suggest that the basal regulation of eNOS transcription by Rac1 is likely mediated by PAK rather than the NADPH oxidase-ROS cascade (Figure 5B). Hence, the mechanism by which Rac1 impacts endothelial function may differ in a context-dependent manner.
Our results suggest that Rac1 modulates eNOS at transcriptional, posttranscriptional, and posttranslational levels. Rac1 has been shown to regulate gene transcription through a diverse set of transcription factors, including NF-κB and AP-1.27,28 NF-κB is responsible for at least part of the shear-induced eNOS gene transcription.29 Assuming that Rac1 physiologically mediates shear-induced eNOS upregulation, Rac1 may induce eNOS gene transcription through NF-κB. Our data suggest that PAK is the direct effector of Rac1 responsible for activation of eNOS promoter. Indeed, recent studies demonstrate a required role for PAK in the activation of NF-κB downstream of distinct stimuli.30 Therefore, it is likely that Rac1-PAK-NF-κB cascade represents an important signaling cascade for basal transcription of eNOS in vivo. The well-known mechanism that affects eNOS mRNA stability is actin cytoskeletal dynamics. Inhibition of actin polymerization by Rho/Rho-kinase inhibitors leads to stabilization of eNOS mRNA.8,9 We have found that Rac1+/- endothelial cells display reorganized actin cytoskeleton such that F-actin is localized in the cell cortical regions (NS and JKL, unpublished data). It is currently under investigation whether this alteration in actin cytoskeleton could also account for the destabilization of eNOS mRNA by inhibition of endothelial Rac1.
The finding that Rac inhibitor acutely worsens endothelial function without changing eNOS expression indicates that Rac1 also regulates eNOS activity at the posttranslational level. Because the reduced eNOS activity was associated with a similar extent of decrease in l-arginine uptake (Figure 6C), we postulate that Rac1 posttranslationally regulates eNOS activity, in part, by enhancing l-arginine transport through CAT-1. The activity of CAT-1 is modulated not only by its expression level, but also by the intracellular localization and interaction with other proteins.4,31 Indeed, localization of CAT-1 to membrane microdomain caveolae and association with eNOS is required for full activation of eNOS. We found that Rac1 effector PAK associates with CAT-1 in a Rac1-dependent manner. The Rac1-triggered formation of PAK-CAT-1 complex may alter the CAT-1 localization in the cell compartments and/or interaction with other proteins and thereby lead to augmented uptake of l-arginine through CAT-1. The relevance of the PAK-CAT-1 interaction in eNOS regulation, however, requires further investigation.
Acknowledgments
We thank Seo-Kyoung Hwang for technical assistance and Bernd Arnold (German Cancer Research Center, Heidelberg, Germany) for providing us with Tie2-CreERT2 mice. We are also grateful to Drs Anne J. Ridley, John G. Collard, and Gary M. Bokoch for the generous gifts of the adenovirus for N17Rac1, pGEX-2T-PAK-CRIB domain construct and K299R PAK1 expression vector, respectively.
Sources of Funding
This work was support by grants from the National Institutes of Health (HL052233 and HL080187). NS was supported by fellowships from the Uehara Memorial Foundation, the Cell Science Research Foundation, and Mochida Memorial Foundation for Medical and Pharmaceutical Research.
Footnotes
Disclosures
None.
References
- 1.Landmesser U, Hornig B, Drexler H. Endothelial function: a critical determinant in atherosclerosis? Circulation. 2004;109(suppl 1):II27–33. doi: 10.1161/01.CIR.0000129501.88485.1f. [DOI] [PubMed] [Google Scholar]
- 2.Tai SC, Robb GB, Marsden PA. Endothelial nitric oxide synthase: a new paradigm for gene regulation in the injured blood vessel. Arterioscler Thromb Vasc Biol. 2004;24:405–412. doi: 10.1161/01.ATV.0000109171.50229.33. [DOI] [PubMed] [Google Scholar]
- 3.Dudzinski DM, Igarashi J, Greif D, Michel T. The regulation and pharmacology of endothelial nitric oxide synthase. Annu Rev Pharmacol Toxicol. 2006;46:235–276. doi: 10.1146/annurev.pharmtox.44.101802.121844. [DOI] [PubMed] [Google Scholar]
- 4.McDonald KK, Zharikov S, Block ER, Kilberg MS. A caveolar complex between the cationic amino acid transporter 1 and endothelial nitric-oxide synthase may explain the ‘arginine paradox.’. J Biol Chem. 1997;272:31213–31216. doi: 10.1074/jbc.272.50.31213. [DOI] [PubMed] [Google Scholar]
- 5.Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–179. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 6.Wojciak-Stothard B, Ridley AJ. Shear stress-induced endothelial cell polarization is mediated by Rho and Rac but not Cdc42 or PI 3-kinases. J Cell Biol. 2003;161:429–439. doi: 10.1083/jcb.200210135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tzima E. Role of small GTPases in endothelial cytoskeletal dynamics and the shear stress response. Circ Res. 2006;98:176–185. doi: 10.1161/01.RES.0000200162.94463.d7. [DOI] [PubMed] [Google Scholar]
- 8.Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem. 1998;273:24266–24271. doi: 10.1074/jbc.273.37.24266. [DOI] [PubMed] [Google Scholar]
- 9.Takemoto M, Sun J, Hiroki J, Shimokawa H, Liao JK. Rho-kinase mediates hypoxia-induced downregulation of endothelial nitric oxide synthase. Circulation. 2002;106:57–62. doi: 10.1161/01.cir.0000020682.73694.ab. [DOI] [PubMed] [Google Scholar]
- 10.Soga N, Namba N, McAllister S, Cornelius L, Teitelbaum SL, Dowdy SF, Kawamura J, Hruska KA. Rho family GTPases regulate VEGF-stimulated endothelial cell motility. Exp Cell Res. 2001;269:73–87. doi: 10.1006/excr.2001.5295. [DOI] [PubMed] [Google Scholar]
- 11.Murohara T, Asahara T, Silver M, Bauters C, Masuda H, Kalka C, Kearney M, Chen D, Symes JF, Fishman MC, Huang PL, Isner JM. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J Clin Invest. 1998;101:2567–2578. doi: 10.1172/JCI1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gregg D, Rauscher FM, Goldschmidt-Clermont PJ. Rac regulates cardiovascular superoxide through diverse molecular interactions: more than a binary GTP switch. Am J Physiol Cell Physiol. 2003;285:C723–734. doi: 10.1152/ajpcell.00230.2003. [DOI] [PubMed] [Google Scholar]
- 13.Glogauer M, Marchal CC, Zhu F, Worku A, Clausen BE, Foerster I, Marks P, Downey GP, Dinauer M, Kwiatkowski DJ. Rac1 deletion in mouse neutrophils has selective effects on neutrophil functions. J Immunol. 2003;170:5652–5657. doi: 10.4049/jimmunol.170.11.5652. [DOI] [PubMed] [Google Scholar]
- 14.Koni PA, Joshi SK, Temann UA, Olson D, Burkly L, Flavell RA. Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J Exp Med. 2001;193:741–754. doi: 10.1084/jem.193.6.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forde A, Constien R, Grone HJ, Hammerling G, Arnold B. Temporal Cre-mediated recombination exclusively in endothelial cells using Tie2 regulatory elements. Genesis. 2002;33:191–197. doi: 10.1002/gene.10117. [DOI] [PubMed] [Google Scholar]
- 16.Lim YC, Garcia-Cardena G, Allport JR, Zervoglos M, Connolly AJ, Gimbrone MA, Jr, Luscinskas FW. Heterogeneity of endothelial cells from different organ sites in T-cell subset recruitment. Am J Pathol. 2003;162:1591–1601. doi: 10.1016/S0002-9440(10)64293-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sander EE, van Delft S, ten Klooster JP, Reid T, van der Kammen RA, Michiels F, Collard JG. Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell-cell adhesion or cell migration and is regulated by phosphatidylinositol 3-kinase. J Cell Biol. 1998;143:1385–1398. doi: 10.1083/jcb.143.5.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Satoh M, Ogita H, Takeshita K, Mukai Y, Kwiatkowski DJ, Liao JK. Requirement of Rac1 in the development of cardiac hypertrophy. Proc Natl Acad Sci U S A. 2006;103:7432–7437. doi: 10.1073/pnas.0510444103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolfrum S, Dendorfer A, Rikitake Y, Stalker TJ, Gong Y, Scalia R, Dominiak P, Liao JK. Inhibition of Rho-kinase leads to rapid activation of phosphatidylinositol 3-kinase/protein kinase Akt and cardiovascular protection. Arterioscler Thromb Vasc Biol. 2004;24:1842–1847. doi: 10.1161/01.ATV.0000142813.33538.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Igarashi J, Bernier SG, Michel T. Sphingosine 1-phosphate and activation of endothelial nitric-oxide synthase. Differential regulation of Akt and MAP kinase pathways by EDG and bradykinin receptors in vascular endothelial cells. J Biol Chem. 2001;276:12420–12426. doi: 10.1074/jbc.M008375200. [DOI] [PubMed] [Google Scholar]
- 21.Salomone S, Yoshimura S, Reuter U, Foley M, Thomas SS, Moskowitz MA, Waeber C. S1P3 receptors mediate the potent constriction of cerebral arteries by sphingosine-1-phosphate. Eur J Pharmacol. 2003;469:125–134. doi: 10.1016/s0014-2999(03)01731-x. [DOI] [PubMed] [Google Scholar]
- 22.Yamahara K, Itoh H, Chun TH, Ogawa Y, Yamashita J, Sawada N, Fukunaga Y, Sone M, Yurugi-Kobayashi T, Miyashita K, Tsujimoto H, Kook H, Feil R, Garbers DL, Hofmann F, Nakao K. Significance and therapeutic potential of the natriuretic peptides/cGMP/cGMP-dependent protein kinase pathway in vascular regeneration. Proc Natl Acad Sci USA. 2003;100:3404–3409. doi: 10.1073/pnas.0538059100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A. 2004;101:7618–7623. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sells MA, Knaus UG, Bagrodia S, Ambrose DM, Bokoch GM, Chernoff J. Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr Biol. 1997;7:202–210. doi: 10.1016/s0960-9822(97)70091-5. [DOI] [PubMed] [Google Scholar]
- 25.Higuchi M, Masuyama N, Fukui Y, Suzuki A, Gotoh Y. Akt mediates Rac/Cdc42-regulated cell motility in growth factor-stimulated cells and in invasive PTEN knockout cells. Curr Biol. 2001;11:1958–1962. doi: 10.1016/s0960-9822(01)00599-1. [DOI] [PubMed] [Google Scholar]
- 26.Hatzoglou M, Fernandez J, Yaman I, Closs E. Regulation of cationic amino acid transport: the story of the CAT-1 transporter. Annu Rev Nutr. 2004;24:377–399. doi: 10.1146/annurev.nutr.23.011702.073120. [DOI] [PubMed] [Google Scholar]
- 27.Ridley AJ. Rho family proteins: coordinating cell responses. Trends Cell Biol. 2001;11:471–477. doi: 10.1016/s0962-8924(01)02153-5. [DOI] [PubMed] [Google Scholar]
- 28.Perona R, Montaner S, Saniger L, Sanchez-Perez I, Bravo R, Lacal JC. Activation of the nuclear factor-kappaB by Rho, CDC42, and Rac-1 proteins. Genes Dev. 1997;11:463–475. doi: 10.1101/gad.11.4.463. [DOI] [PubMed] [Google Scholar]
- 29.Davis ME, Grumbach IM, Fukai T, Cutchins A, Harrison DG. Shear stress regulates endothelial nitric-oxide synthase promoter activity through nuclear factor kappaB binding. J Biol Chem. 2004;279:163–168. doi: 10.1074/jbc.M307528200. [DOI] [PubMed] [Google Scholar]
- 30.Frost JA, Swantek JL, Stippec S, Yin MJ, Gaynor R, Cobb MH. Stimulation of NFkappa B activity by multiple signaling pathways requires PAK1. J Biol Chem. 2000;275:19693–19699. doi: 10.1074/jbc.M909860199. [DOI] [PubMed] [Google Scholar]
- 31.Li C, Huang W, Harris MB, Goolsby JM, Venema RC. Interaction of the endothelial nitric oxide synthase with the CAT-1 arginine transporter enhances NO release by a mechanism not involving arginine transport. Biochem J. 2005;386:567–574. doi: 10.1042/BJ20041005. [DOI] [PMC free article] [PubMed] [Google Scholar]