

Abstract
It remains uncertain whether megalin participates in the reabsorption of filtered proteins with large molecular size and, in so doing, contributes to the development of tubule injury in glomerular diseases. To find answers to these questions, we utilized mosaic megalin knockout (KO) mice, which lack megalin expression in 60% of proximal tubule cells (PTCs), and were subjected to experimental induction of non-selective proteinuria. Megalin KO mice were mated with another transgenic line, NEP25. In the latter, glomerular injury can be induced by immunotoxin, LMB2, which has a specific affinity to a transgene product expressed selectively on podocytes. Megalin-KO/NEP25 mice (n=11) were injected with LMB2. Ten days after the injection, megalin-KO/NEP25 mice showed massive non-selective proteinuria and mild glomerular and tubular injury. Comparison of megalin-intact (+) vs. deficient (−) PTCs within each megalin-KO/NEP25 mouse kidney revealed that albumin, immunoglobulin light chain, IgA and IgG were preferentially accumulated in megalin (+) PTCs. Moreover, tubule injury markers, namely heme-oxygenase-1, monocyte chemoattractant protein-1 and apoptosis, were preferentially expressed in megalin (+) PTCs. These results collectively indicate that megalin plays a pivotal role in the reabsorption of small to large molecular size proteins. The study also provided direct in vivo evidence that reabsorption of filtered proteins triggers events that can lead to tubule injury.
INTRODUCTION
Many glomerular diseases accompany proteinuria, and structural damage develops along the downstream tubule. This tubule injury is closely correlated with progressive loss of kidney function (1, 2). Clinical studies indicated that the level of proteinuria predicts the risk of developing into end-stage renal failure (3, 4). In that, when compared to selective proteinuria, non-selective proteinuria has a significantly greater chance of tubulointerstitial injury and progression of renal diseases (5–7).
Although, in the past, many in vitro studies have been designed and executed to demonstrate the causal role of proteinuria for progressive renal cell injury (8–10), Zandi-Nejad and Brenner et al. pointed to uncertainties underlying those studies. The uncertainties stem from nonphysiologic exposure of apical tubule cell surfaces to proteins in vitro, the extremely high concentrations of various proteins tested in vitro, and nonuniform end points measured (5). Also noted are a lack of correlation often seen between in vitro and in vivo findings and a lack of uniformity of results, even for seemingly similar in vitro experiments (5). Therefore, despite the abundance of in vitro findings suggesting the pathogenic role of urinary proteins in tubule damage, it appeared warranted to design an in vivo study which tests the effect of proteinuria occurring in glomerular disease.
Megalin, a member of LDL receptor family, is expressed intensely on the apical membrane of proximal tubule cells (PTCs) and works as a scavenger of filtered proteins by endocytotic process. Most systemic megalin null-mutant mice die perinataly due to defective forebrain development or lung dysfunction (11). A strain of transgenic mice carrying a megalin null allele selectively in renal cells was engineered with a use of Cre-loxP system recently (12). This carries two loxP sites in introns 71 and 74 of the megalin gene and the Cre transgene. The Cre gene is regulated by 3.0-kb promoter fragment of the human ApoE gene, which drives remarkably intensive expression selectively in the kidney, but not in the liver or other organs (13, 14).
Of note, megalin gene deletion in the above model occurs in a mosaic pattern. As a consequence, the proximal tubule consists of cells with and without intact megalin gene, thereby allowing a direct comparison of megalin-knockout vs. megalin-intact PTCs that belong to the same nephron, and hence are exposed to the same quality and quantity of filtered proteins.
Using this as a unique opportunity to identify the role of filtered proteins for the tubule injury in glomerular diseases in vivo, Theilig et al. induced anti-glomerular basement membrane (GBM) nephritis, a model of rapidly progressive glomerulonephritis, in this transgenic strain (15). They found that enhancement in protein endocytosis and upregulation of profibrotic mediators, including intracellular adhesion molecule (ICAM)-1, vascular cellular adhesion molecule (VCAM)-1 and TGF-β, occurred in a megalin-dependent manner. However, tubule degeneration and interstitial fibrosis were associated with glomerular crescentic lesions, but not with megalin expression, thus it appeared that endocytosis does not play a pathogenic role for the tubulointerstitial injury. Of interest, in this regard, the proteinuria of the anti-GBM nephritis studied was not in nephrotic range and selective, i.e. primarily of albumin (16).
We therefore studied the effect of massive, non-selective proteinuria on PTCs, using the above transgenic model, with the goals to verify whether megalin is involved in the reabsorption of filtered proteins of large molecular size, and if so, whether megalin participates in tubule injury. To induce massive non-selective proteinuria, the kidney-specific megalin knockout (hereafter designated as megalin-KO) mice were mated with another strain of transgenic mouse (NEP25), which expresses human (h) CD25 selectively in podocytes. After injection of hCD25-targeted recombinant immunotoxin, anti-Tac (Fv)-PE38 (LMB2) (17), NEP25 mice develop nephrotic syndrome, focal segmental glomerular sclerosis (FSGS) and secondary tubulointerstitial injury. These injuries are dependent on the dosage of LMB2 and the duration after LMB2 injection, but not on gender (18).
RESULTS
Characterization of megalin-KO/NEP25 mice without LMB2
We examined the extent of megalin expression in the kidney of megalin-KO/NEP25 mice by two methods. Western blot analysis on whole kidneys revealed that the amount of megalin in megalin-KO/NEP25 mice was decreased, on average, to 40% compared with that of control mice (megalinloxP/loxP; ApoE-Cre(−); NEP25, hereafter designated as megalin-intact/NEP25) (Figure 1). Analysis of immunostained sections showed that megalin was uniformly and intensively expressed on the apical membrane of the PTCs and the glomerular parietal epithelial cells in megalin-intact/NEP25 mice. By contrast, megalin staining was defective in mosaic pattern in PTCs and glomerular parietal epithelial cells in megalin-KO/NEP25 mice. Assessment of megalin staining in five randomly selected fields revealed that the proportion of megalin expressing cells to the whole PTCs was variable among megalin-KO/NEP25 mice, ranging from 34.7 to 50.1%, and on average 41.4±6.4%, while that in megalin-intact/NEP25 mice was 100%.
Figure 1. Western blotting of megalin in megalin-KO/NEP25 (KO) and megalin –intact/NEP25 mice (intact).

The amount of megalin expression in megalin-KO/NEP25 mice was decreased, on average, to 40% of that of megalin-intact/NEP25 mice.
Without LMB2, urinary total protein/creatinine ratio of megalin-KO/NEP25 mice was, on average, 33.0±13.1 in male and 5.4±3.3 in female mice, which were comparable to those of megalin-intact/NEP25 mice, 26.3±7.2 (male), 4.5±4.0 (female) (NS). However, SDS-PAGE analysis of the urine confirmed that megalin-KO/NEP25 mice excreted higher amount of low molecular weight proteins although the value varied among animals (Figure 2).
Figure 2. SDS-PAGE analysis of urine collected from megalin-KO/NEP25 (KO) and megalin-intact/NEP25 mice (intact).

The analysis confirmed that megalin-KO/NEP25 mice excreted predominantly low molecular weight proteins before LMB2 injection. The analysis also confirmed that proteinuria after LMB2 injection was non-selective, i.e., not only low but also intermediate and high molecular-weight proteins were excreted in large quantities.
The intense bands at ~20 kDa of mouse major urinary protein before LMB2 injection diminished after LMB2 injection by a mechanism beyond the scope of the current investigation.
Both megalin-KO/NEP25 and megalin-intact/NEP25 mice showed normal renal histology without LMB2.
Induction of proteinuria and tubule injury
We performed a pilot study to determine the appropriate dosage of LMB2 and the timing of analysis using NEP25 mice with wild-type megalin gene. We found that 10 days after the injection of 1.25 ng/g BW of LMB2, when mild but significant tubule injury developed, megalin remained readily detectable in 100% of the PTCs. The identity of the latter was verified by their localization in the cortex, presence of brush border and negative staining for both Tamm-Horsfall Protein (THP), a marker of the thick ascending limb, and thiazaide-sensitive NaCl cotransporter (TSH), a marker of the distal tubule, on the adjacent sections. With higher doses of LMB2, or at later time points, more severe tubule injury developed. However, megalin was downregulated in severely injured PTCs. To be able to determine megalin genotype of PTCs in megalin-KO/NEP25 mice by megalin staining, we employed the above experimental conditions in the subsequent studies.
After injection of 1.25 ng/g BW of LMB2, megalin-KO/NEP25 and megalin-intact/NEP25 mice similarly showed massive proteinuria. Urinary protein/creatinine ratio was, on average, 125.0±43.0 in male and 91.1±38.1 in female megalin-KO/NEP25 mice, which were comparable to those in megalin-intact/NEP25 mice (113.8±64.2 in male and 78.6±79.1 in female, NS). SDS-PAGE analysis confirmed that proteinuria was non-selective, i.e. not only low but also intermediate and high molecular weight proteins were excreted, and the quantity was large (Figure 2). At the time of assessment, both megalin-KO/NEP25 and megalin-intact/NEP25 mice similarly showed mild edema and ascites with body weight gain, on average, 16.8±22.3% in megalin-KO/NEP25 mice and 15.6±17.1% in megalin-intact/NEP25 mice (NS).
Light microscopic analyses revealed that both megalin-KO/NEP25 and megalin-intact/NEP25 mice mostly developed mild glomerular injury, and less than 5.3% of glomeruli were modestly or severely damaged (Figure 3). Semiquantification of glomerular injury revealed that the indices for glomerular mesangial and epithelial injury were, on average, 1.04±0.76 and 1.05±0.97, respectively, in megalin-KO/NEP25 mice, and 0.96±0.80 and 0.93±0.82, respectively, in megalin-intact/NEP25 mice. There was no significant difference between the two groups, indicating that similar mild glomerular injury was induced in both groups.
Figure 3. Histology of the kidney of a megalin-KO/NEP25 mouse given LMB2.

The histology shows mild to severe glomerular injury, casts and protein reabsorption droplets in proximal tubule cells (depicted by black arrows).
Both types of mice similarly showed mild tubulointerstitial changes with occasional tubule dilatation and rare fibrosis.
Protein reabsorption in megalin (+) and (−) PTCs in megalin-KO/NEP25 mice
As approximately 40% of PTCs still express megalin in megalin-KO/NEP25 mice, we compared protein reabsorption by megalin-intact (+) vs. megalin-deficient (−) PTCs within the same megalin-KO/NEP25 mouse kidney. Without LMB2, small albumin droplets were seen in a small fraction of PTCs whereas immunoglobulin κ light chain (IgL), IgA (α heavy chain) and IgG (whole molecule) were rarely stained in PTCs.
After LMB2 injection, small to large albumin droplets were seen in PTCs in variable degrees in megalin-KO/NEP25 mice (Figure 4b). Immunostaining for IgL (Figure 4d), IgA (Figure 4f) and IgG (Figure 4h) showed small to large reabsorption droplets in a fashion similar to albumin in megalin-KO/NEP25 mice.
Figure 4. Accumulation of proteins in proximal tubule cells of megalin-KO/NEP25 mouse given LMB2.

In (a)–(h), black arrows depict megalin (+) proximal tubule cells (PTCs), and yellow arrows depict megalin (−) PTCs. (a) and (b) are from adjacent sections stained for megalin and albumin, respectively. (c) and (d) are from adjacent sections stained for megalin and immunoglobulin light chain (IgL), respectively. (e) and (f) are from adjacent sections stained for megalin and IgA, respectively. (g) and (h) are from adjacent sections stained for megalin and IgG, respectively. (b), (d), (f) and (h) show immunostained droplets of respective protein in megalin (+) PTCs.
(i)–(l) are from serial sections of megalin-KO/NEP25 mouse kidney stained for megalin (i), IgL (j), IgA (k) and IgG (l), respectively. Note that megalin (+) PTCs (black arrows) contain IgL, IgA and IgG, but megalin (−) PTCs (yellow arrows) do not.
Figures 4a and 4b are from adjacent sections of a megalin-KO/NEP25 mouse kidney stained for megalin and albumin. In Figure 4a, megalin (+) (black arrows) and (−) (yellow arrows) PTCs are next to each other. The albumin staining in the adjacent section (Figure 4b) demonstrates that megalin (+) PTCs contain albumin droplets whereas megalin (−) PTCs do not.
Morphometrical analysis revealed that, on average, 25.2±14.6% of megalin (+) PTCs accumulated albumin in the cytoplasm whereas only 5.0±4.6% of megalin (−) cells showed albumin accumulation (p 0.004, Figure 5a). Analysis for IgL (Figure 4c and d), IgA (Figure 4e and f) and IgG (Figure 4g and h) demonstrated that, likewise, these proteins were preferentially accumulated in megalin (+) PTCs. Thus, the ratio of protein accumulation in megalin (+) vs. (−) cells was, on average, 14.5±8.7% vs. 1.3±1.4% (p 0.003) for IgL (Figure 5b), 18.8±7.3% vs. 2.1±3.2% (p 0.003) for IgA (Figure 5c), and 7.3±4.6% vs. 0.1±0.3 (p 0.005) for IgG (Figure 5d), respectively.
Figure 5. Percentage of protein-accumulating cells in megalin (+) vs. (−) proximal tubule cells of megalin-KO/NEP25 mice given LMB2.
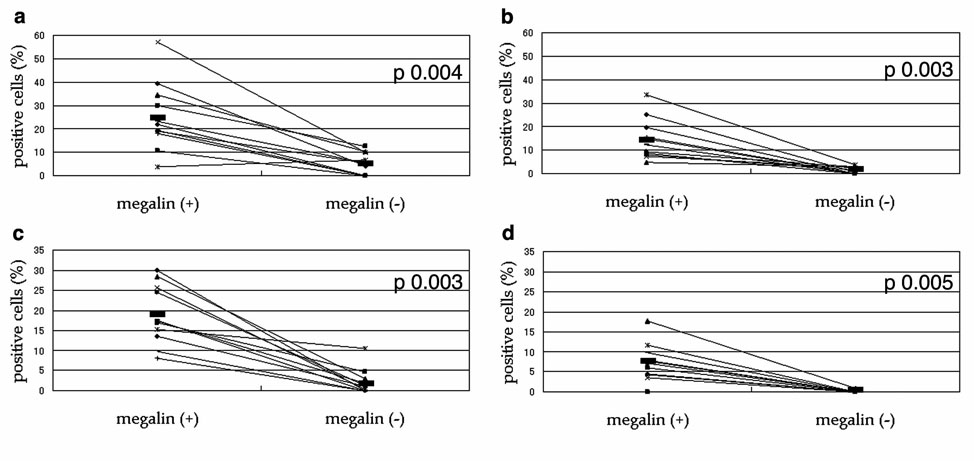
In these figures, each pair of dots connected with a line represents average values for megalin (+) vs. (−) proximal tubule cells (PTCs) from the same mosaic mouse. (a), percentage of albumin accumulating cells. (b), percentage of immunoglobulin light chain (IgL) accumulating cells. (c), percentage of IgA accumulating cells. (d), percentage of IgG accumulating cells. All four species of protein assessed were accumulated preferentially in megalin (+) PTCs.
Relatively low percentage of IgG accumulation likely reflects low affinity of the antibody used for staining of the antigen.
Serial section analysis revealed that protein reabsorption droplets in megalin (+) cells of megalin-KO/NEP25 mice (Figure 4i, black arrows) contain IgL (Figure 4j), IgA (Figure 4k) and IgG (Figure 4l).
Assessment of proximal tubule cell injury in megalin (+) and (−) PTCs in megalin-KO/NEP25 mice
We next compared tubule insult/injury markers in megalin (+) and (−) PTCs within the same megalin-KO/NEP25 mouse kidney. Thus, we assessed expressions of heme-oxygenase-1 (HO-1), monocyte chemoattractant protein-1 (MCP-1) and aquaporin-1 (AQP-1) and the evidence of apoptosis.
As shown in Figure 6b, HO-1 staining shows numerous small round-shape materials in the cytoplasm of PTCs, which resemble protein reabsorption droplets on PAS-stained tissues. All of the HO-1 expressing cells shown in this figure are megalin positive (Figure 6a). Quantitative analysis in all megalin-KO/NEP25 mice revealed that, on average, 2.32±2.26% of megalin (+) PTCs expressed HO-1 whereas only 0.24±0.20% of megalin (−) PTCs expressed HO-1 (p 0.003, Figure 7a).
Figure 6. Expression of early tubule injury markers in proximal tubule cells of megalin-KO/NEP25 mouse given LMB2.

In each picture, black arrows depict megalin (+) proximal tubule cells (PTCs), and yellow arrows depict megalin (−) PTCs. (a) and (b) are from adjacent sections stained for megalin and heme-oxygenase-1 (HO-1), respectively. HO-1 is selectively expressed in megalin (+) PTCs. (c) and (d) are from adjacent sections stained for megalin and monocyte chemoattractant protein-1 (MCP-1), respectively. MCP-1 is selectively expressed in megalin (+) PTCs. (e) is a section double-stained for megalin and TUNEL. Only megalin (+) PTCs are undergoing apoptosis. (f) and (g) are from adjacent sections stained for megalin and aquaporin-1 (AQP-1), respectively. Expression of AQP-1 is diminished in megalin (+) and (−) PTCs similarly.
Figure 7. Percentage of early injury marker-expressing cells in megalin (+) vs. (−) proximal tubule cells of megalin-KO/NEP25 mice given LMB2.

In (a), (b) and (c) , each pair of dots connected with a line represents total values for megalin (+) vs. (−) cells from each mosaic mouse. (a), percentage of heme-oxygenase-1 (HO-1) expressing cells. (b), percentage of monocyte chemoattractant protein-1 (MCP-1) expressing cells. (c), percentage of apoptotic cells. HO-1 expression, MCP-1 expression and apoptosis were observed preferentially in megalin (+) proximal tubule cells (PTCs). (d), percentage of aquaporin-1 (AQP-1) diminishing cells. Each pair of dots connected with a line represents average values for megalin (+) vs. (−) cells from each mosaic mouse. Diminished AQP-1 was observed similarly in megalin (+) and (−) PTCs.
Likewise, most MCP-1 expressing cells and apoptotic cells were positive for megalin staining (Figure 6d and e). Thus, on average, 0.37±0.57% of megalin (+) PTCs expressed MCP-1 whereas only 0.08±0.12% of megalin (−) PTCs expressed MCP-1 (p 0.043, Figure 7b). Moreover, on average, 0.43±0.52% of megalin (+) PTCs underwent apoptosis whereas only 0.13±0.19% of megalin (−) PTCs underwent apoptosis (p 0.026, Figure 7c).
AQP-1 is normally expressed on both apical and basolateral membranes of PTCs. In a variety of glomerular diseases including immunofluorescence-negative mesangial proliferative glomerulonephritis, IgA nephropathy and Lupus nephritis, expression of AQP-1 is diminished along the basolateral membrane (19). As shown in Figure 6g, PTCs in megalin-KO/NEP25 mice injected with LMB2 showed diminution of basolateral AQP-1 expression. However, AQP-1 diminution was not limited to megalin (+) PTCs. Quantitative analysis revealed that, on average, 34.7±25.4% of megalin (+) PTCs and 38.0±14.8% of megalin (−) PTCs showed diminution of basolateral AQP-1 (NS, Figure 7d).
DISCUSSION
The present study explored the function of megalin in PTCs exposed to massive non-selective proteinuria following glomerular podocyte injury. Analysis in megalin-KO/NEP25 mice, which lack tubule megalin in mosaic fashion, revealed that uptake of albumin, IgL, IgA and IgG was dependent on megalin. In addition, megalin (+) PTCs preferentially expressed three injury markers, namely HO-1, MCP-1 and apoptosis more intensely, compared to megalin (−) PTCs within the same kidney.
Previous studies showed that albumin and IgL are ligands of megalin (20), however, it was not known whether macromolecules including immunoglobulin heavy chains are ligands of megalin. The anti-IgA antibody used in the present study was specific for α heavy chain. The finding that most IgA staining was in megalin (+) PTCs indicates that uptake of α heavy chain is dependent on megalin. As our study did not directly test that α heavy chain has significant affinity to megalin, it is conceivable that heavy chain may be incorporated into PTCs through interaction between light chain and megalin.
Of importance, in our study, megalin (+) PTCs showed more HO-1, i.e., an oxidative stress marker, more MCP-1, i.e., an inflammatory marker, and more apoptosis than megalin (−) PTCs within the same kidney under massive non-selective protein overload. Expression of these markers has been shown to be closely associated with tubule injury in a variety of settings including protein overload (21, 22, 23, 24). Our study indicated that there is a causal relationship between macromolecule reabsorption through megalin and the injury markers. Since HO-1 activation has been shown to cause NF-κB activation (8, 21, 24), possible mechanism underlying tubule injury is that massive macromolecule uptake somehow induces oxidative stress, which then causes NF-κB activation and inflammatory responses, leading to tubule injury. However, further studies are necessary to clarify precise molecular mechanisms.
Our findings are in accordance with previous reports showing close association between high molecular weight proteinuria and tubule injury. Thus, in the remnant kidney model and passive Heymann nephritis model, interstitial infiltrates are observed in the vicinity of tubule segment that contains casts or PTCs reabsorbing IgG (25). An in vitro study utilizing cultured PTCs showed that high concentration IgG induced expression of inflammatory and fibrogenic mediators, including MCP-1 and RANTES (26). Moreover, Morais et al. documented a relationship between high molecular weight proteins and apoptosis in a study with human PTCs (27). Exposure to 100–440 and 30–440 kDa plasma protein fractions produced significant increases in apoptosis whereas PTCs exposed to a 30–100 kDa fraction were not significantly different from control cells. Some clinical studies have also described that non-selective proteinuria, but not selective proteinuria, is closely associated with tubule injury and poor prognosis (6, 7).
In the recent study by Theilig et al. discussed earlier, evidence was lacking to indicate that endocytosis plays a pathogenic role in the development of the tubulointerstitial damage in anti-GBM nephritis, a model for rapidly progressive glomerular nephritis (15). In that, proteinuria was mild and presumed to be primarily albuminuria, and tubule injury was closely associated with an encroachment of the crescents into glomerulotubular transition and tubule obstruction (15). In contrast, NEP25 is characterized by massive non-selective proteinuria, in which focal segmental glomerular sclerosis and tubule damage develop without obstruction of glomerulotubular junction. It appears, then, that, when compared to low molecular weight proteins, high molecular weight proteins have substantially greater injurious impact on PTCs, and in nephrosis when compared to nephritis, proteinuria has a greater pathogenic significance in the development of tubule damage.
When LMB2 is administered into wild-type mice, it is accumulated in the kidney and excreted into the urine, suggesting that LMB2 is filtered through glomeruli and reabsorbed by tubule cells (28). One may concern that reabsorption of LMB2 may be mediated through megalin, so that megalin (+) PTCs are preferentially injured through reabsorption of LMB2. However, this is unlikely since LMB2 was previously shown to have no toxic effect on tubule cells in wild-type mice. (18). We verified this aspect in the present study, also. Thus, we injected 1.25 ng/g BW of LMB2 into megalin-KO and megalin-intact mice without hCD25 transgene. Again, none of these mice showed proteinuria, abnormal histology, expression of HO-1, MCP-1 or apoptosis in the tubule.
AQP-1 is expressed in both apical and basolateral membranes of PTCs and works as a water channel. Diminution of its expression was shown previously in cisplatin nephritis (29), unilateral ureteral obstruction (30) and several human renal diseases (19). Although we did observe AQP-1 diminution in basolateral membrane dominantly, there was no significant correlation with megalin expression or protein reabsorption by PTCs. The finding is consistent with the existence of tubule injury processes independent of protein reabsorption, such as one triggered by tubule obstruction (31).
Of note, our study was limited to assessment of early stages of tubule injury, i.e., at stages when the genotype marker, megalin, still remains intact and appreciable, and the damage of PTCs of a given genotype has yet to bring about secondary injury to the PTCs of the other genotype. In fact, in our study, when studied in late stages, severe tubule damage was always accompanied by lack/loss of megalin expression. In that, one may erroneously conclude that lack of megalin brought about injury instead of the other way around. In any case, further studies are required to determine whether the effect of megalin on the PTCs demonstrated in the present study by several early injury markers continues to be significant throughout the development of histologically full-blown tubulointerstitial changes, and whether reabsorption of filtered macromolecules alone can bring about the histological changes.
In summary, at least at the stage we analyzed, when glomeruli are injured and a large quantity of proteins is filtered, a variety of proteins are reabsorbed by PTCs via megalin, the latter leading to expression of early injury markers in PTCs. It should be noted that the model we studied is unique, namely it is characterized by massive non-selective proteinuria, and the assessment was made shortly after the onset of proteinuria. It is, therefore, entirely conceivable that other mechanisms also operate in tubule injuries, which take place more chronically with less severe proteinuria. The present study does not determine the quantitative importance of those mechanisms.
METHODS
Animal experiment
The Animal Experimentation Committee at Tokai University Medical School approved the protocol, in accordance with the principles and procedures outlined in the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
Both kidney-specific megalin knockout mice (megalinloxP/loxP;ApoE-Cre(+)) and NEP25 mice were maintained on C57BL/6 strain. These were mated more than twice to obtain mice homozygous for megalin-loxP and heterozygous for ApoE-Cre and NEP25 (megalinloxP/loxP; ApoE-Cre(+); NEP25).
Eleven (4 males and 7 females) megalin-KO/NEP25 mice and 10 (5 males and 5 females) megalin-intact/NEP25 mice, 6 to 22 weeks of age, were weighed and housed in metabolic cages for 24 hours for urine collection. Then, under anesthesia with diethyl ether, mice were intravenously injected with 1.25 ng/g BW LMB2 diluted in 0.1ml of 0.1% BSA/ phosphate-buffered-saline (PBS). Nine days after the injection, mice were weighed, and 24-h urine was collected. Ten days after the injection, mice were euthanized, and kidney samples were harvested. For Western blot analysis, kidney samples were snap-frozen in liquid nitrogen and stored at −80°C. For histological analysis, kidney samples were fixed in buffered 4% paraformaldehyde and embedded in paraffin.
Concentration of total protein and creatinine were determined in an outside laboratory using an automatic analyzer (SRL, Tokyo, Japan). SDS-PAGE was performed on urine samples (2 µl) using 2 to 15% gradient gel.
Western blotting for megalin
Kidney samples from megalin-KO/NEP25 and megalin-intact/NEP25 mice were homogenized in a lysis buffer, which is one tablet of Complete Protease Inhibitor Cocktail (Roche Diagnostic, Basel, Switzerland) dissolved in 25ml PBS, and the protein concentration was determined using RC DC Protein Assay Kit II (Bio-Rad, CA, USA). Samples, each containing 50 µg protein, were separated in NuPAGE® 3–8% Tris-Acetate gel (Invitrogen, CA, USA) under a denatured condition. After electrophoresis, proteins were transferred onto PVDF membrane (Immobilon-P, Millipore, Tokyo, Japan) using a wet transfer system at 15V, overnight. Then, the membrane was blocked in 3%BSA/0.1%NaN3/PBS for one hour at 37°C, incubated with anti-rat megalin antibody (32) diluted at 1:5000 at 37°C overnight. The membrane was then, washed in 0.05% Tween20/PBS for 60 minutes 3 times, incubated in HRP-anti rabbit IgG (1:5000, GE Healthcare, Chalfont St Giles, UK) for one hour, washed in 0.05% Tween20/PBS again. Signals were detected using ECL Plus Western Blotting Detection Reagents (GE Healthcare).
Assessment of glomerular injury
To quantify glomerular damage, injuries of epithelial cells and mesangial cells were graded separately for each glomerulus on PAS-stained 2-µm sections, using a score of 0 to 4, with 0 representing no lesion and 4 representing severest lesion as we described previously (18).
Immunostaining
Immunostaining was performed on 2-µm serial paraffin sections. Primary antibodies used were polyclonal rabbit anti-rat megalin antibody (1:1000), polyclonal rabbit anti-human THP antibody (1:600, Biomedical Technologies, MA, USA), polyclonal rabbit anti-mouse TSC antibody (1:1000, Chemicon, CA, USA), polyclonal rabbit anti-mouse albumin antibody (1:2500, Nordic Bioscience, Herlev, Denmark), polyclonal goat anti-mouse Kappa light chain (IgL) antibody (1:500, Cappel, Ohio, USA), polyclonal goat anti-mouse IgA (alpha heavy chain) antibody (1:500, Cappel), polyclonal goat anti-mouse IgG (whole molecule) antibody (1:100, Cappel), polyclonal rabbit anti-rat AQP-1 antibody (1:300, Chemicon), polyclonal rabbit anti-HO-1 antibody (1:1000, Stressgen, MI, USA) and polyclonal goat anti-mouse MCP-1 antibody (1:70, R&D systems, MN, USA). For albumin, IgL, IgG and HO-1, sections were heated in citrate buffer by autoclave or microwave. For TSC and MCP-1, sections were treated with 0.1% trypsin for 10 min at 37°C. Immunoreactive signals were visualized by biotin-secondary antibodies, Vectastain ABC Reagent (Vector Laboratories, CA, USA) and diaminobentidine (DAB, Nichirei Bioscience, Tokyo, Japan). Sections stained for megalin was counterstained with PAS, and for others with hematoxylin.
For detection of apoptosis, TUNEL staining was performed utilizing a detection kit (ApopTag Peroxidase In Situ Apoptosis Detection Kit S7100, Chemicon) and visualized with DAB. For analysis, TUNEL staining was performed following megalin staining performed on the same sections, visualized with Vector VIP Substrate Kit for Peroxidase (Vector Laboratories).
Morphometrical analysis
In sections stained for megalin, five non-overlapping high power fields were randomly chosen in the cortex and photographed. In each field, all PTCs (approximately 100 per a field) were assessed, and the megalin genotype was determined by its staining. On the adjacent sections stained for albumin, IgL, IgA or IgG, each PTC was assessed for cytoplasmic accumulation of these proteins. The ratio of protein-accumulating vs. non-accumulating cells was determined for megalin (+) and (−) PTCs separately. Similarly, AQP-1 expression was assessed on the sections adjacent to megalin-stained sections. The ratio of AQP-1 diminishing vs. non-diminishing cells was determined for megalin (+) and (−) PTCs.
For HO-1 examination, all PTCs expressing HO-1 on tissue sections were assessed and megalin genotype was determined for each cell on the adjacent sections stained for megalin. Assessment for MCP-1 was performed in a similar fashion. Apoptosis was assessed on sections double-stained for megalin and TUNEL. All PTCs positive for TUNEL staining were assessed, and megalin genotype was determined. For these analyses, the proportion of megalin (+) or (−) PTCs to total PTCs was determined in five randomly selected representative fields. It was utilized to calculate the proportion of HO-1 or MCP-1 expressing cells or apoptotic cells in megalin (+) and (−) PTCs, separately.
Statistical analysis
Results are expressed as means±SD. Mann-Whitney U test was used to compare megalin-KO/NEP25 and megalin-intact/NEP25 groups. Wilcoxon signed-rank test was used to compare megalin (+) vs. (−) cells within megalin-KO/NEP25 mouse. Values were considered significant at p<0.05. It is indicated as NS if there was no statistical difference.
ACKNOWLEDGMENTS
We thank Mr. Tsukamoto, Ms. Sakurai and Ms. Imai for their expert technical assistance.
This study was supported by Grant-in Aid for Scientific Research of the Japan Society for the Promotion of Science, 16109005, and in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
A part of this study was presented in abstract form at the International Pediatric Nephrology Association meeting (Budapest, Hungary, August 31 to September 4, 2007) and the American Society of Nephrology meeting (San Francisco, CA, November 2 to 5, 2007).
Footnotes
DISCLOSURE None
REFERENCES
- 1.Bruzzi I, Benigni A, Remuzzi G. Role of increased glomerular protein traffic in the progression of renal failure. Kidney Int. 1997;52 Suppl 62:S29–S31. [PubMed] [Google Scholar]
- 2.Birn H, Christensen EI. Renal albumin absorption in physiology and pathology. Kidney Int. 2006;69:440–449. doi: 10.1038/sj.ki.5000141. [DOI] [PubMed] [Google Scholar]
- 3.Abbate M, Zoja C, Remuzzi G. How does proteinuria cause progressive renal damage? J Am Soc Nephrol. 2006;17:2974–2984. doi: 10.1681/ASN.2006040377. [DOI] [PubMed] [Google Scholar]
- 4.Ruggenenti P, Perna A, Remuzzi G. Retarding progression of chronic renal disease: The neglected issue of residual proteinuria. Kidney Int. 2003;63:2254–2261. doi: 10.1046/j.1523-1755.2003.00033.x. [DOI] [PubMed] [Google Scholar]
- 5.Zandi-Nejad K, Eddy AA, Glassock RJ, et al. Why is proteinuria an ominous biomarker of progressive kidney disease? Kidney Int. 2004;66 Suppl 92:S76–S89. doi: 10.1111/j.1523-1755.2004.09220.x. [DOI] [PubMed] [Google Scholar]
- 6.Bazzi C, Petrini C, Rizza V, et al. A modern approach to selectivity of proteinuria and tubulointerstitial damage in nephrotic syndrome. Kidney Int. 2000;58:1732–1741. doi: 10.1046/j.1523-1755.2000.00334.x. [DOI] [PubMed] [Google Scholar]
- 7.Bakoush O, Grubb A, Rippe B, et al. Urine excretion of protein HC in proteinuric glomerular diseases correlates to urine IgG but not to albuminuria. Kidney Int. 2001;60:1904–1909. doi: 10.1046/j.1523-1755.2001.00018.x. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y, Rangan GK, Tay YC, et al. Induction of monocyte chemoattractant protein-1 by albumin is mediated by Nuclear Factor κB in proximal tubule cells. J Am Soc Nephrol. 1999;10:1204–1213. doi: 10.1681/ASN.V1061204. [DOI] [PubMed] [Google Scholar]
- 9.Erkan E, De Leon M, Devarajan P. Albumin overload induces apoptosis in LLC-PK(1) cells. Am J Physiol Renal Physiol. 2001;280:F1107–F1114. doi: 10.1152/ajprenal.2001.280.6.F1107. [DOI] [PubMed] [Google Scholar]
- 10.Sengul S, Zwizinski C, Simon EE, et al. Endocytosis of light chains induces cytokines through activation of NK-kappaB in human proximal tubule cells. Kidney Int. 2002;62:1977–1988. doi: 10.1046/j.1523-1755.2002.00660.x. [DOI] [PubMed] [Google Scholar]
- 11.Willnow TE, Hilpert J, Armstrong SA, et al. Defective forebrain development in mice lacking gp330/megalin. Proc Natl Acad Sci USA. 1996;93:8460–8464. doi: 10.1073/pnas.93.16.8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leheste JR, Melsen F, Wellner M, et al. Hypocalcemia and osteopathy in mice with kidney-specific megalin gene defect. FASEB J. 2003;17:247–269. doi: 10.1096/fj.02-0578fje. [DOI] [PubMed] [Google Scholar]
- 13.Simonet WS, Bucay N, Lauer SJ, et al. In the absence of a downstream element, the Apolipoprotein E gene is expressed at high levels in kidneys of transgenic mice. J Biol Chem. 1990;265:10809–10812. [PubMed] [Google Scholar]
- 14.Simonet WS, Bucay N, Pitas E, et al. Multiple tissue-specific elements control the Apolipoprotein E/C-I gene locus in transgenic mice. J Biol Chem. 1991;266:8651–8654. [PubMed] [Google Scholar]
- 15.Theilig F, Kriz W, Jerichow T, et al. Abrogation of protein through megalin-deficient proximal tubules does not safeguard against tubulointerstitial injury. J Am Soc Nephrol. 2007;18:1824–1834. doi: 10.1681/ASN.2006111266. [DOI] [PubMed] [Google Scholar]
- 16.Feith GW, Assmann KJ, Bogman MJ, et al. Lack of albuminuria in the early heterologous phase of anti-GBM nephritis in beige mice. Kidney Int. 1993;43:824–827. doi: 10.1038/ki.1993.116. [DOI] [PubMed] [Google Scholar]
- 17.Kreitman RJ, Bailon P, Chaudhary VK, et al. Recombinant immunotoxins containing anti-Tac(Fv) and derivatives of Pseudomonas exotoxin procedure complete regression in mice of an interleukin-2 receptor-expressing human carcinoma. Blood. 1994;83:426–434. [PubMed] [Google Scholar]
- 18.Matsusaka T, Xin J, Niwa S, et al. Genetic engineering of glomerular sclerosis in the mouse via control of onset and severity of podocyte-specific injury. J Am Soc Nephrol. 2005;16:1013–1023. doi: 10.1681/ASN.2004080720. [DOI] [PubMed] [Google Scholar]
- 19.Bedford JJ, Leader JP, Walker RJ. Aquaporin expression in normal human kidney and in renal disease. J Am Soc Nephrol. 2003;14:2581–2587. doi: 10.1097/01.asn.0000089566.28106.f6. [DOI] [PubMed] [Google Scholar]
- 20.Christensen EI, Birn H. Megalin and cubilin: multifunctional endocytic receptors. Nature reviews. 2002;3:258–268. doi: 10.1038/nrm778. [DOI] [PubMed] [Google Scholar]
- 21.Murali NS, Ackerman AW, Croatt AJ, et al. Renal upregulation of HO-1 reduces albumin-driven MCP-1 production: implications for chronic kidney disease. Am J Physiol Renal Physiol. 2007;292:F837–F844. doi: 10.1152/ajprenal.00254.2006. [DOI] [PubMed] [Google Scholar]
- 22.Shimizu M, Ohta K, Yang Y, et al. Glomerular proteinuria induces Heme Oxygenase-1 gene expression within renal epithelial cells. Pediatric Research. 2005;58:666–671. doi: 10.1203/01.PDR.0000180557.68222.5A. [DOI] [PubMed] [Google Scholar]
- 23.Kuusniemi AM, Lapatto R, Holmberg C, et al. Kidneys with heavy proteinuria show fibrosis, inflammation, and oxidative stress, but no tubular phenotypic change. Kidney Int. 2005;68:121–132. doi: 10.1111/j.1523-1755.2005.00386.x. [DOI] [PubMed] [Google Scholar]
- 24.Erkan E, Garcia CD, Patterson LT, et al. Induction of renal tubular cell apoptosis in focal segmental glomerulosclerosis: Roles of proteinuria and Fas-dependent pathways. J Am Soc Nephrol. 2005;16:398–407. doi: 10.1681/ASN.2003100861. [DOI] [PubMed] [Google Scholar]
- 25.Abbate M, Zoja C, Corna D, et al. In progressive nephropathies, overload of tubular cells with filtered proteins translates glomerular permeability dysfunction into cellular signals of interstitial inflammation. J Am Soc Nephrol. 1998;9:1213–1224. doi: 10.1681/ASN.V971213. [DOI] [PubMed] [Google Scholar]
- 26.Zoja C, Donadelli R, Colleoni S, et al. Protein overload stimulates RANTES production by proximal tubular cells depending on NF-kappa B activation. Kidney Int. 1998;53:1608–1615. doi: 10.1046/j.1523-1755.1998.00905.x. [DOI] [PubMed] [Google Scholar]
- 27.Morais C, Westhuyzen J, Metharom P, et al. High molecular weight plasma proteins induce apoptosis and Fas/FasL expression in human proximal tubular cells. Nephrol Dial Transplant. 2005;20:50–58. doi: 10.1093/ndt/gfh561. [DOI] [PubMed] [Google Scholar]
- 28.Kreitman RJ, Pastan I. Accumulation of a Recombinant Immunotoxin in a Tumor in vivo: Fewer than 1000 molecules per cell are sufficient for complete responses. Cancer Research. 1998;58:968–975. [PubMed] [Google Scholar]
- 29.Kishore BK, Krane CM, Iulio DD, et al. Expression of renal aquaporins 1, 2, and 3 in a rat model of cisplatin-induced polyuria. Kidney Int. 2000;58:701–711. doi: 10.1046/j.1523-1755.2000.00216.x. [DOI] [PubMed] [Google Scholar]
- 30.Li C, Wang W, Knepper MA, et al. Downregulation of renal aquaporins in response to unilateral ureteral obstruction. Am J Physiol Renal Physiol. 2003;284:F1066–F1079. doi: 10.1152/ajprenal.00090.2002. [DOI] [PubMed] [Google Scholar]
- 31.Kriz W, Hosser H, Hähnel B, et al. From segmental glomerulosclerosis to total nephron degeneration and interstitial fibrosis: a histopathological study in rat models and human glomerulopathies. Nephrol Dial Transplant. 1998;13:2781–2798. doi: 10.1093/ndt/13.11.2781. [DOI] [PubMed] [Google Scholar]
- 32.Saito A, Kazama JJ, Iino N, et al. Bioengineered implantation of megalin-expressing cells: a potential intracorporeal therapeutic model for uremic toxin protein clearance in renal failure. J Am Soc Nephrol. 2003;14:2025–2032. doi: 10.1097/01.asn.0000078804.98322.4a. [DOI] [PubMed] [Google Scholar]