

Abstract
A wide variety of bioluminescent luciferase proteins are available for use in transcriptional or biochemical reporter assays. However, spectral overlap normally prevents them from being monitored simultaneously. To address this problem, a Java plug-in for ImageJ was written to deconvolute bioluminescent images composed of signals from multiple luciferases. The methodology was validated by testing the program with both simulated and real luciferase images. Bioluminescent images were acquired using a CCD camera equipped with optical filters, and the images were deconvoluted using the ImageJ plug-in. HeLa cells were transfected with either click beetle red luciferase (CBR), click beetle green luciferase (CBG99), or Renilla luciferase (Rluc), and mixed lysates were imaged in varying proportions in a 96-well plate to biochemically validate the methodology. After spectral deconvolution, the predicted, pure luciferase signals could be recovered with maximal cross-talk errors of ±1.5%. In addition, live cells expressing CBR, CBG99, and Rluc were simultaneously imaged and deconvoluted in 96-well plates to demonstrate the feasibility of applying this methodology to high-throughput applications. Finally, multicolor transcriptional and posttranslational modification reporters were simultaneously imaged and shown to deconvolute normalized IκB kinase activity in longitudinal assays. Thus, our software provided a rapid, simple, and accurate method for simultaneously measuring multiple bioluminescent reporters in living cells.
Bioluminescent assays provide a platform for the study of numerous cellular processes, including promoter activity, cisacting elements, protein degradation, protein-protein interactions, and cell viability.1-7 Since one photon of light is emitted by the enzymatic turnover of a single substrate, bioluminescence reporters can provide a noninvasive, quantitative, real-time readout of cellular activities.
Luciferase activity can be assayed under selected conditions in 96-well plate formats with either live cells or cell lysates and can be monitored by luminometers or cooled CCD cameras. While luciferases are convenient biosensors,2-6,8 variations in photon signal from bioluminescence reporter constructs may reflect changes in cell mass, viability, or transfection efficiency rather than changes in reporter activity. However, a convenient method to normalize reporter data in a single, facile step does not exist. Current protocols call for normalizing photon emission to total cell protein or DNA, each of which requires cell lysis9,10 and limits the ability to conduct repetitive longitudinal studies. Other normalization procedures use mitochondrial activity, e.g., 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide as a reflection of mass, cell viability, or both.11 These procedures cause a permanent color change in cells or lysates and hence can be utilized only once upon termination of the protocol to avoid spectral overlap and nonlinear absorption artifacts. Investigators also can normalize for DNA transfection by cotransfecting with a second plasmid, often encoding for Renilla luciferase. Quantifying the Renilla luciferase activity for each transfection allows the researcher to normalize for differences in DNA delivery and cell viability.12 However, this method utilizes expensive reagents, such as coelenterazine, is laborious, and also often requires cell lysis.
Spectral unmixing is a technique that uses filtered images to separate and quantify individual components comprising a composite image. This method has a long history in remote sensing and fluorescence microscopy. For example, satellites equipped with multiple band-pass filters have been used to monitor mineral content and vegetation coverage on earth.13 Many of the problems and proposed solutions for spectral unmixing in remote sensing have been extensively reviewed by Keshava and Mustard.14 In addition, multiple fluorescent species with overlapping emission spectra have been readily separated by using spectral unmixing techniques that are now included in many commercial optics software packages.15 Furthermore, quantum dots with different fluorescent spectra have been used to tag and resolve cells in vivo.16 We herein describe the theory, a software algorithm, and several applications of a new spectral unmixing technique applicable to bioluminescence.
MATERIALS AND METHODS
Imaging
All images were acquired on an IVIS 100 (Xenogen, Alameda, CA) equipped with a stage heated to 37 °C unless otherwise noted. Images were acquired for the time indicated at binning 8 × 8 pixels (spatial x-y), f/stop 1, 10-cm field of view, and with the indicated filter sets. All plates were black-walled with clear bottoms. Images are corrected for camera noise and lens distortion by the Xenogen software. The 32-bit bioluminescence output image calculated by Living Image v 2.5 was used for all spectral unmixing calculations.
Reagents
Acetyl CoA (Sigma, St. Louis, MO) stock solutions (1 mM) were prepared in Milli-Q water and stored at -80 °C. Sodium phosphate monobasic, sodium phosphate dibasic, and sodium ATP were purchased from Sigma and stored as recommended. Promega Passive Lysis Buffer 5× was purchased from Promega (Fitchburg, WI) and stored at -20 °C. Firefly d-luciferin (Biosynth, Naperville, IL) was dissolved in Milli-Q water (100 mM final) and stored at -20 °C. Coelenterazine (Biotium, Hayward, CA) was dissolved in acidic methanol (4.7 mM final). Gelatin type A from porcine skin was purchased from Sigma and dissolved to 20 mg/mL in Milli-Q water. This stock was stored at 4 °C until immediately before use and then melted at 37 °C before dilution into assay buffer or lysis buffer. Fugene 6 was purchased from Roche (Alameda, CA).
pCBR, pCBG99, and phRluc plasmids were all purchased from Promega. pIkB-CBG99 was constructed exactly as the pIkB-Fluc construct published by Gross and Piwnica-Worms, except pCBG99 was used in place of pGL3 as the source for the luciferase.17
Tissue Culture
All cells were cultured in vented flasks at 37 °C (Fisher Scientific, Pittsburgh, PA) in a 5% CO2, 20% O2, 75% N2, water-saturated atmosphere. HeLa and 293T cells were cultured in DMEM (Invitrogen, Carlsbad, CA) supplemented with 10% v/v heat-inactivated fetal bovine serum and 2 mM glutamine. Imaging assays were conducted in 96-well plates with black walls and clear bottoms to minimize photon cross talk between wells (Fisher Scientific Int., Hampton, NH). Imaging medium refers to DMEM lacking phenol red and supplemented with 10% v/v heat-inactivated fetal bovine serum and 2 mM glutamine. MEBSS refers to a minimal salt solution containing 144 mM NaCl, 5.4 mM KCl, 0.8 mM MgSO4, 0.8 mM NaH2PO4, 1.2 mM CaCl2, 5.6 mM α-d-glucose, and 4.0 mM HEPES buffer at pH 7.4.
Biolumunmixing v. 1.0 Software Development
The graphical user interface was built using code from the region of interest (ROI) Manager of ImageJ. The program will thus interface with any ROI analysis supported by ImageJ. Furthermore, any ROI used in Biolumunmixing v. 1.0 can be saved by the ROI Manager for later analysis.18 The linear unmixing algorithm utilizes the pseudoinverse function of the JAMA matrix algebra library, available for free download from the National Institutes of Standards and Technologies, to determine the least-squares solution of the unmixing problem.19 To determine the spectral response of each luciferase, the user draws a ROI around a control well expressing only one luciferase and adds it to the list of luciferases present in the image. This is repeated until one control well has been added to the list for each luciferase present in the image. After either the iterative or linear unmixing option has been selected, the spectral unmixing calculations are performed completely internally by the program. The program outputs a stack of images containing the deconvoluted pure light for each luciferase present in the image, as well as a map of the residuals. Furthermore, the conditional value of the spectral matrix is returned to quantitatively assess the error of the experiment.
The mathematics of the spectral unmixing are completely invisible to the end user but are calculated as follows. K is determined from the control wells indicated by the user. First, a set of normalized images is calculated by dividing each filtered image by the unfiltered image. The average value of Kij is calculated for each filter (i) and each luciferase (j) based upon the average intensity of the normalized images within the user-defined control well. The conditional value of K is then calculated and returned by the JAMA matrix algebra package.
Next, for the linear method, the images are converted into a matrix of dimensions number of filters × number of pixels. The JAMA matrix algebra package then calculates the pseudoinverse of K, Kinv, and multiplies this by Pixel, the image matrix, to obtain the least-squares solution for Luciferase, the luciferase image matrix:
| (1) |
The residuals are calculated by summing the intensities of all luciferases for a given pixel and subtracting this from the unfiltered image. This matrix is also returned as an image. If the algorithm were perfect and if no noise were present in any image, the residuals would be zero. For the iterative unmixing option, the least-squares solution found above is used as the initial estimation of Luciferase for further iterations of the EMML algorithm.20 Finally, for both methods, the matrices are converted back into images.
The Java code is given in the Supporting Information S1. The plug-in and instructions will be available publicly on the NIH’s ImageJ web site.
Testing the Software with Virtual Images
The sensitivity of Biolumunmixing v. 1.0 was assessed using virtual luciferase images with varying degrees of Gaussian noise. Three images were created in ImageJ each with a circle representing a “control well” and the words “RED”, “GREEN”, or “BLUE”. These images were combined into a stack of images. This stack of images was then multiplied by K containing values similar to those resulting from Renilla luciferase (blue), click beetle green (green), or click beetle red (red) spectral emissions. This results in a stack of images similar to one that may be acquired from a set of filtered bioluminescence images. Then, the original three images were overlaid on the top of the stack to represent the unfiltered image. Finally, Gaussian noise was added at a signal-to-noise ratio (S/N) of 1000:1, 100:1, 10:1, and 1:1 relative to the intensity in the original “unfiltered” image. These noisy images were then unmixed using Biolumunmixing v. 1.0. Furthermore, the iterative unmixing algorithm was tested against images with 10:1 and 1:1 S/N.
Spectral Unmixing of Lysates
The 293T cells were transfected with either pCBG99, pCBR, or phRluc-N3 using Fugene 6 as per the manufacturer’s protocol (1 μg of plasmid DNA per well in a 6-well plate). Briefly, confluent 293T cells from a T-25 flask were trypsinized, diluted 1/10 into media (DMEM, 10% v/v heat-inactivated fetal bovine serum, 2 mM glutamine), and 2 mL were plated per well in a six-well plate. One well was not transfected as a negative control.
Transfection mixture (100 μL) was added dropwise to each well, and the plates were incubated for 1 day. The medium was then removed, and the cells were washed twice with 2 mL of solution A (0.2 mg/mL potassium chloride, 0.2 mg/mL potassium phosphate monobasic, 8 mg/mL sodium chloride, 1.15 mg/mL sodium phosphate dibasic provide by Washington University Tissue Culture Support Center, St. Louis, MO). Cells were then stored at -80 °C. For analysis, cells were lysed for 10 min at room temperature with 0.5 mL of 1× Promega Passive Lysis Buffer containing an additional 2 mg/mL gelatin and 1× Complete Protease Inhibitor Mix without EDTA (Roche). Cell debris was pelleted by centrifugation at 14 000 rpm for 3 min at 4 °C.
The linear combinations of luciferases may be represented as a “luciferase space”. For three luciferases, this space can be represented as a triangle with each vertex corresponding to 100% pure luciferase and the center of the triangle representing a mixture of 1/3 of each luciferase. To sample all regions of luciferase space, one generally assays several critical areas as laid out in Figure 1.14
Figure 1.

Representation of luciferase image space. This triangle and interior points represent all possible combinations of three different luciferases. By sampling the triangle at the points indicated by either a rectangle or a circle, one has sampled each possible region in luciferase image space.
Each luciferase was diluted to a similar level of activity to ensure that any error in one luciferase channel did not overwhelm signals from the others. Each midpoint of a side of the triangle was then sampled in triplicate by adding 5 μL of one luciferase lysate to 5 μL of lysate from another luciferase in a black-walled 96-well plate. Similarly, to test the center of the triangle, 3.3 μL aliquots of all three luciferases were added together. Furthermore, as a control to determine how much activity should be present in the pure light from each luciferase in the mixed wells, each luciferase was diluted 2- and 3-fold with untransfected cell lysate. Finally, as a negative control, three wells received 10 μL of untransfected cell lysate.
A 90-μL aliquot of luciferase assay buffer (4.7 μM coelenterazine, 500 μM d-luciferin, 1.5 mM MgCl2, 300 μM ATP, 1 mM d/l-dithiothreitol, 10 μM acetyl CoA, 2 mg/mL gelatin, 100 mM sodium phosphate, pH 7.9) was then added to each lysate mixture on the plate using a 12-channel pipet. The plate was then imaged at 25 °C for 10 s per filter, with sequential filters: open, <510-nm short pass, 540 ± 20 nm band-pass, and >650-nm long pass. The robustness of the unmixing was tested in two independent sets of transfections and images.
Triple Color Imaging for High-Throughput Screening in Live Cells
Confluent HeLa cells were split 1/7 into 11 mL of culture media. The 100-μL aliquots of the diluted cells were plated into wells A1-A4 in a black-walled 96-well plate. Wells A1-A3 were transfected using Fugene 6 with pCBR, pCBG99, or phRluc-N3 as per the manufacturer’s protocol; A4 served as an untransfected control for background coelenterazine chemiluminescence. Diluted cells (10 mL) were transfected with the following mix: 1 mL of DMEM, 45 μL of Fugene 6, 5 μg of pCBR, 5 μg of pCBG99, and 0.5 μg of phRluc-N3. The 100-μL samples of these cells were then plated using an electronic 12-channel pipet into the remaining wells of the 96-well plate. After 24 h, the wells were washed twice with 100 μL of prewarmed (37 °C) MEBSS. A separate plate was treated as above but was not transfected with any luciferases in wells A5-H12 in order to serve as the negative control for determining S/N and the z-factor (Z’; used for evaluating the robustness of high-throughput screens).21
Immediately prior to imaging, 100 μL of prewarmed (37 °C) MEBSS with 200 nM coelenterazine and 500 μM d-luciferin was added to the wells using a 12-channel pipet. The plate was immediately imaged for 30 s per filter with sequential filters: open, <510-nm short pass, 540 ± 20 nm band-pass, and >650-nm long pass. The images were then unmixed using Biolumunmixing v. 1.0. Light intensity from each well was quantified by ROI analysis using ImageJ and statistical analysis was performed in Microsoft Excel.
Dual Color Imaging of Live Cells
HeLa cells were split 1/10 into two 100-mm dishes and grown until ∼70% confluent. HeLa cells were then transfected with 10 μg of pCBR or pCBG99 using Fugene 6 as per the manufacturer’s protocol. Sixteen hours later, cells were trypsinized and resuspended in imaging media. The cells were then counted on a hemocytometer and diluted in imaging media to a concentration of 750 000 cells/mL. d-Luciferin stock was added such that the final concentration was 500 μM. Then, using an 8-channel pipet, a gradient of cells transfected with pCBR was plated into 6 columns of a 96-well plate. Similarly, a gradient of cells transfected with pCBG99 was plated quickly into six columns of the same plate. Three of these columns overlapped with CBR transfected cells. Imaging media with 500 μM d-luciferin was added to appropriate wells to maintain a volume of 100 ± 2 μL across the entire plate.
The plate was then placed in the IVIS 100 for 10 min to equilibrate to 37 °C. The plate was then imaged for 30 s per filter with sequential filters: open, 540 ± 20 nm band-pass, and >650-nm long pass. The images were unmixed using Biolumunmixing v. 1.0 and quantified using ImageJ. GraphPad Prizm v. 4.03 was used to plot the data and check for statistical differences in the slope of the spectrally unmixed gradients. This assay was repeated with independent transfection and imaging sessions to validate the robustness of the assay.
Imaging IKK Activity Using an IκBα-CBG99 Fusion Construct
HeLa cells were diluted 1/10 into culture media and plated into 24-well plates. The next day, the medium was changed and cells were immediately transfected. A pool of 2.1 μg of pCBR and 2.1 μg of pIμBα-CBG99 was added to 440 μL of DMEM and 12 μL of Fugene 6. Twenty microliters of this transfection mix was added to each well to be assayed in the plate. Finally, 0.2 μg of either pCBR or pCBG99 was transfected into control wells using Fugene 6 as per the manufacture’s protocol.
After a 24-h incubation, the medium was again changed and replaced with 500 μL of MEBSS containing 500 μM d-luciferin. After equilibrating the plate in the IVIS 100, a set of images was acquired. Wells were treated in triplicate with either 0.5 μL of TNFα (10 μg/mL) in solution A with 1% BSA, 0.5 μL of solution A with 1% BSA as a vehicle control, or no treatment. Sets of images were then acquired at the time points indicated in Figure 7. Each time point was then unmixed using Biolumunmixing v. 1.0. At each time point, the CBG99/CBR ratio was calculated, and data were processed as in Gross and Piwnica-Worms.17
Figure 7.

Effects of signaling ligands on IKK activity. HeLa cells were transiently transfected with IκB-CBG99 and CBR and were then treated with either TNFα, TNFα vehicle, or not treated. They were then imaged at the indicated time points with no filter, 540-nm band-pass filter, or >650-nm long-pass filter. After unmixing, the images were quantified, and the CBG/CBR ratio was normalized to time 0 and expressed as percent of untreated control.
RESULTS
In theory, the photon output captured by a CCD camera from a multiple luciferase assay can be represented by a set of linear equations:
| (2) |
| (3) |
| (4) |
The measured intensity in each pixel of a bioluminescent image contains the sum of the light from all the different luciferases emitting in that pixel (eq 2). If the image is reacquired a short time later with a filter of certain bandwidth, each pixel remains the sum of the light from all of the luciferases present emitting within that bandwidth (eq 3). Thus, a constant, K(filter,luciferase), defines the fractional light from a given luciferase transmitted through a given filter. The emission spectrum of each luciferase, the spectral response of the instrument, and the transmittance of the optical filter all contribute to the value of Kij. Each image acquired with a new filter will generate a new equation. Equation 3 can be rewritten in matrix format (eq 4). Pixel is a matrix defined to contain a row for each filtered image and a column for each pixel in the image; the elements contain the intensity of detected light. K is a spectral response matrix with a row for each different filter used to acquire the images and a column for each different luciferase present in the image. The elements of this matrix represent the fraction of the total light from an individual luciferase which is detected in the image with an individual filter. Luciferase is a matrix of unknown light intensity. The rows correspond to each luciferase present in the image, and the columns represent the pixels in the image. The sum of the elements in an individual column of Luciferase is equal to the light intensity from an unfiltered image for a given pixel. Error is a matrix that contains the stochastic uncorrelated noise from the detector.
Virtual luciferase images were created using the words “Red”, “Green”, and “Blue” to test the unmixing algorithm. Each word was given spectral characteristics similar to those found in real luciferases and then overlaid to form a composite image. The program was tested against these images with varying degrees of Gaussian noise added to the open and filtered images (Figure 2). The algorithm readily deconvoluted images down to 10:1 S/N but failed at 1:1.
Figure 2.
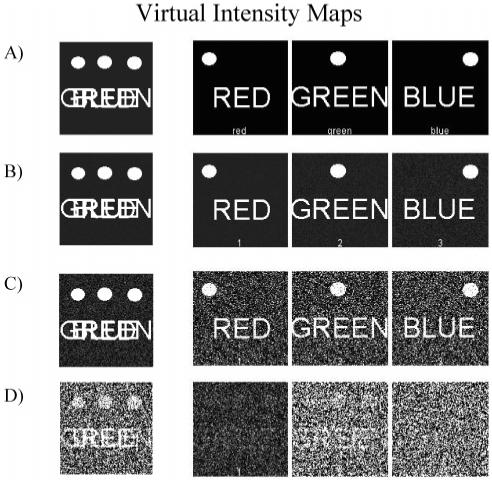
Unmixing of virtual intensity maps. The “unfiltered” images are on the left. The unmixed images are on the right. Each image is labeled with a number corresponding to the “luciferase” by the program to facility image identification. (A) The starting set of images that should be recaptured by the software. (B-D) The unmixed images after Gaussian noise added at 100:1, 10:1, and 1:1 S/N. The results in each column should match (A).
Due to the sensitivity of commercially available CCD cameras, a S/N of 10:1 is readily achieved in a laboratory setting. Therefore, the software and unmixing algorithm should be compatible with standard luciferase assays. When virtual images were unmixed using the iterative algorithm, calculation time was increased significantly from under a second to several minutes with no improvement in the image. Although the iterative algorithm will remain with the software, for most luciferase assays, the linear unmixing algorithm should suffice, and it is advisable to acquire images for a length of time such that the lowest expected signal has a S/N of 10:1.
In an actual bioluminescence experiment, several other variables will affect the ability to accurately deconvolute images. First, the bursting kinetics of Renilla luciferase photon emission are often more rapid than those of the click beetle luciferases; thus, one must either assay both at room temperature to slow bursting activity or, for live cells, transfect significantly less Renilla luciferase. Second, coelenterazine chemiluminesces in the presence of BSA or in micelles, so appropriate assay and cell lysis buffers must be chosen.22 Next, dispensing viscous cell lysates adds variability to the assays. Other sources of variability should be taken into account by the experimental controls used to calculate K. For example, the emission spectra of luciferase enzymes are known to shift with temperature. However, because temperatures are nearly uniform in all of the areas within a plate, any shifts in the spectrum should be taken into account. Furthermore, if there is a global time-dependent decrease or increase in bioluminescence from one filtered frame to the next, this will be normalized by K as well.
293T cells were transfected with plasmids encoding CBR, CBG99, or Renilla luciferase plasmids to determine whether spectral unmixing could work in a real laboratory setting that would include the described sources of variance. Lysates with similar levels of luciferase activity (within 2-fold) were mixed in a 96-well plate as shown in Figure 3. The images for each luciferase were then calculated by Biolumunmixing v. 1.0 using the linear unmixing algorithm. Upon visual inspection of the unmixed images, the relative proportions of each luciferase were discerned immediately. The ability to visually inspect the images for activity is potentially useful in large screening assays where time is more critical than calculating precise photon output from each well.
Figure 3.

Unmixing lysates. (A) Layout of the 96-well plate used to biochemically validate the spectral unmixing program. The colors around each well indicate the predicted color of the light output from those wells. (B) Bioluminescent images of lysates acquired with no filter, a <510-nm short-pass filter, a 540-nm band-pass filter, or a >650-nm long-pass filter. The next row contains spectrally unmixed images of CBR, CBG99, or Renilla. (C) A false color image generated by mapping click beetle red to the red channel, click beetle green to the green channel, and Renilla to the blue channel. Note that the colors in the mixed wells correspond almost exactly to those of the predicted colors.
Although visually pleasing, full quantification of bioluminescence images is important. The calculated intensity of light from each well in the luciferase images was quantified using ROI analysis in ImageJ (Figure 4). Within the variance of the experiment, there is no difference between the unmixed wells and predicted wells. Interestingly, there is a small (1.5%), but consistent, cross talk between the Renilla channel and the CBG99 channel. The differences between the predicted and the unmixed wells range from 0 to 20%.
Figure 4.
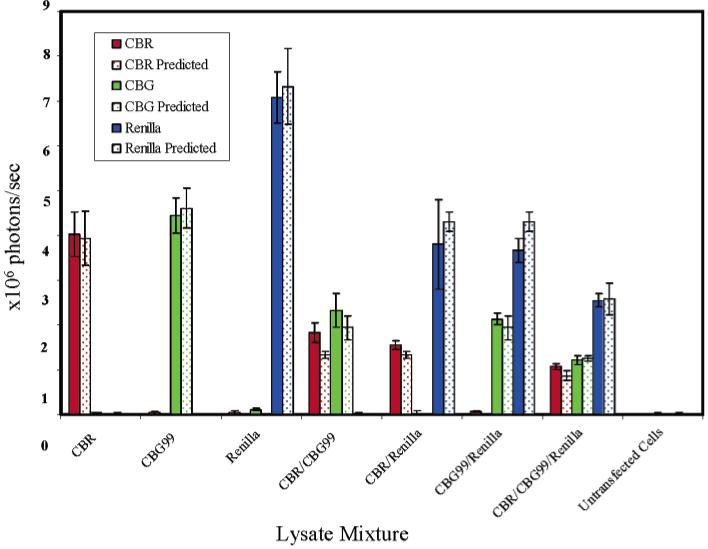
Quantification of spectral unmixing of CBR, CBG99, and Renilla lysates. Error bars are given at one standard deviation. If the delivery of the lysates to the wells and the spectral unmixing were perfect, the solid bars (unmixed) would exactly match their correspondingly colored dotted bars (control).
Another standard use for luciferase assays is to calculate promoter activity in living cells. Thus, HeLa cells were transiently transfected with plasmids encoding CBG99 and CBR, under the control of the SV40 promoter, and Renilla luciferase under control of the cytomegalorvirus promoter. Under control of these promoters, the luciferases are constitutively expressed. A plate of untransfected cells was treated in parallel as a negative control. Across the 92 assayed wells, there were no areas of either systematically high or low activity, and the S/Ns (when compared to an untransfected plate) after unmixing were as follows: CBR, 291; CBG99, 68; Renilla, 320. The percent standard deviation and the z-factor (Z’) value were as follows: CBR, SD = 14.6%, Z’=0.54; CBG99, SD = 13.4%, Z’ = 0.53; Renilla, SD = 25.7%, and Z’ = 0.22. While Renilla luciferase has the highest value of S/N, the signal itself is also much more variable than either the CBG or the CBR luciferases. For assays requiring maximum sensitivity, the Renilla family of luciferases may be advantagous. However, for those assays where precision is critical, the click beetle family luciferases are more appropriate. Thus, by utilizing the spectral unmixing software, 92 wells × 3 assays (one for each luciferase) were imaged in under 3 min, a significant advance over current luciferase assays.
Biolumunmixing v. 1.0 also was utilized to enumerate living cells in a mixed population. There are a number of reasons to study multiple cell types in coculture in the same well. For example, culturing endothelial cells in the presence of neurons and astrocytes cells induces further differentiation.23 Others may wish to culture cancer cells with stromal cells to more accurately represent in vivo biology in vitro.24,25 For these assays, the viability of each cell type might be quantified after addition of drug or receptor ligand. However, standard assays for either protein or mitochondrial viability cannot discriminate between cell type. By transfecting one cell line with a plasmid encoding CBR and another a plasmid encoding CBG99, the researcher may be able to quantify the viability of each cell type simultaneously in living coculture.
The feasibility of this assay was demonstrated using live HeLa cells transfected with either CBR or CBG99. Two gradients of CBR and two gradients of CBG99 were seeded on the same 96-well plate in opposite directions. One of the CBR gradients overlapped with one of the CBG99 gradients as shown in Figure 5. Thus, the gradients of CBR and CBG99 transfected cells alone serve as a control for the mixed gradient. After spectral unmixing, one can clearly identify the two gradients of cells in the control wells and in the mixed population. After quantification of these images, a standard curve was generated for the light output per cell from the control wells. The slopes of the lines (photons/s per cell) from the standard curves were not significantly different from the slopes obtained from the unmixed coculture wells, p = 0.52 and p = 0.83 (Figure 6). Thus, the number of cells from a mixed population may be quantified using spectral unmixing.
Figure 5.
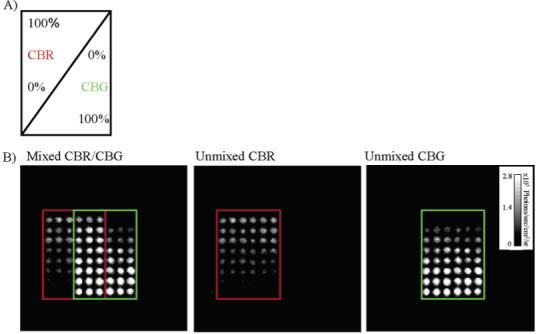
Resolving mixed population of cells. (A) HeLa cells transiently transfected with either CBG99 or CBR plasmids were counted and plated in crossing gradients on a 96-well plate. In one set of wells, the gradients were crossing each other as indicated above. In another set of wells, the identical gradient was plated alone to serve as a control. (B) Left: unfiltered bioluminescence image. Center and right: images after spectral unmixing. Boxes indicate where the gradients of CBR and CBG99 were plated.
Figure 6.
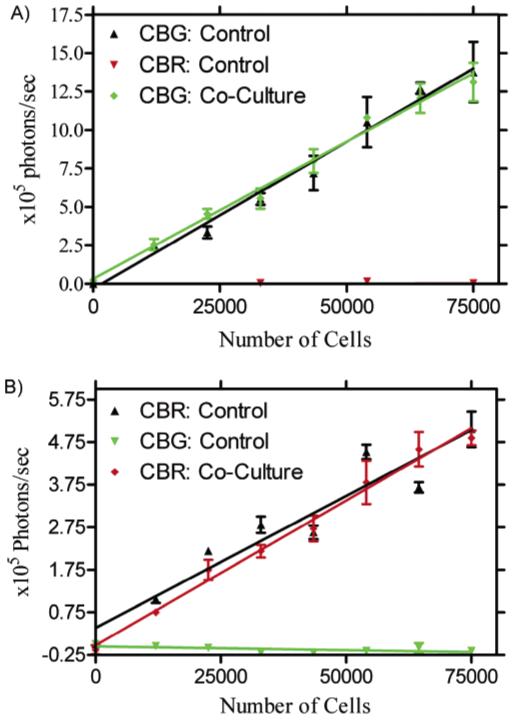
Quantification of the unmixed luciferase images for the crossing gradients of HeLa cells transfected with either CBR or CBG99. (A) Comparison of CBG99 photon output in control and coculture. (B) Comparison of CBR photon output in control and coculture.
Another application of bioluminescent imaging is the ability to study signal transduction pathways nondestructively over time. Recently, IκBα fused to firefly luciferase was characterized as a reporter of IκBα kinase (IKK) activity in living cells and animals.17 When cells are treated with an activator of IKK such as tumor necrosis factor alpha (TNFα), the IκBα firefly luciferase fusion is targeted for proteasome-mediated degradation in a phosphoserine-dependent manner, and the luciferase signal decreases relative to untreated cells. A rapid and facile method to normalize for both transfection efficiency and cell viability would further improve the accuracy and precision of this assay. Thus, IκBα fused to CBG99 and unfused CBR were cotransfected into HeLa cells for timeresolved spectral unmixing following TNFα activation (Figure 7). After spectral deconvolution by biolumunmixing v. 1.0, there was a significant increase in the precision of the assay over the original publication. For example, the propagated standard error of the mean at time of maximal degradation of IκBα in the original publication was 10%,17 but utilizing spectral unmixing it was only 3.2%. Thus, one can use spectral unmixing to rapidly and quickly normalize for transfection efficiency, or perhaps more interestingly, one may assay multiple pathways simultaneously with multiple luciferases.
DISCUSSION
Spectral deconvolution of bioluminescence images provides opportunities for quantitation not possible with other modalities. The primary problem in remote sensing is determining the number of chemical species to be detected. Satellite sensing suffers acutely from this problem, as virtually an unlimited number of chemical species could be detected in any field of view. This is also the case in fluorescence microscopy, where several endogenous fluorophores could potentially be present within an image.26,27 Researchers have eliminated much of the autofluorescence problem by overdetermining the system with multiple band-pass filters15 and utilizing quantum dots, which remove the photobleaching artifacts that plagued earlier attempts at spectral unmixing of fluorescence signals.28 However, in bioluminescence, the number of species (luciferase enzymes) is known a priori because the number of luciferase enzymes present in the system is set by the investigator. Another difficulty in remote sensing is calculating the spectral matrix K. Automated algorithms are required to search for “pure pixels” in other remote sensing applications. Yet in bioluminescence assays, control wells are added for each luciferase enzyme in the image to allow for facile and accurate determination of K. Thus, a sample of “pure” light from each luciferase can be manually found and quantified in each filtered image. Since all values of K are determined simultaneously, small time-dependent changes in the bioluminescence are also normalized by K. However, since bioluminescence is an enzymatic process, large kinetic differences between different enzymes may confound K because one substrate could be depleted before the other substrate. Thus, care must be taken to choose the appropriate sets of enzymes and substrate concentrations to minimize or eliminate this confounding factor. This is analogous to the problem of photobleaching confounding the spectral unmixing of some organic dyes.29 Furthermore, changes in the bioluminescent spectrum with different media or pH are also accounted for since K is redetermined in each experiment.
Dual color and, recently, tricolor luciferase assays have been reported as methods to normalize reporter assays. However, both analyses assume a constant luciferase spectrum, are not easily implemented absent skill with linear algebra, and are not amenable to high-throughput screening. The commercial dual color assay methodology cannot be extended to multiple luciferases and does not readily account for any changes in detector response from day to day.30 Recently, Nakajima and co-workers simultaneously assayed three different firefly luciferases.31 Since only one substrate needs to be administered to their system, kinetic differences in coelenterazine-utilizing enzymes and firefly luciferin-utilizing enzymes are minimized. However, the researchers did not demonstrate deconvolution of all possible combinations of luciferases. Furthermore, the filters used to calculate the unmixing were all overlapping, thereby exacerbating errors introduced during deconvolution. From a practical perspective, the assay is time intensive, as it would take ∼5 h to assay a 96-well plate. Finally, both of these methodologies require investigators to have a sound understanding of linear algebra, making an otherwise useful technique cumbersome to those not mathematically trained. Through the use of our protocol and software program, we have alleviated the need for laborious mathematical analyses and make spectral unmixing of bioluminescent signals amenable to standard laboratory assays as well as high-throughput screening.
As the number of luciferase assays increases, spectral unmixing of luminescent reporters will allow a rapid, accurate, and cost-effective means of measuring these targets and their responses to various ligands and small molecule libraries.
CONCLUSION
A facile and freely available method for deconvoluting bioluminescence images has been demonstrated in cell lysates and live cells. This method has also been demonstrated to be applicable for 96-well plate format for high-throughput screening.
ACKNOWLEDGMENT
This work was supported by NIH P50 CA94056 (D.P.-W.), NIH GM36262 (G.W.G.), and Training Grant Fellowships T32 GM08785 (W.M.L.). We thank Dr. Vijay Sharma for many useful discussions.
Footnotes
SUPPORTING INFORMATION AVAILABLE
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Gross S, Piwnica-Worms D. Cancer Cell. 2005;7:5–15. doi: 10.1016/j.ccr.2004.12.011. [DOI] [PubMed] [Google Scholar]
- (2).Luker KE, Smith MC, Luker GD, Gammon ST, Piwnica-Worms H, Piwnica-Worms D. Proc. Natl. Acad. Sci. U. S. A. 2004;101:12288–12293. doi: 10.1073/pnas.0404041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ikariyama Y, Nishiguchi S, Koyama T, Kobatake E, Aizawa M, Tsuda M, Nakazawa T. Anal. Chem. 1997;69:2600–2605. doi: 10.1021/ac961311o. [DOI] [PubMed] [Google Scholar]
- (4).Tauriainen S, Karp M, Chang W, Virta M. Biosens. Bioelectron. 1998;13:931–938. doi: 10.1016/s0956-5663(98)00027-x. [DOI] [PubMed] [Google Scholar]
- (5).Pastorino L, Disawal S, Nicolini C, Lvov YM, Erokhin VV. Biotechnol. Bioeng. 2003;84:286–291. doi: 10.1002/bit.10761. [DOI] [PubMed] [Google Scholar]
- (6).Michelini E, Leskinen P, Virta M, Karp M, Roda A. Biosens. Bioelectron. 2005;20:2261–2267. doi: 10.1016/j.bios.2004.10.018. [DOI] [PubMed] [Google Scholar]
- (7).Doyle TC, Burns SM, Contag CH. Cell Microbiol. 2004;6:303–317. doi: 10.1111/j.1462-5822.2004.00378.x. [DOI] [PubMed] [Google Scholar]
- (8).Kaihara A, Kawai Y, Sato M, Ozawa T, Umezawa Y. Anal. Chem. 2003;75:4176–4181. doi: 10.1021/ac0300800. [DOI] [PubMed] [Google Scholar]
- (9).Bradford MM. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- (10).Pichler A, Prior J, Piwnica-Worms D. Proc. Natl. Acad. Sci. U. S. A. 2004;101:1702–1707. doi: 10.1073/pnas.0304326101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Cory AH, Owen TC, Barltrop JA, Cory JG. Cancer Commun. 1991;3:207–212. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]
- (12).Behre G, Smith LT, Tenen DG. Biotechniques. 1999;26:24–26. 28. doi: 10.2144/99261bm03. [DOI] [PubMed] [Google Scholar]
- (13).Smith M, Johnston P, Adams J. J. Geophys. Res. 1985;90:797–804. [Google Scholar]
- (14).Keshava N, Mustard JF. IEEE Signal Process. Mag. 2002;19:44–57. [Google Scholar]
- (15).Dickinson ME, Bearman G, Tille S, Lansford R, Fraser SE. Biotechniques. 2001;31:1272, 1274–1276, 1278. doi: 10.2144/01316bt01. [DOI] [PubMed] [Google Scholar]
- (16).Levenson R, Mansfield J. Mol. Imaging. 2004;3:227. [Google Scholar]
- (17).Gross S, Piwnica-Worms D. Nat. Methods. 2005;2:607–614. doi: 10.1038/nmeth779. [DOI] [PubMed] [Google Scholar]
- (18).Rasband W. National Institutes of Health; Bethesda, MD: 2005. http://rsb.info.nih.gov/ij/ [Google Scholar]
- (19).Miller B, Hicklin J, Moler C, Webb P, Boisvert R, Pozo R, Remington K. The MathWorks and the National Institute of Standards and Technology. 1.0.1 ed. 2005. [Google Scholar]
- (20).Byrne CL. IEEE Trans. Image Process. 1998;7:100–109. doi: 10.1109/83.650854. [DOI] [PubMed] [Google Scholar]
- (21).Zhang JH, Chung TD, Oldenburg KR. J. Biomol. Screening. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- (22).Zhao H, Doyle T, Wong R, Cao Y, Stevenson D, Piwnica-Worms D, Contag C. Mol. Imaging. 2004;3:43–54. doi: 10.1162/15353500200403181. [DOI] [PubMed] [Google Scholar]
- (23).Schiera G, Sala S, Gallo A, Raffa MP, Pitarresi GL, Savettieri G, Di Liegro I. J. Cell. Mol. Med. 2005;9:373–379. doi: 10.1111/j.1582-4934.2005.tb00362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Tanaka R, Saito T, Ashihara K, Nishimura M, Mizumoto H, Kudo R. Gynecol. Oncol. 2003;90:297–304. doi: 10.1016/s0090-8258(03)00335-4. [DOI] [PubMed] [Google Scholar]
- (25).Heneweer M, Muusse M, Dingemans M, de Jong PC, van den Berg M, Sanderson JT. Toxicol. Sci. 2005;83:257–263. doi: 10.1093/toxsci/kfi025. [DOI] [PubMed] [Google Scholar]
- (26).Andersson-Engels S, Klinteberg C, Svanberg K, Svanberg S. Phys. Med. Biol. 1997;42:815–824. doi: 10.1088/0031-9155/42/5/006. [DOI] [PubMed] [Google Scholar]
- (27).Chorvat D, Jr, Kirchnerova J, Cagalinec M, Smolka J, Mateasik A, Chorvatova A. Biophys. J. 2005 doi: 10.1529/biophysj.105.073866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Gao X, Cui Y, Levenson RM, Chung LW, Nie S. Nat. Biotechnol. 2004;22:969–976. doi: 10.1038/nbt994. [DOI] [PubMed] [Google Scholar]
- (29).Tsurui H, Nishimura H, Hattori S, Hirose S, Okumura K, Shirai T. J. Histochem. Cytochem. 2000;48:653–662. doi: 10.1177/002215540004800509. [DOI] [PubMed] [Google Scholar]
- (30).2003. pp. 1–20. Promega. Technical Manual No. 062. [Google Scholar]
- (31).Nakajima Y, Kimura T, Sugata K, Enomoto T, Asakawa A, Kubota H, Ikeda M, Ohmiya Y. Biotechniques. 2005;38:891–894. doi: 10.2144/05386ST03. [DOI] [PubMed] [Google Scholar]