

SUMMARY
Two transglutaminases (TGs), Factor XIIIA (FXIIIA) and TG2, undergo physiologic upregulation in growth plate hypertrophic chondrocytes and pathologic upregulation in osteoarthritic cartilage. Extrenalization of guanine nucleotide-bound TG2 drives chondrocyte maturation to hypertrophy, a state linked to matrix remodeling and calcification. TGs have been demonstrated to interact with certain integrin subunits. During osteoarthritis (OA) α1β1 integrin is up-regulated and associated with hypertrophic chondrocytes. Here we examined FXIIIA in cartilage to test the hypothesis that FXIIIA promotes hypertrophic differentiation together with TG2. Using human articular chondrocytes, we determined that extracellular FXIIIA induced chondrocyte hypertrophy associated with rapid movement of TG2 to the cell surface. Site directed mutants revealed the FXIIIA endoproteolytic Pro37 site and the integrity of the Ca2+-binding residue Ala457, but not intrinsic TG catalytic activity were necessary for FXIIIA to induce chondrocyte hypertrophy. The α1β1 integrin was critical for both FXIIIA to induce both TG2 mobilization to the cell surface and phosphorylation of the chondrocyte differentiation mediator p38 MAP kinase. Our results identify a unique functional network between 2 cartilage TG isoenzymes that accelerates chondrocyte maturation without requirement for TG-catalyzed transamidation by either TG.
Keywords: Transglutaminases, chondrocyte, hypertrophy, type X collagen
INTRODUCTION
Transglutaminases (TGs) catalyze a calcium-dependent transamidation reaction that generates covalent crosslinks of available substrate glutamine residues with primary amino groups (EC 2.3.2.13), thereby modifying proteins and protein-protein interactions (Lorand and Graham, 2003). Expression and cellular release of the most widely expressed TG isoenzyme (TG2), and of the TG isoenzyme FXIIIA, the homodimeric tissue form of the heterotetrameric plasma coagulation protein Factor XIII, have been identified in bone and cartilage (Aeschlimann et al., 1996; Nurminskaya and Linsenmayer, 1996; Nurminskaya et al., 1998). Moreover, FXIIIA and TG2 have both been implicated in extracellular matrix modification that modulates the capacity of osteoblasts to mature and form bone mineral (Nurminskaya and Kaartinen, 2006). Changes in FXIIIA and TG2 expression and release also have been identified in the physiologic maturation of growth plate chondrocytes, a process that occurs in a temporal and spatially organized manner that progresses through resting, proliferative and pre-hypertrophic differentiation, to terminal hypertrophic differentiation and cell death (Aeschlimann et al., 1993; Borge et al., 1996).
The growth plate chondrocyte hypertrophy gene expression program promotes remodeling of the extracellular matrix, partly through a shift in cartilage-specific collagen expression from type II to type X and by enhanced MMP-13 expression (Drissi et al., 2005). Functional consequences of chondrocyte hypertrophy in the growth plate include calcification mediated partly by matrix vesicle shedding and angiogenesis mediated in part by VEGF expression (Kirsch et al., 1997). Though articular cartilage chondrocytes are in a physiologic, maturity-arrested differentiation state, pathologic hypertrophic differentiation is observed to develop among osteoarthritic (OA) chondrocytes in situ and has the potential to promote OA progression through dysregulated matrix repair (Tchetina et al., 2006) and pathologic calcification (Kirsch et al., 2000). Significantly, both FXIIIA and TG2 are molecular markers of chondrocyte hypertrophic differentiation in the growth plate (Aeschlimann et al., 1993; Nurminskaya and Linsenmayer, 1996). Furthermore, TG2 and FXIIIA expression, as well as total TG catalytic activity, are up-regulated in human knee OA cartilage chondrocytes (Johnson et al., 2001).
Despite lacking a signal peptide, TG2 and FXIIIA both are released by chondrocytes and osteoblasts (Nurminskaya and Kaartinen, 2006). Transamidation of proteins in the extracellular matrix by secreted TGs has the potential to alter cell differentiation and function, specifically exemplified by TG2-catalyzed transamidation of extracellular matrix collagen to promote calcification (Chau et al., 2005). TG2 is essential to accelerate chondrocyte maturation to hypertrophy in response to signals provided by retinoic acid and CXCL1 (Johnson et al., 2003; Merz et al., 2003). Moreover, exogenous nanomolar TG2 is sufficient to directly induce hypertrophic differentiation in chondrocytes in articular cartilage organ culture (Johnson and Terkeltaub, 2005). In addition, paracrine and juxtacrine effects of TG2 released from chondrocytes modulate osteoblast differentiation through extracellular TG2-induced PKA signaling (Nurminskaya et al., 2003; Nurminskaya and Kaartinen, 2006).
Cardinal aspects of the multifunctionality of TG2 include the potential for TG2 to function as an unconventional GTPase and as a cell adhesion protein. TG2 interconverts in a reciprocally regulated manner between a TG catalytically latent guanine nucleotide-bound state and TG catalytically active calcium-bound state (Fesus and Piacentini, 2002). Cell surface TG2 serves as an integrin co-receptor for fibronectin (Hang et al., 2005) and extracellular TG2 promotes integrin clustering that induces RhoA activation (Janiak et al., 2006). In this context, exogenous TG2 must be in a guanine nucleotide-bound state and employ β1 integrin mediated signaling and rapid activation of p38 MAPK pathway signaling to induce chondrocyte hypertrophic differentiation in vitro (Johnson and Terkeltaub, 2005). TG2 does not require transamidation activity, GTPase activity, or fibronectin binding to promote chondrocyte maturation to hypertrophy (Johnson and Terkeltaub, 2005).
FXIIIA, like TG2, exerts both transamidation-dependent and transamidation-independent activities, binds integrins and functions to modulate both cell adhesion and differentiation. For example, FXIIIA binds αVβ1 and αVβ3 integrin molecules that also are potential substrates for transamidation (Dardik et al., 2002). Moreover, FXIIIA, coordinately with vascular endothelial growth factor receptor 2 (VEGFR-2), modulates cell signaling that drives angiogenesis (Dardik et al., 2005). Significantly, increases in FXIIIA expression and FXIIIA extrusion coincide with and directly stimulate matrix calcification in chondrocytes in situ and in vitro (Johnson et al., 2001; Nurminskaya et al., 1998; Nurminskaya et al., 2002). Therefore, this study examined molecular structure-function of FXIIIA in chondrocyte differentiation and tested the hypothesis that FXIIIA assists TG2 with the development of chondrocyte hypertrophy. Our results identify a unique functional network between two TGs that accelerates chondrocyte maturation without the requirement for either TG isoenzyme to catalyze transamidation.
RESULTS
Molecular FXIIIA structure-function in the induction of type X collagen
To test the hypothesis that extracellular FXIIIA can enhance chondrocyte hypertrophy, we first treated cultured human articular chondrocytes with soluble recombinant FXIIIA (sFXIIIA) in comparison with recombinant TG2 (sTG2). Treatment with either of the TG isoenzymes was sufficient to increase expression of vascular endothelial growth factor (VEGF) and matrix metalloproteinase-13 (MMP-13) (genes typically expressed by hypertrophic chondrocytes), and to markedly augment the ratio of type X collagen (the stereotypic marker of chondrocyte hypertrophy) to type II collagen mRNA within 8 hours (Fig. 1A). In bovine knee articular cartilage in organ culture, sTG2 and sFXIIIA both induced type X collagen (Fig. 1B). Under these conditions, sTG2 but not sFXIIIA stimulated enlargement of the chondrocyte-containing lacunae in articular cartilage explants (Fig. 1B), which suggested differential effects on chondrocyte hypertrophy.
Fig 1.
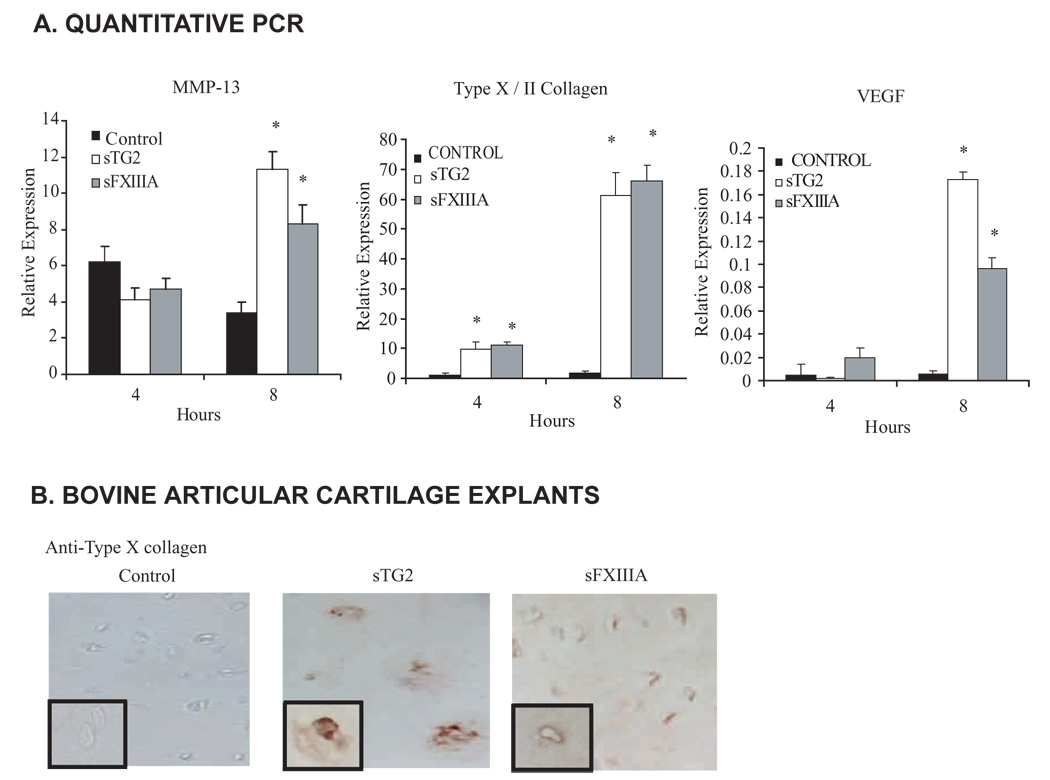
Both exogenous FXIIIA and TG2 induce chondrocyte hypertrophic differentiation. We studied induction by TG2 and FXIIIA of MMP-13, VEGF, type X collagen and Syndecan-3 in cultured chondrocytes. Normal human knee articular chondrocytes plated in 12 well dishes (at 1 × 105 cells/well) were incubated 4 or 8 hours with 100 ng/ml of recombinant wild type TG2 or FXIIIA in the ascorbate-containing medium A described in the Methods. (A) Using quantitative RT-PCR, we determined chondrocyte mRNA expression levels relative to GAPDH for MMP-13 and VEGF, and the mRNA expression ratio of type X : type II collagen, studying data collected from three separate human donors (n=9, *p<0.05). (B) Assessment of the induction by TG2 and FXIIIA of Type X collagen in cartilage organ culture. Normal bovine articular cartilage explants in organ culture were incubated with 100 ng/ml recombinant wild type TG2 or FXIIIA for 5 days in medium A, and 10 µm frozen sections were stained for type X collagen expression (representative of 5 donors).
To test if TG2 stimulated chondrocyte hypertrophy in a manner mediated by FXIIIA or vice versa, we studied cultured knee articular chondrocytes from Tgm2 and F13A knockout mice and congenic wild type controls. We observed that sFXIIIA failed to induce expression of type X collagen in Tgm2−/− chondrocytes, whereas sTG2 did not require FXIIIA to induce type X collagen, sFXIIIA did require endogenous FXIIIA (Fig. 2). In parallel studies, al-trans retinoic acid (ATRA) required both TG2 and FXIIIA to induce type X collagen (Fig. 2). In contrast, chondrocytes did not require FXIIIA expression to develop type X collagen expression in response to CXCL8, under conditions where TG2 was necessary (Fig. 2).
Fig 2.
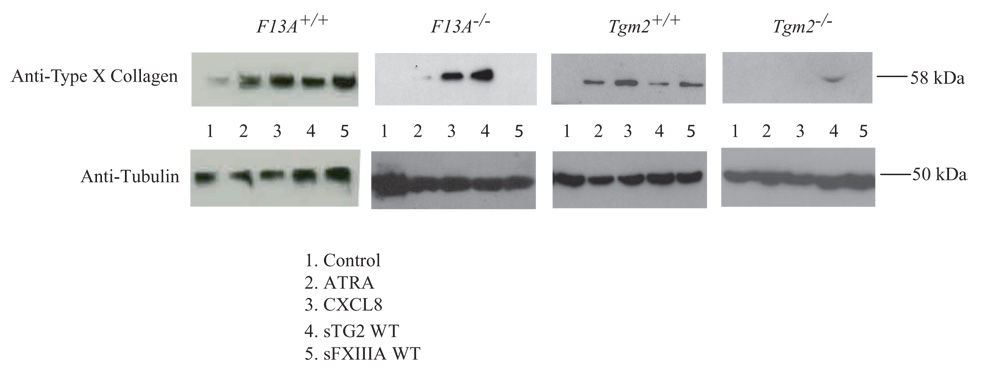
FXIIIA-stimulated type X collagen expression is dependent upon TG2. Assessment of TG2 and FXIIIA knockout mouse cells. Primary knee chondrocytes were removed from F13A+/+, F13A−/− Tgm2+/+, and Tgm2−/− mice. After two weeks in culture, aliquots of 5×103 cells in Medium A were stimulated for 5 days with 10 nM ATRA, 10 ng/ml CXCL8, or 100 ng/ml of sTG2 or sFXIIIA, and then type X collagen was examined by SDS-PAGE/Western blotting, as described in the Methods. Representative of three experiments.
To elucidate FXIIIA structure-function in chondrocyte hypertrophy induction, we designed a site mutagenesis strategy that factored in the constitutive latency of FXIIIA as a TG. Specifically, binding of Ca2+to FXIIIA is required for TG catalytic activity, triggered by endoproteolysis of FXIIIA at the Arg38–G1y39 bond by thrombin or alternatively by excess Ca2+ alone (Aeschlimann et al., 1996). Mutation of the Pro37 alters the conformation and accessibility of this bond. This process removes a 4 kDa peptide to expose the conserved TG family catalytic triad that includes Cys314 FXIII TG activity is subsequently inactivated by thrombin-catalyzed proteolysis at Met513, or alternatively by a decrease in ambient Ca2+ (Hettasch and Greenberg, 1994). We observed catalytic activity of recombinant WT FXIIIA protein, most likely due to the presence of Ca2+ during amplification (K.A. Johnson, unpublished). We next made recombinant forms of each FXIIIA site mutants previously characterized to functionally compromise the "activation site" (P37A), the catalytic site (C314A), and the "inactivation site" (M513G) (Hettasch and Greenberg, 1994) as schematized in Fig. 3A. Initial comparison of the human FXIIIA mutants for TG catalytic activity, in the presence and absence of thrombin, verified that individual mutants possessed the predicted, relative changes in TG activity (K.A. Johnson, unpublished).
Fig 3.
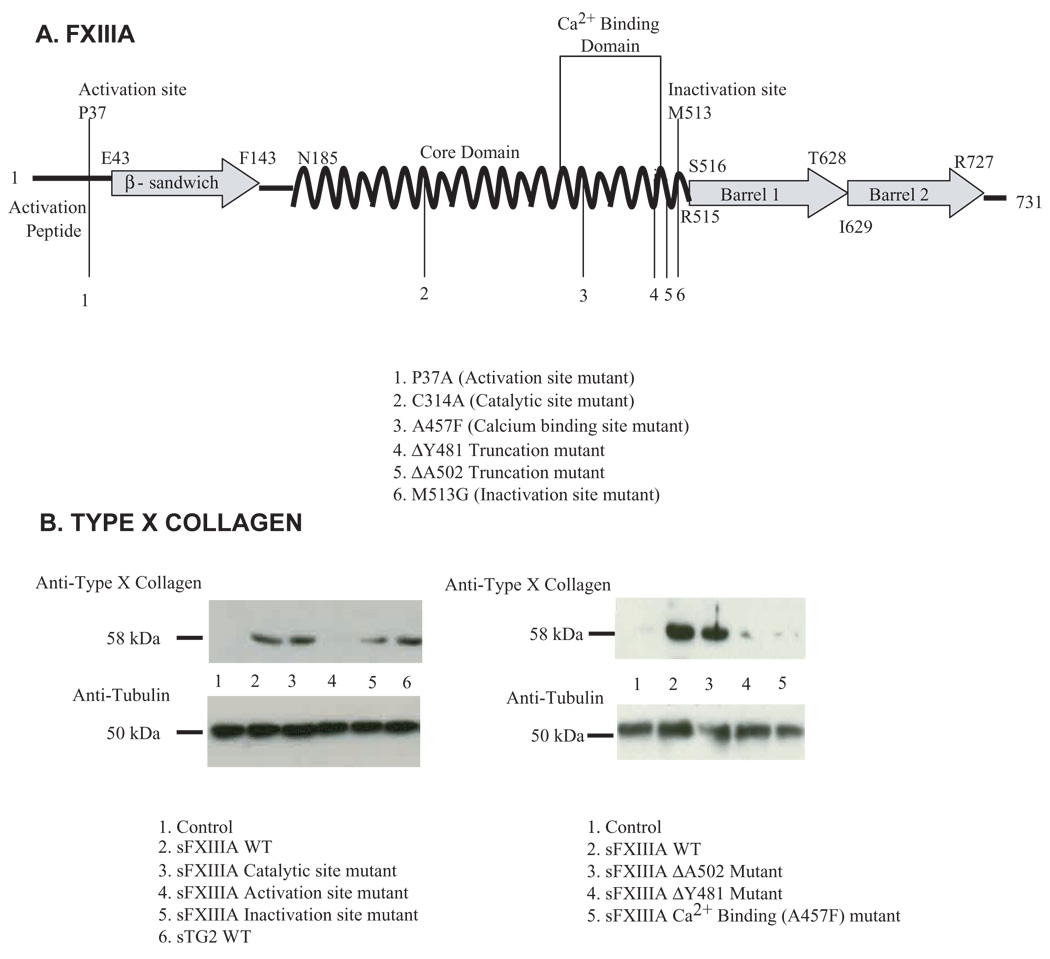
(A) FXIIIA site directed mutants generated for structural analysis examination. The schematic depicts features of the primary structure of wild type (WT) FXIIIA and the panel of FXIIIA site directed mutants generated and studied. (B) Aliquots of human articular chondrocytes (1 × 105 cells) were incubated with 100 ng/ml of each recombinant protein in medium A for 72 hours, and type X collagen assessed in cell lysates by SDS-PAGE/ Western blotting. Representative of three donors in three separate experiments (n=9).
Incubation of chondrocytes with recombinant forms of FXIIIA mutants revealed dependence on the "activation site" P37 residue for extracellular sFXIIIA to induce type X collagen expression (Fig. 3B). To assess if sFXIIIA must bind Ca2+ in order to modulate chondrocyte differentiation, we generated and characterized a FXIIIA ΔA502 truncation mutant that retains the entire Ca2+ binding domain and a FXIIIA ΔY481 truncation mutant designed to functionally interrupt the Ca2+ binding domain. Additionally, we generated a mutant at Ala457, one of the five amino acids which is theorectically critical for FXIIIA to bind Ca2+ (Fox et al., 1999). We ascertained that the amino acid ΔY481 truncation and the Ca2+binding domain point mutation at Ala457 significantly suppressed the catalytic activity of FXIIIA (K.A. Johnson, unpublished). Treating chondrocytes with sFXIIIA, we observed that the amino acid ΔY481 truncation mutant and the Ala457 mutant failed to induce type X collagen (Fig. 3B). By contrast, the capacity of FXIIIA to induce type X collagen was preserved after mutation of Asp270 involved in the binding of cations that inhibit the catalytic activity of FXIIIA (Fox et al., 1999), or mutation of Arg310 involved in the formation of FXIIIA isomers that appear to stabilize FXIIIA (K.A. Johnson, unpublished) (Weiss et al., 1998).
FXIIIA induces p38 MAPK pathway signaling dependent on rapid externalization of TG2
Release of TG2 from articular chondrocytes is required for TG2 dependent hypertrophic differentiation, and recombinant TG2 induces phosphorylation of p38 MAP Kinase within minutes (Johnson and Terkeltaub, 2005). Signaling through the p38 MAP kinase pathway plays a central role in transducing maturation to hypertrophy under certain conditions in cultured chondrocytes (Merz et al., 2003; Wang and Beier, 2005; Zhang et al., 2006). Because FXIIIA appeared to require TG2 to induce hypertrophic differentiation, we hypothesized that FXIIIA may mediate the localization of TG2 to the cell surface to promote the rapid initiation of chondrocyte hypertrophic differentiation.
To detect TG2 on the cell surface, chondrocytes were starved and then stimulated with sFXIIIA. The non-permeabilized and fixed cells were incubated with a biotinylated monoclonal antibody to TG2 (Balklava et al., 2002). First, a 200-fold change in the amount of TG2 localized on the cell surface was detected within 5 minutes of treatment of primary human articular chondrocytes with wild type sFXIIIA (Fig. 4A). These results were not due to transcriptional regulation of TG2, as there was no quantitative change in the mRNA expression for TG2 (K.A. Johnson, unpublished). Second, the activation site, the Ala457 Ca2+ binding and ΔY481 truncation mutants of FXIIIA, all were able to induce an increase in the cell surface TG2 expression but at significantly lower levels than that of WT sFXIIIA or the other mutants (Fig. 4A). These data correlated with the lack of induction of type X collagen (Fig. 3B). Next, we determined that sFXIIIA stimulated phosphorylation of p38 MAP kinase within minutes, an activity not shared by the activation site mutant or the sFXIIIA Ala457 Ca2+ binding mutant of FXIIIA (Fig. 4B).
Fig 4.

Rapid mobilization of TG2 by Ca2+-bound FXIIIA is essential for stimulation of p38 phosphorylation. (A) Aliquots of 5 × 103 human articular chondrocytes were starved in serum-free DMEM high glucose medium for 2 hours and then stimulated with WT or mutant sFXIIIA. TG2-specific antibodies were used to detect membrane bound TG2, quantified through successive incubations with biotin anti-rabbit and strepavidin-AP antibodies as described in the Methods. (n=9) (B) Aliquots of 3 × 105 human articular chondrocytes were starved in serum free DMEM high glucose medium for 2 hours and then stimulated with WT or mutant sFXIIIA for the time indicated. Cell lysates were analyzed by Western blotting for p-p38 and total p38. Representative of three donors in three separate experiments (n=9).
FXIIIA engagement of the α1 integrin subunit is critical for induction of p38 MAPK pathway signaling in chondrocytes
After application of both recombinant TGs to the chondrocytes, we observed binding (within minutes) of the TGs to the chondrocytes, where they remained cell-associated (Fig 5A). In vitro binding assays did not reveal a direct association between TG2 and FXIIIA, and in transfection experiments, there was no clear correlation between the extracellular levels of the two TG isoenzymes (K.A. Johnson, unpublished). As such, there was no evidence that TG2 mobilization to the cell surface by FXIIIA was caused by direct mutual interaction.
Fig 5.
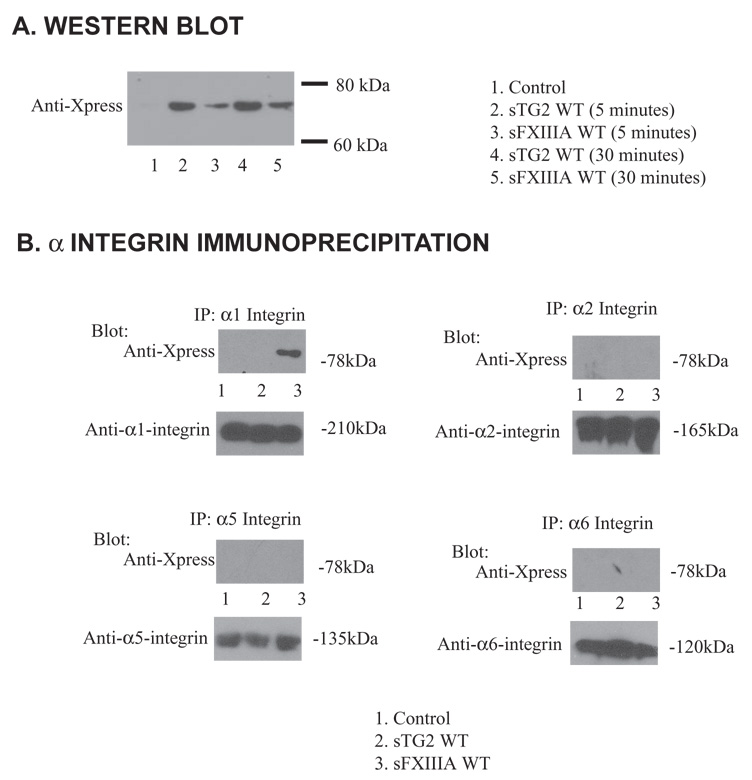
Interaction of recombinant FXIIIA with human chondrocytes through α1 integrin subunit. (A) Aliquots of 1 × 105 human articular chondrocytes were starved for 2 hours in serum free DMEM and then incubated for the indicated times with 100 ng/ml of sTG2 or sFXIIIA. Washed cell lysates were examined for the presence of the Xpress epitope on the recombinant proteins by SDS-PAGE/Western blotting. (B) Aliquots of 1 × 106 human articular chondrocytes were incubated for three days in medium A containing 100 ng/ml of sTG2 or sFXIIIA where indicated. Cell lysates (200 µg protein) were immunoprecipitated using 1 µg/ml of α1 (clone TS2/7), α2, α5, or α6 integrin subunit-specific antibody, and precipitated proteins were analyzed for the Xpress tag or each respective α integrin subunit by Western blotting. Representative of 3 separate experiments using 3 different donors.
In fibroblasts, TG2 can bind to the extracellular domain of β1 and β3 integrin subunits (Akimov et al., 2000). Activated FXIIIA can bind to αVβ3 in HUVECs (Dardik et al., 2002). Chondrocytes themselves express multiple integrins. These include α1β1, α2β1, α5β1, α6β1, α10β1, αVβ3 (Loeser, 2000). Previously, β1 integrin subunit appeared critical for TG2-induced hypertrophy while inhibition of β3 integrin subunit had no effect (Johnson and Terkeltaub, 2005). Therefore, we treated chondrocytes with recombinant TG2 and FXIIIA and assessed for interaction between TGs and four α integrin subunits. sTG2 did not detectably bind to α1, α2, α5 or α6 integrin, but sFXIIIA interacted robustly with the α1 integrin subunit (Fig 5B).
To assess if the binding of α1 integrin subunit is critical in FXIIIA-induced hypertrophy, chondrocytes were pretreated with the α1 integrin subunit blocking antibody FB12, which inhibited the capacity of sFXIIIA but not sTG2 to induce type X collagen (Fig. 6A). In contrast, the blocking antibody to the promiscuous β1 integrin subunit, suppressed type X collagen induction in response to both TG isoenzymes under these conditions (Fig. 6A). Additionally, sFXIIIA-induced p38 MAP kinase activation, along with FAK phosphorylation assessed as a readout for integrin signaling, were inhibited by pretreatment of chondrocytes with α1 integrin subunit blocking antibody FB 12 (Fig. 6B).
Fig 6.
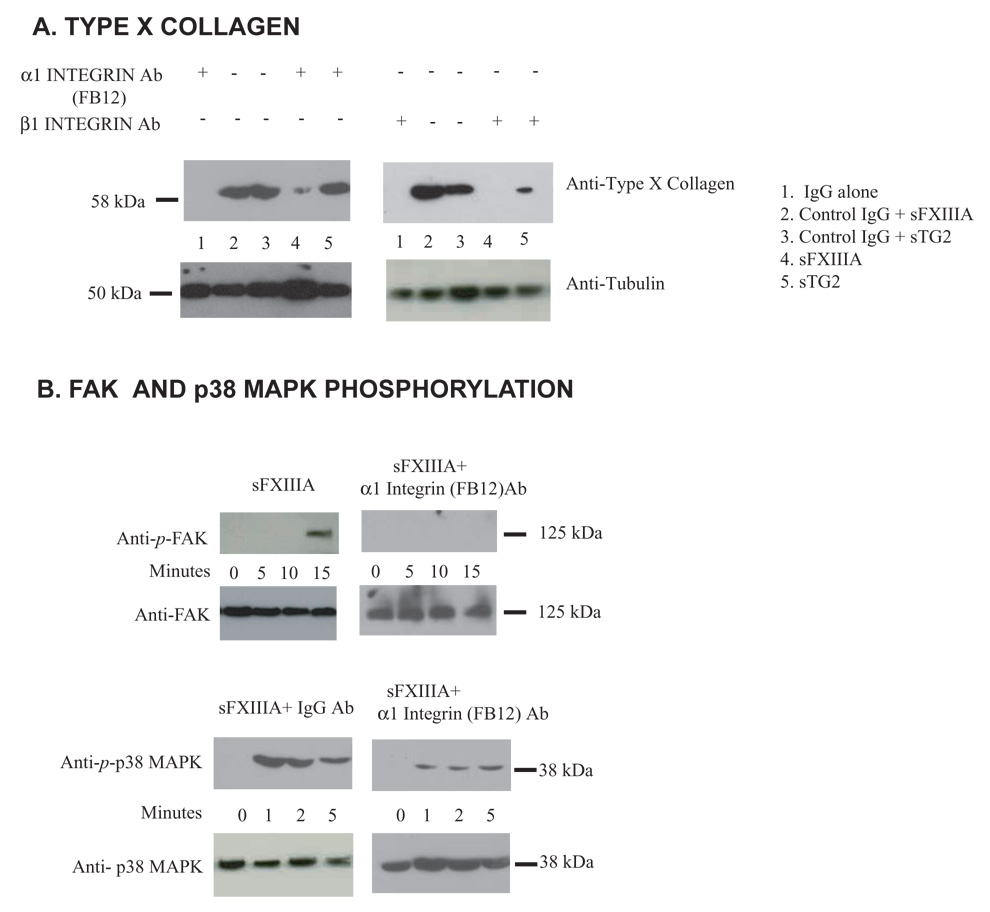
FXIIIA induction of type X collagen is associated with FAK and p38 MAP kinase phosphorylation and dependent upon interaction with the α1 integrin subunit. (A) Aliquots of 1 × 105 human articular chondrocytes were starved and pretreated for 2 hours with 1 µg/ml of IgG control, β1 integrin subunit or α1 integrin subunit blocking (FB12) antibodies in serum free DMEM and then incubated for the indicated times with 100 ng/ml of sTG2 or sFXIIIA, with type X collagen assessed by Western blotting of cell lysates as above. (B) Aliquots of 1 × 105 cells were starved for 2 hours in serum-free DMEM and then incubated for the indicated times with 100 ng/ml of sTG2 or sFXIIIA and 1 µg/ml of α1 integrin subunit-blocking antibody (FB12) where indicated, with cell lysates examined for FAK and p38 phosphorylation by Western blotting. Representative of results from 3 experiments employing 3 separate donors.
In OA articular cartilage, α1β1 integrin expression is augmented, particularly in the hypertrophic chondrocytes (Zemmyo et al., 2003). Crosslinking of the α1 integrin subunit antibody (TS2/7) with a mouse IgG mimicked the enhanced movement of TG2 to the plasma membrane seen with sFXIIIA WT (Fig. 7). Furthermore, pretreating chondrocytes with the α1 integrin subunit blocking antibody FB12 inhibited the ability of FXIIIA to increase TG2 membrane expression (Fig 7). Hence, interaction of FXIIIA and α1β1 integrin was critical to FXIIIA-induced chondrocyte hypertrophy.
Fig 7.
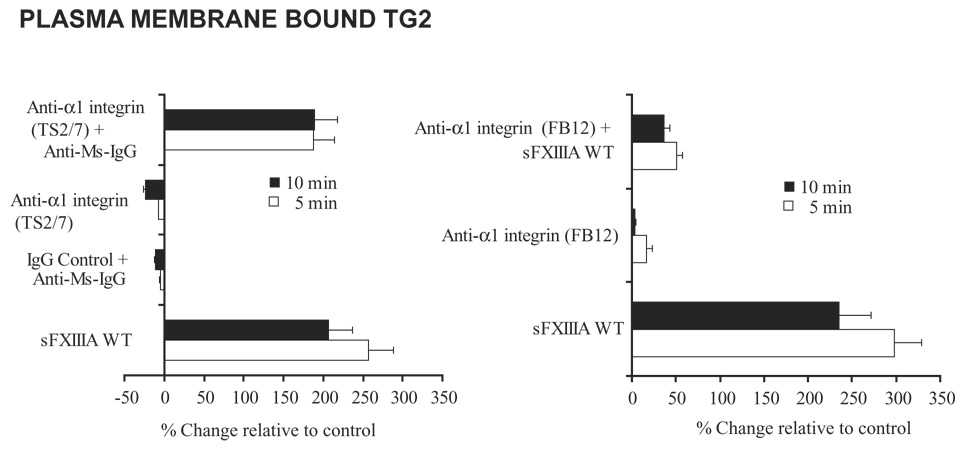
Rapid mobilization of TG2 to the cell surface by sFXIIIA and antibody crosslinking of α1 integrin subunit. To determine if FXIIIA-induced movement of TG2 to the cell surface is integrin dependent, aliquots of 5 × 103 human articular chondrocytes were starved in serum-free DMEM high glucose medium for 2 hours and then stimulated with sFXIIIA, the α1 integrin subunit antibody, TS2/7 (with and without crosslinking by anti-mouse IgG) versus an IgG control antibody. Additionally, after starvation the chondrocytes were pretreated with the blocking α1 integrin subunit antibody, FB12 and then stimulated for 5 and 10 minutes with WT sFXIIIA. After fixation of the cells, TG2-specific antibodies were used to detect membrane bound TG2, quantified through successive incubations with biotin anti-rabbit and strepavidin-AP antibodies as described in the Methods.
DISCUSSION
In this study, we identified novel functional implications of up-regulation of FXIIIA expression in the growth plate chondrocyte hypertrophic differentiation program and in OA cartilage chondrocytes (Linsenmayer et al., 1998; Nurminskaya and Linsenmayer, 1996). Chondrocytes have the capacity to release FXIIIA (Nurminskaya et al., 1998; Nurminskaya et al., 2002; Rosenthal et al., 2001). Our data reveal interaction between FXIIIA and the α1 subunit of α1β1 integrin critical for chondrocyte hypertrophy and TG2 mobilization. These findings are pertinent in part because up-regulation of α1β1 integrin develops in the superficial and upper mid-zone in murine OA cartilage, a condition in which α1β1 integrin appears to mediate cartilage matrix remodeling (Zemmyo et al., 2003).
We found that stimulation of chondrocytes with recombinant FXIIIA increased the movement and release of TG2. We previously demonstrated TG2 effects on chondrocyte maturation to be dependent on TG2 externalization (Johnson and Terkeltaub, 2005). In the absence of TG2 or lack of the promotion of TG2 externalization, FXIIIA was unable to induce hypertrophy. Both mineralizing hypertrophic growth plate chondrocytes and mineralizing osteoblasts robustly release FXIIIA (Nurminskaya et al., 1998; Nurminskaya and Kaartinen, 2006). Our results suggest that one of the functions of FXIIIA release by bone-forming cells is to fine tune TG2 release and thereby regulate TG2-dependent effects on chondrocyte and osteoblast differentiation (Aeschlimann et al., 1996; Al-Jallad et al., 2006; Heath et al., 2001) and transamidation-catalyzed extracellular matrix remodeling in the skeleton (Aeschlimann et al., 1993; Nurminskaya and Kaartinen, 2006).
In this study, recombinant sTG2 was able to induce chondrocyte hypertrophy in the absence of FXIIIA expression, but not vice versa. These findings indicated TG2 to be the driving force of the functional network of two TG isoenzymes that stimulated chondrocyte maturation. Once outside the cell, FXIIIA rapidly mobilized TG2, evidenced by ~200-fold enrichment of TG2 on the surface of chondrocytes within minutes of sFXIIIA addition. Changes in subcellular localization of TG2 modulate wound healing, partly through interactions of plasma membrane-bound TG2 with fibronectin and certain integrins that regulate cell adhesion and migration (Akimov and Belkin, 2001). Additionally, plasma membrane-associated TG2 and FXIIIA, in conjunction with increased FXIIIA expression (Johnson et al., 2001), may contribute to the up-regulated activity of the p38 MAP kinase signaling pathway in OA cartilage chondrocytes in situ (Chun, 2004).
Significantly, both sFXIIIA and sTG2 were observed to directly induce type X collagen in non-proliferating single chondrocytes within lacunae in articular explants in this study. Thus, robust release of TG2 and FXIIIA in OA cartilage potentially bypasses the conventional growth plate chondrocyte maturation sequence. The capacity of TG2 but not FXIIIA to promote enlargement of the chondrocytes in the lacunae could be attributed to the fact that TG2 and FXIIIA preferentially mediate the transamidation of distinct protein sequences (Sugimura et al., 2006).
In cultured chondrocytes, activation of the p38 signaling pathway in response to signals including sTG2, and certain calgranulins and chemokines, plays a central role in promoting maturation to hypertrophy (Johnson and Terkeltaub, 2005; Merz et al., 2003; Wang and Beier, 2005; Zhen et al., 2001). However, it is noted that p38 signaling does increase the transcriptional activity of the chondrogenic master transcription factor Sox9, an inhibitor of chondrocyte maturation to hypertrophy in vivo and in vitro. Moreover, constitutively activated p38 signaling in MKK6 transgenic mice is associated with reduced chondrocyte proliferation and inhibition of hypertrophic chondrocyte differentiation in growth plates in situ (Zhang et al., 2006).
Further studies will be needed to elucidate which downstream signaling pathways, beyond FAK and p38, transduce chondrocyte maturation in response to FXIIIA and TG2. The functional role of FXIIIA-induced chondrocyte p38 pathway activation within growth plate and articular cartilages may critically depend on the timing and spatial organization of up-regulated FXIIIA release by chondrocytes, as well as the concurrent expression of TG2. In addition, internalization of secreted TG2 followed by nuclear localization of TG2 and its receptor could mediate signaling by TG2 in chondrocytes, as demonstrated with VEGFR-2 in endothelial cells (Dardik and Inbal, 2006).
FXIIIA induced TG2-dependent chondrocyte hypertrophy through its interaction with α1β1 integrin. TG2 has been shown to cluster cell surface integrins and lead to integrin-dependent signaling and activation (Janiak et al., 2006). Thus, it is conceivable that FXIIIA may act to cluster α1β1 integrin and thereby prime chondrocytes for TG2-dependent induction of hypertrophy. Our observation that crosslinking α1β1 integrin through an antibody complex induced TG2 mobilization supports this notion. Alternatively, it remains possible that FXIIIA might function indirectly by binding to an integrin-associated protein that binds or clusters α1β1 integrin. The latter scenario merits further study in chondrocytes, as ternary complex formation of FXIIIA with αVβ3 integrin and VEGFR-2 on the endothelial cell surface mediates angiogenesis through VEGF-independent VEGFR-2 activation (Dardik et al., 2005).
In both the F13A−/− and Tgm2−/− mouse chondrocytes, sFXIIIA was not able to induce an increase in the type X collagen expression. This suggests that in addition to TG2, other mediators may be required for FXIIIA induction of chondrocyte hypertrophy, which are lacking in these chondrocytes. This finding also suggests that FXIIIA exerts significant signaling effects intracellularly in chondrocytes. FXIIIA dimerization of the type 1 angiotensin II receptor via crosslinking of receptor cytosolic tails is a notable example of such FXIIIA intracellular activity (AbdAlla et al., 2004). Alternatively, the F13A−/− chondrocytes could have decreased levels of α1 integrin subunit and therefore may not respond to sFXIIIA stimulation. Experiments to address the expression of α1 integrin subunit in both TG knockout mouse chondrocytes along with an analysis of the α1 integrin subunit knockout chondrocytes would reveal a more detailed explanation of our proposed mechanism.
Limitations in this study included use of monolayer culture conditions for these experiments, necessary not only because of the low yields of primary mouse articular chondrocytes but also to allow subsequent comparative studies with human chondrocytes. Monolayer culture conditions may have imposed significant cell-cell and cell-matrix interactions that would not take place in growth plate and articular cartilages in vivo. Another limitation within much of this study was treatment of cultured chondrocytes with nanomolar amounts of exogenous recombinant FXIIIA and TG2 to assess mechanisms for TG effects on differentiation. Endogenous chondrocyte TG2 and FXIIIA typically reach only high picomolar extracellular concentrations in chondrocytes (K. A. Johnson, unpublished). However, the movement of secreted endogenous TG isoenzymes to the cell surface is likely to be more efficient than for exogenous TGs. This study did not attempt to define specific effects on chondrocyte differentiation of endogenous intracellular TG2 or FXIIIA. In this light, guanine-nucleotide bound TG2 does functionally engage several α integrin subunit cytosolic tails (Kang et al., 2004). We also did not specifically test potential roles, in mediating FXIIIA and TG2 effects on chondrocyte differentiation, of binding or transamidation of TGFβ (Verderio E et al., 1999), soluble integrin ligands, or other extracellular matrix proteins. Last, we did not assess for potential internalization of exogenous TG isoenzymes by chondrocytes, or signaling effects by intracellular TG2 (Dardik and Inbal, 2006). Additionally, the Ala457 which has been hypothesized to be involved in the Ca2+ binding by FXIIIA (Fox et al., 1999) has not been thoroughly examined for its ability to either modulate the conformation of FXIIIA or its significance in reducing the actual Ca2+ binding.
In summary, we have established that two TG isoenzymes network to promote and accelerate chondrocyte maturation, but do so in a transamidation-independent manner. Our results add to growing evidence that direct interactions between certain TG isoenzymes and integrins alter cell signaling and differentiation. Our results point to the biologic significance of the robust expression of two TG isoenzymes in association with chondrocyte hypertrophy in vivo, that may lead to a rapid signal amplification and progression of the disease state. Our results identify relative timing of FXIIIA and TG2 up-regulation in cartilages as a novel mechanism for modulation of chondrocyte differentiation under both physiologic and pathologic conditions.
MATERIALS AND METHODS
Reagents
All chemicals and other reagents were obtained from Sigma (St. Louis, MO), unless otherwise indicated.
Generation of FXIIIA cDNA Mutants and Soluble Recombinant Forms of FXIIIA
Human FXIIIA cDNA in pcDNA4/HisMax was the template for generation of the FXIIIA mutants and recombinant forms. The recombinant TG2 and FXIIIA were prepared through overexpression in mammalian cells (HEK 293 cells) and harvested under sterile conditions. After purification through binding to nickel columns, dialysis and concentration, the preparation was tested with Limulus Amebocyte Lysate QCL 1000 assay (Cambrex, Baltimore, MD) and determined to have <0.1EU/ml (or below the detection limits) of endotoxin (Johnson and Terkeltaub, 2005). Where indicated, purified, soluble, recombinant TG2 protein was used after treatment to generate Mg2+ nucleotide complexes of GTP, as described (Johnson and Terkeltaub, 2005).
Tgm2−/− and F13A−/− mice
Tgm2+/+ and Tgm2−/− mice were originally provided by Dr. R. Graham (Nanda, et al., 2001). F13A−/− mice and wild type congenic littermate controls were bred from F13A+/− mice previously generated, characterized, and generously provided by Dr. G. Dickneite and colleagues (Aventis Behring GMBH, Germany) (Lauer, et al., 2002). All animal housing, experimentation and protocols were approved by the IACUC (Institutional Animal Care and Use Committee) of the San Diego Veteran’s Administration Medical Center.
Cell and explant culture isolations, conditions and shRNA design
Primary articular chondrocytes were isolated by dissection of the tibial plateaus and femoral condyles of the Tgm2+/+, Tgm2−/−, F13A+/+ and F13A −/− mice at two months of age, as described (Johnson et al., 2003). Human articular chondrocytes from normal donor knees were isolated as described (Merz et al., 2003). First passage human articular chondrocytes and mouse chondrocytes were cultured in DMEM high glucose supplemented with 10% FCS, 1% glutamine, 100 U/ml Penicillin, 50 µg /ml Streptomycin (Mediatech, Herndon, VA) and maintained at 37°C. Studies on differentiation and function were performed in Medium A (DMEM high glucose supplemented with 1% FCS, 1% glutamine, 100 U/ml Penicillin, 50 µg/ ml Streptomycin, and 50 µg/ml of ascorbic acid) with 100 ng/ml of sFXIIIA and sTG2 added where indicated.
Ambion’s web-based shRNA design program was used to identify 21-mer regions within TG2 and FXIIIA effective for shRNA targeting. Five sequences were originally tested to find an optimal sequence. The 21-mers were then used to generate the 55bp oligos, which included two 19bp regions specific to human TG2 or FXIIIA complementary to each other to form the hairpin, a loop sequence separating the complementary domains and a dinucleotide overhang that can hybridize with the RNA target (part of the original 21-mer). The two 55bp complementary oligos were annealed and then ligated into the pSilencer 4.1-CMV neo vector (Ambion, Austin, TX). The scrambled TG2 and FXIIIA shRNAs were randomly generated with the same basepairs as the siTG2. After sequence confirmation, the vectors were transfected into human articular chondrocytes, using the AMAXA as described. The optimal 19 by sequences for human TG2, 5’-GAGCGAGAT GATCTGGAAC-3’(1116–1132) and for human FXIIIA, 5’-GAGTTTCTTAATGTCACGA-3’ (214–232).
For cartilage organ culture studies, two millimeter by two millimeter slices of articular cartilage were removed from the patellar groove and femoral condyles of normal bovine knees (Animal Technologies, Tyler, TX). Explants were cultured, treated, sectioned and stained as previously described (Johnson and Terkeltaub, 2005).
For immunocytochemical analysis of human articular chondrocytes, aliquots of 1 × 105 cells were plated on 18 mm circular glass coverslips and in medium A. The cells were then fixed for 20 minutes at room temperature with 4 % paraformaldehyde and washed with PBS. All primary antibodies were used at a 1:100 dilution. For light microscopy, bound antibodies were detected by the ABC method. All light microscopy images were visualized on a Nikon microscope using the 4X and 10X objective lenses and with 10X binoculars, and Nikon digital camera images were captured using ACT-2U software. The camera images were captured as TIFF files, cropped and arranged using Adobe Photoshop and Illustrator software. All imaging was performed at room temperature.
SDS PAGE/Western Blotting, and RT-PCR
For SDS-PAGE / Western blotting analyses, conditioned media and/or cell lysates were collected and treated as described (Johnson and Terkeltaub, 2005). Anti-type X collagen (Calbiochem, San Diego, CA), anti-TG2 and anti-FXIIIA (Neomarkers, Freemont, CA), anti-p-FAK (Try 567,577), anti-FAK, anti-p-p38, anti-p38 (Cell Signaling, Beverly, MA), anti-Xpress (Invitrogen, San Diego, CA) and anti-tubulin primary antibodies were used at 1:1000 dilution in Western blotting studies and detected as described (Johnson et al., 2003) The monoclonal α1 integrin subunit antibody (TS2/7) (Genetex, San Antonio, TX) was used for immunoprecipitation in addition to immunofluorescent staining. The FB12 α1 integrin subunit antibody (Chemicon / Millipore, Billerica, MA) a validated blocking antibody was used to pretreat the chondrocytes for 1 hour prior to stimulations where indicated.
Total RNA was isolated as described (Johnson et al., 2003). For quantitative RT-PCR, 1 µl of a 5-fold dilution of the cDNA from reverse transcription reactions was amplified using the LightCycler FastStart DNA MasterPlus SYBR Green I kit (Roche Diagnostics, Indianapolis, IN) with addition of 0.5 µM of each primer in the LightCycler 2.0 (Roche Diagnostics, Indianapolis, IN). Following amplification, a monocolor relative quantification of the target gene and reference (GAPDH) analysis determined the normalized target gene: GAPDH mRNA copy ratios by the manufacturer's LightCycler Software (Version 4.0). The primers employed where designed using LightCycler Probe software, version 2.0 (Roche, Diagnostics, Indianapolis, IN).
Assays of Transamidation Activity and TG externalization
TG transamidation activity was determined as previously described (Johnson et al., 2003). For qualitative evaluation of TG2 or FXIIIA release, SDS PAGE /Western blotting analyses were performed on concentrated conditioned media. TG2 or FXIIIA was additionally quantified in the conditioned media by direct ELISA, using biotin-labeled TG2-specific antibody CUB7402 or FXIIIA specific antibody (Neomarkers, Freemont, CA). Alternatively, TG2 on the plasma membrane was quantified in cells fixed with 4% PFA for 15 minutes, followed by detection by direct ELISA as above.
Statistical Analyses
Statistical analyses were performed using the Student's t test (paired 2-sample testing for means). Error bars, where indicated, represented s.d.
Acknowledgements
Supported by the Research Service of the Department of Veterans Affairs, and by grants from the NIH (NIAMS, NIA, NHLBI).
REFERENCES
- AbdAlla S, Lother H, Langer A, el Faramawy Y, Quitterer U. Factor XIIIA transglutaminase crosslinks AT1 receptor dimers of monocytes at the onset of atherosclerosis. Cell. 2004;119:343–354. doi: 10.1016/j.cell.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Aeschlimann D, Wetterwald A, Fleisch H, Paulsson M. Expression of tissue transglutaminase in skeletal tissues correlates with events of terminal differentiation of chondrocytes. J Cell Biol. 1993;20:1461–1470. doi: 10.1083/jcb.120.6.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aeschlimann D, Mosher D, Paulsson M. Tissue transglutaminase and factor XIII in cartilage and bone remodeling. Semin Thromb Hemost. 1996;22:437–443. doi: 10.1055/s-2007-999043. [DOI] [PubMed] [Google Scholar]
- Akimov SS, Belkin AM. Cell-surface transglutaminase promotes fibronectin assembly via interaction with the gelatin-binding domain of fibronectin: a role in TGFbeta-dependent matrix deposition. J Cell Sci. 2001;114:2989–3000. doi: 10.1242/jcs.114.16.2989. [DOI] [PubMed] [Google Scholar]
- Akimov SS, Krylov D, Fleischman LF, Belkin AM. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J Cell Biol. 2000;148:825–838. doi: 10.1083/jcb.148.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Jallad HF, Nakano Y, Chen JL, McMillan E, Lefebvre C, Kaartinen MT. Transglutaminase activity regulates osteoblast differentiation and matrix mineralization in MC3T3-E1 osteoblast cultures. Matrix Biol. 2006;25:135–148. doi: 10.1016/j.matbio.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Balklava Z, Verderio E, Collighan R, Gross S, Adams J, Griffin M. Analysis of tissue transglutaminase function in the migration of Swiss 3T3 fibroblasts: the active-state conformation of the enzyme does not affect cell motility but is important for its secretion. J Biol Chem. 2002;277:16567–16575. doi: 10.1074/jbc.M109836200. [DOI] [PubMed] [Google Scholar]
- Borge L, Demignot S, Adolphe M. Type II transglutaminase expression in rabbit articular chondrocytes in culture: relation with cell differentiation, cell growth, cell adhesion and cell apoptosis. Biochim Biophys Acta. 1996;312:117–124. doi: 10.1016/0167-4889(96)00028-6. [DOI] [PubMed] [Google Scholar]
- Chau DY, Collighan RJ, Verderio EA, Addy VL, Griffin M. The cellular response to transglutaminase-cross-linked collagen. Biomaterials. 2005;26:6518–6529. doi: 10.1016/j.biomaterials.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Chun JS. Expression, activity, and regulation of MAP kinases in cultured chondrocytes. Methods Mol Med. 2004;100:291–306. doi: 10.1385/1-59259-810-2:291. [DOI] [PubMed] [Google Scholar]
- Dardik R, Inbal A. Complex formation between tissue transglutaminase II (tTG) and vascular endothelial growth factor receptor 2 (VEGFR-2): proposed mechanism for modulation of endothelial cell response to VEGF. Exp Cell Res. 2006;312:2973–2982. doi: 10.1016/j.yexcr.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Dardik R, Shenkman B, Tamarin I, Eskaraev R, Harsfalvi J, Varon D, Inbal A. Factor XIII mediates adhesion of platelets to endothelial cells through alpha(v)beta(3) and glycoprotein IIb/IIIa integrins. Thromb Res. 2002;105:317–323. doi: 10.1016/s0049-3848(02)00014-2. [DOI] [PubMed] [Google Scholar]
- Dardik R, Loscalzo J, Eskaraev R, Inbal A. Molecular mechanisms underlying the proangiogenic effect of factor XIII. Arterioscler Thromb Vasc Biol. 2005;25:526–532. doi: 10.1161/01.ATV.0000154137.21230.80. [DOI] [PubMed] [Google Scholar]
- Drissi H, Zuscik M, Rosier R, O'Keefe R. Transcriptional regulation of chondrocyte maturation: potential involvement of transcription factors in OA pathogenesis. Mol Aspects Med. 2005;26:169–179. doi: 10.1016/j.mam.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Fesus L, Piacentini M. Transglutaminase 2: an enigmatic enzyme with diverse functions. Trends Biochem Sci. 2002;27:534–539. doi: 10.1016/s0968-0004(02)02182-5. [DOI] [PubMed] [Google Scholar]
- Fox BA, Yee VC, Pedersen LC, Le Trong I, Bishop PD, Stenkamp RE, Teller DC. Identification of the calcium binding site and a novel ytterbium site in blood coagulation factor XIII by x-ray crystallography. J Biol Chem. 1999;274:4917–4923. doi: 10.1074/jbc.274.8.4917. [DOI] [PubMed] [Google Scholar]
- Hang J, Zemskov EA, Lorand L, Belkin AM. Identification of a novel recognition sequence for fibronectin within the NH2-terminal beta-sandwich domain of tissue transglutaminase. J Biol Chem. 2005;280:23675–23683. doi: 10.1074/jbc.M503323200. [DOI] [PubMed] [Google Scholar]
- Heath DJ, Downes S, Verderio E, Griffin M. Characterization of tissue transglutaminase in human osteoblast-like cells. J Bone Miner Res. 2001;16:1477–1485. doi: 10.1359/jbmr.2001.16.8.1477. [DOI] [PubMed] [Google Scholar]
- Hettasch JM, Greenberg GS. Analysis of the catalytic activity of human factor XIIIa by site-directed mutagenesis. JBiol Chem. 1994;269:28309–28313. [PubMed] [Google Scholar]
- Janiak A, Zemskov EA, Belkin AM. Cell Surface Transglutaminase Promotes RhoA Activation via Integrin Clustering and Suppression of the Src-pl90RhoGAP Signaling Pathway. Mol Biol Cell. 2006;17:1606–1619. doi: 10.1091/mbc.E05-06-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KA, Terkeltaub RA. External GTP-bound transglutaminase 2 is a molecular switch for chondrocyte hypertrophic differentiation and calcification. J Biol Chem. 2005;280:15004–15012. doi: 10.1074/jbc.M500962200. [DOI] [PubMed] [Google Scholar]
- Johnson K, Hashimoto S, Lotz M, Pritzker K, Terkeltaub R. Interleukin-1 induces pro-mineralizing activity of cartilage tissue transglutaminase and factor XIIIa. Am J Pathol. 2001;159:149–163. doi: 10.1016/S0002-9440(10)61682-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KA, van Etten D, Nanda N, Graham RM, Terkeltaub RA. Distinct transglutaminase 2-independent and transglutaminase 2-dependent pathways mediate articular chondrocyte hypertrophy. J Biol Chem. 2003;278:18824–18832. doi: 10.1074/jbc.M301055200. [DOI] [PubMed] [Google Scholar]
- Kang SK, Yi KS, Kwon NS, Park KH, Kim UH, Baek KJ, Im MJ. Alpha1B-adrenoceptor signaling and cell motility: GTPase function of Gh/transglutaminase 2 inhibits cell migration through interaction with cytoplasmic tail of integrin alpha subunits. J Biol Chem. 2004;279:36593–36600. doi: 10.1074/jbc.M402084200. [DOI] [PubMed] [Google Scholar]
- Kirsch T, Nah HD, Shapiro IM, Pacifici M. Regulated production of mineralization-competent matrix vesicles in hypertrophic chondrocytes. J Cell Biol. 1997;137:1149–1160. doi: 10.1083/jcb.137.5.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch T, Swoboda B, Nah H. Activation of annexin II and V expression, terminal differentiation, mineralization and apoptosis in human osteoarthritic cartilage. Osteoarthritis Cartilage. 2000;8:294–302. doi: 10.1053/joca.1999.0304. [DOI] [PubMed] [Google Scholar]
- Lauer P, Metzner HJ, Zetthneissl G, Li M, Smith AG, Lathe R, Dickneite G. Targeted inactivation of the mouse locus encoding coagulation factor XIII-A: hemostatic abnormalities in mutant mice and characterization of the coagulation deficit. Thromb Haemost. 2002;88:967–974. [PubMed] [Google Scholar]
- Linsenmayer TF, Long L, Nurminskaya M, Chen Q, Schmid TM. Type X collagen and other up-regulated components of the avian hypertrophic cartilage program. Prog Nucleic Acid Res Mol Biol. 1998;60:79–109. doi: 10.1016/s0079-6603(08)60890-9. [DOI] [PubMed] [Google Scholar]
- Loeser RF. Chondrocyte integrin expression and function. Biorheology. 2000;37:109–116. [PubMed] [Google Scholar]
- Lorand L, Graham RM. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol. 2003;4:140–156. doi: 10.1038/nrm1014. [DOI] [PubMed] [Google Scholar]
- Merz D, Liu R, Johnson K, Terkeltaub R. IL-8/CXCL8 and growth-related oncogene alpha/CXCL1 induce chondrocyte hypertrophic differentiation. J Immunol. 2003;171:4406–4415. doi: 10.4049/jimmunol.171.8.4406. [DOI] [PubMed] [Google Scholar]
- Nanda N, Iismaa SE, Owens WA, Husain A, Mackay F, Graham RM. Targeted inactivation of Gh/tissue transglutaminase II. J Biol Chem. 2001;276:20673–20678. doi: 10.1074/jbc.M010846200. [DOI] [PubMed] [Google Scholar]
- Nurminskaya M, Kaartinen MT. Transglutaminases in mineralized tissues. Front Biosci. 2006;11:1591–1606. doi: 10.2741/1907. [DOI] [PubMed] [Google Scholar]
- Nurminskaya M, Linsenmayer TF. Identification and characterization of up-regulated genes during chondrocyte hypertrophy. Dev Dyn. 1996;206:260–271. doi: 10.1002/(SICI)1097-0177(199607)206:3<260::AID-AJA4>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Nurminskaya M, Magee C, Nurminsky D, Linsenmayer TF. Plasma transglutaminase in hypertrophic chondrocytes: expression and cell-specific intracellular activation produce cell death and externalization. J Cell Biol. 1998;142:1135–1144. doi: 10.1083/jcb.142.4.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurminskaya MV, Recheis B, Nimpf J, Magee C, Linsenmayer TF. Transglutaminase factor XIIIA in the cartilage of developing avian long bones. T. Dev Dyn. 2002;223:24–32. doi: 10.1002/dvdy.1230. [DOI] [PubMed] [Google Scholar]
- Nurminskaya M, Magee C, Faverman L, Linsenmayer TF. Chondrocyte-derived transglutaminase promotes maturation of preosteoblasts in periosteal bone. Dev Biol. 2003;263:139–152. doi: 10.1016/s0012-1606(03)00445-7. [DOI] [PubMed] [Google Scholar]
- Rosenthal AK, Masuda I, Gohr CM, Derfus BA, Le M. The transglutaminase, Factor XIIIA, is present in articular chondrocytes. Osteoarthritis Cartilage. 2001;9:578–581. doi: 10.1053/joca.2000.0423. [DOI] [PubMed] [Google Scholar]
- Sugimura Y, Hosono M, Wada F, Yoshimura T, Maid M, Hitomi K. Screening for the preferred substrate phage-displayed peptide library: identification of peptide substrates for TGASE 2 and Factor XIIIA. J Biol Chem. 2006;281:17699–17706. doi: 10.1074/jbc.M513538200. [DOI] [PubMed] [Google Scholar]
- Tchetina EV, Antoniou J, Tanzer M, Zukor DJ, Poole AR. Transforming Growth Factor-{beta}2 Suppresses Collagen Cleavage in Cultured Human Osteoarthritic Cartilage, Reduces Expression of Genes Associated with Chondrocyte Hypertrophy and Degradation, and Increases Prostaglandin E2 Production. Am J Pathol. 2006;168:131–140. doi: 10.2353/ajpath.2006.050369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verderio E, Gaudry C, Gross S, Smith C, Downes S, Griffin M. Regulation of cell surface tissue transglutaminase: effects on matrix storage of latent transforming growth factor-beta binding protein-1. J Histochem Cytochem. 1999;47:1417–1432. doi: 10.1177/002215549904701108. [DOI] [PubMed] [Google Scholar]
- Wang G, Beier F. Racl/Cdc42 and RhoA GTPases antagonistically regulate chondrocyte proliferation, hypertrophy, and apoptosis. J Bone Miner Res. 2005;20:1022–1031. doi: 10.1359/JBMR.050113. [DOI] [PubMed] [Google Scholar]
- Weiss MS, Metzner HJ, Hilgenfeld R. Two non-proline cis peptide bonds may be important for factor XIII function. FEBS Lett. 1998;423:291–296. doi: 10.1016/s0014-5793(98)00098-2. [DOI] [PubMed] [Google Scholar]
- Zemmyo M, Meharra EJ, Kuhn K, Creighton-Achermann L, Lotz M. Accelerated, aging-dependent development of osteoarthritis in alphal integrin-deficient mice. Arthritis Rheum. 2003;48:2873–2880. doi: 10.1002/art.11246. [DOI] [PubMed] [Google Scholar]
- Zhang R, Murakami S, Coustry F, Wang Y, de Crombrugghe B. Constitutive activation of MKK6 in chondrocytes of transgenic mice inhibits proliferation and delays endochondral bone formation. Proc Natl Acad U S A. 2006;103:365–370. doi: 10.1073/pnas.0507979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen X, Wei L, Wu Q, Zhang Y, Chen Q. Mitogen-activated protein kinase p38 mediates regulation of chondrocyte differentiation by parathyroid hormone. J Biol Chem. 2001;276:4879–4885. doi: 10.1074/jbc.M004990200. [DOI] [PubMed] [Google Scholar]