

Abstract
Systemic lupus erythematosus (SLE) is an autoimmune disease mediated by autoantibodies (AAbs) and preferentially affecting women of childbearing age. Since the offspring of mothers with SLE exhibit a high frequency of learning disorders1-5, we hypothesized that maternally transferred AAbs that bind DNA and the N-methyl-D-aspartate receptor (NMDAR)6-12 could play a pathogenic role during fetal brain development. Here we describe a maternal SLE murine model wherein pregnant dams harbored DNA-specific, NMDAR-specific AAbs throughout gestation. High titers of these AAbs in maternal circulation led to histological abnormalities in fetal brain and subsequent cognitive impairments in adult offspring. These data support a paradigm in which in utero exposure to neurotoxic AAbs causes abnormal brain development with long-term consequences. This paradigm may apply to multiple congenital neuropsychiatric disorders.
Children born to mothers with SLE display high incidence of learning disorders compared to children born to healthy mothers1-5. The mechanisms for learning disorder remain unknown, but prematurity, low birth weight, maternal disease activity, and medications during pregnancy have not emerged as causative factors. Remarkably, a single study showed that children born from SLE fathers did not exhibit learning disorders1. This led us to ask whether maternally derived factors, particularly antibodies (Abs), might alter fetal brain development in utero, resulting in long-term cognitive changes in the offspring1-5.
Previous work has identified SLE AAbs that bind DNA and cross-react with native NMDAR6,7. Indeed, 30–60% of SLE patients exhibit these cross-reactive AAbs, which are found in serum, cerebrospinal fluid (CSF) and brain parenchyma8-13. Moreover, titers of these AAbs in CSF correlate significantly with manifestations of neuropsychiatric SLE (NPSLE)8,14. Our studies on mouse models of NPSLE show that NMDAR-specific AAbs mediate cognitive and behavioral abnormalities only following breaches in the blood-brain barrier (BBB)15,16. The nature of the brain injury depends on the regional specificity of the compromise of the BBB. When the BBB is disrupted by systemic administration of lipopolysaccharide, mimicking infection, neuronal injury occurs within hippocampus and is associated with memory impairment15. When epinephrine is used to breach the BBB, predominant neuronal death in amygdala leads to abnormal fear responses16.
Intrauterine and perinatal exposure to maternal immunoglobulin (IgG) is a physiologic event. During pregnancy, IgG is transported from mother to fetus across the placenta17. Appreciable rates of IgG transfer begin by the end of the first trimester. At term, the IgG concentration in fetal circulation can exceed that in maternal circulation. Postnatally, maternally acquired Abs are essential in the newborn’s immunity. They can, however, mediate tissue injury. For example, neonatal lupus, characterized by cutaneous lesions and congenital heart block, is mediated by Ro-specific AAbs18.
We exploited a mouse model in which dams harbored NMDAR-specific AAbs throughout gestation. We examined how in utero AAb exposure affected fetal brain development and behavioral outcome in adult offspring (Supplementary Fig. 1). The D/E W D/E Y S/G peptide is a mimetope of DNA; this consensus sequence is present within the NR2A and NR2B subunits of the NMDAR. Mice immunized with DWEYS pentapeptide included within a decapeptide sequence that is octamerized on a polylysine backbone, termed “MP”, developed either high or low titers of DNA-specific, NMDAR-specific AAbs, as previously described19, while female mice that were immunized with polylysine backbone alone, termed “MC”, did not (Fig. 1a). Three weeks later, MP and MC females became pregnant. We collected serum from MP and MC mice just prior to timed pregnancy and at the time of harvesting E15 fetal brains, or just prior to timed pregnancy and at postnatal day 0 (P0). The presence of NMDAR-specific AAbs in maternal circulation was assessed by peptide ELISA. Stable titers of NMDAR-specific AAbs were present throughout pregnancy in MP dams (Fig. 1a). Given the intrinsic variability in AAb production across animals, we were able to segregate MP dams into high AAb titer (MPH) and low AAb titer (MPL) groups. There was no appreciable reactivity to DNA or NMDAR in MC dams (Fig. 1a).
Figure 1.
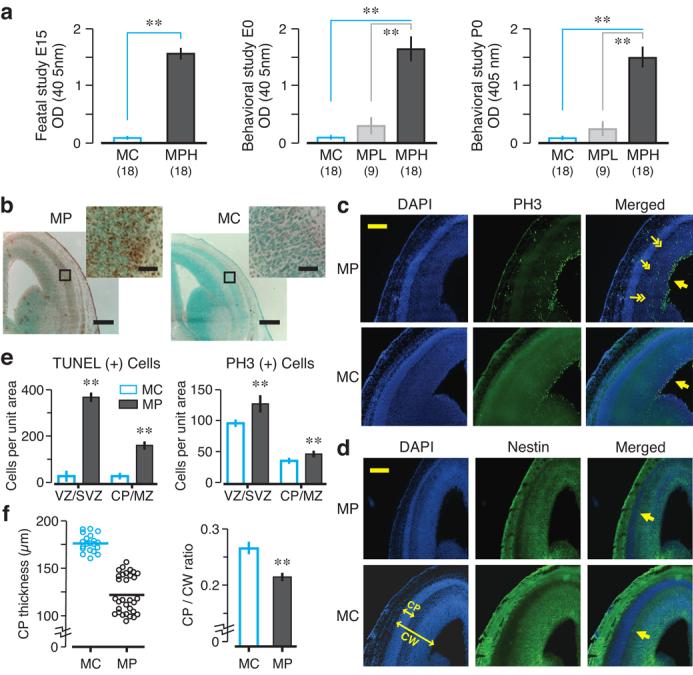
MP brains exposed to maternal NMDAR-specific AAbs in utero display morphological abnormalities at E15. (a) Serology of BALB/c adult females (n = 5 per group, 3 cohorts per group) used in fetal and behavioral studies, expressed as optical density (OD, means ± s.d.). Mice were immunized with DWEYSVWLSN peptide (MP, 100 μg) or polylysine core (MC, 100 μg). MP dams were segregated into high (MPH) and low (MPL) AAb titer groups. (b) TUNEL assays, in coronal sections, show increased cell death in MP brains. Insets (top right) correspond to the boxed areas. (c) DAPI (left panels) detects cell nuclei and PH3 (middle panels) assesses mitotic activity. Most PH3+ cells are located near the ventricular zone (VZ, single arrows), corresponding to the expected labeling pattern. In MP brain, PH3+ cells are also detected in the marginal zone (MZ, double arrows in merged MP panel). (d) The cortical plate (CP) is visualized as a DAPI-dense cellular band lacking nestin (arrows in merged panels). CW, cortical width. (e) TUNEL+ cells and PH3+ cells per unit area (means ± s.d., ** P < 0.0001, Mann-Whitney U test). (f) CP thickness (at left), measured from merged sections, is significantly reduced in MP neocortex (P < 0.0001, Mann-Whitney U test). Ratio of CP to CW, at right, is significantly lower in MP neocortex (means ± s.e.m, ** P < 0.0001, Mann-Whitney U test). For each assay, 2-3 fetal brains per dam were analyzed; n = 3–4 dams by group. Scale bar = 250 μm (inset = 25 μm).
To confirm that maternal Abs accessed fetal brain we conjugated R4A, a monoclonal NMDAR-specific AAb, to europium (Eu). On embryonic day 14 (E14), we gave Eu-labeled R4A to non-immunized pregnant dams and, 24 h later, we detected Eu-labeled R4A in fetal neocortex outside of blood vessels (Supplementary Fig. 2a), demonstrating transport of NMDAR-specific AAbs through fetal circulation and binding to fetal brain. AAbs were present primarily in the ventricular zone (VZ), probably reflecting the transport route into brain. We performed this analysis for each of our monoclonal Abs (Supplementary Fig. 2b) and found 60–70-fold more Ab in fetal brain than maternal brain per mg of tissue (Supplementary Fig. 2c). To demonstrate NMDAR antigen in fetal brain, we stained E15 brains directly with R4A or human monoclonal AAb with similar specificity (Supplementary Fig. 2d). Reactivity was greatest in the neocortex reflecting NMDAR expression at this stage of brain development.
To determine whether fetal neurons were vulnerable to AAb-mediated excitotoxicity, as we had shown for adult neurons6, we compared brains from E15 fetuses of MP and MC dams. In utero exposure to maternal NMDAR-specific AAbs caused increased neocortical cell death in E15 MP fetuses, assessed by TUNEL assay (Fig. 1b). Thus, the maternal AAbs were toxic to the developing brain.
We reasoned that a toxic effect of NMDAR-specific AAbs might be counterbalanced by increased neurogenesis. Thus, we looked for compensatory cell proliferation in E15 brains using phosphohistone-3 (PH3) reactivity. Expression of the M-phase cell cycle marker PH3 is widely used for assessing mitotic activity20. In MP and MC fetuses, PH3 positive (PH+) cells were appropriately localized near the apical surface of the lateral ventricle (Fig. 1c). In MP neocortex, there were more PH3+ cells distant from the germinal zones of the developing brain, suggesting both increased proliferation and perhaps a dysregulated cell cycle progression or abnormal migration of proliferating cells.
Neocortical development is a highly orchestrated process beginning with migration of postmitotic neurons into the cortical plate (CP)21. After entering the CP, postmitotic neurons organize into cortical lamina21. Coronal brain sections were probed for expression of nestin, an intermediate filament protein present in neuronal precursors. DAPI was used to stain cell nuclei. The CP was visualized as a cellular band, rich in DAPI staining and negative for nestin. On E15, the CP was significantly thinner in MP brains, and the ratio of CP to total cerebral width was significantly reduced (Fig. 1d–f), suggesting that proper organization of postmitotic neurons was disrupted by maternal NMDAR-specific AAbs. To further control for Ab specificity, dams were immunized with a scrambled (WSDYEVWLSN) or an alanine-rich sequence (AAAAAVWLSN). These dams did not exhibit NMDAR-specific or DNA-specific reactivity and the brains of their fetuses were indistinguishable from MC brains (Supplementary Fig. 3).
To confirm that NMDAR-specific AAbs alone could be responsible for the pathology, we passively administered R4A, or control IgG2b, to non-immunized gestating dams on E12 and examined fetal brains on E15. All preparations contained <100 pg endotoxin which, by itself, caused no histopathology. R4A bound DWEYS and DNA while control IgG2b did not (Fig. 2a). R4A-exposed brains displayed more apoptotic cells (data not shown), increased mitotic cells (Fig. 2b–c) and a thinned CP (Fig. 2d–e). To confirm that R4A-mediated damage was related to antigenic specificity, we treated pregnant dams with soluble D-peptide (DWEYSVWLSN), non-blocking D-peptide (NEMQSSRLRE) or saline during R4A administration and every 24 h thereafter. Fetal brains from dams given DWEYSVWLSN were normal while fetal brains given NEMQSSRLRE or saline displayed a thinned CP (Supplementary Fig. 4). We also administered human monoclonal Abs derived from B-cells of SLE patients to gestating dams22. In utero exposure to G11, cross-reactive with DNA and NMDAR (Fig. 3a), resulted in neocortical abnormalities (Fig. 3b–d). Exposure to control Abs (B1, binding neither DNA nor NMDAR, and D9, binding DNA but not NMDAR) produced no histopathology (Fig. 3 and Supplementary Fig. 4). Thus, both mouse and human NMDAR-specific AAbs were sufficient by themselves to interfere with normal fetal brain development, and neurotoxicity was unrelated to DNA binding.
Figure 2. Morphological abnormalities in E15 brains exposed to murine anti-NMDAR AAbs in utero.
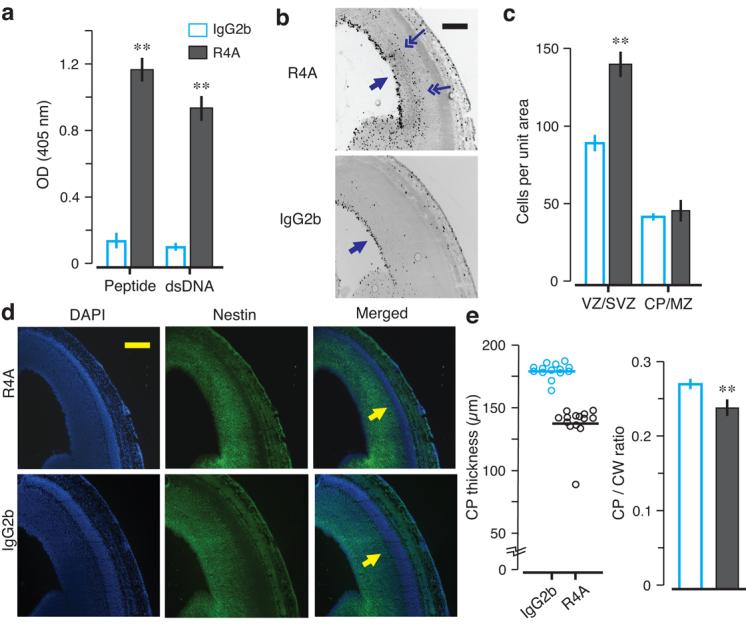
The monoclonal AAb R4A or the control IgG2b was intravenously administered to pregnant dams (200 μg per dam), at E12. Embryos were harvested at E15 for analysis. Coronal sections of E15 fetal brains were stained with PH3, DAPI and nestin. (a) R4A shows reactivity to MP (peptide) and double-stranded DNA (dsDNA) by ELISA (mean ± s.d., ** P < 0.0001, Mann-Whitney U test). (b) Mitotic activity is assessed with PH3 expression. The VZ harbors most PH3+ cells (single arrows) in both groups, but PH3+ cells are found outside of the VZ in R4A-exposed neocortex (double arrows). (c) VZ and (SVZ) of R4A-exposed fetal brains show significantly more PH+ cells (mean ± s.d., ** P < 0.0001, Mann-Whitney U test). (d) Combined DAPI/nestin stained sections of R4A or IgG2b neocortex are used to measure the CP (arrows in the merged panels). Fetal brains exposed to R4A in utero show CP thinning. (e) CP thickness is significantly decreased in the neocortex exposed to R4A (P < 0.0001, Mann-Whitney U test). CP to CW ratio is significantly reduced in R4A-treated neocortex (mean ± s.e.m., ** P < 0.0001, Mann-Whitney U test), demonstrating that passive transfer of R4A from maternal circulation to the fetal neocortex disrupts proper development of the CP. For each assay, 2 fetal brains per dam were analyzed; n = 3 dams for each group. Scale bar = 250 μm.
Figure 3. Morphological abnormalities in E15 brains exposed to human lupus anti-NMDAR AAbs in utero.
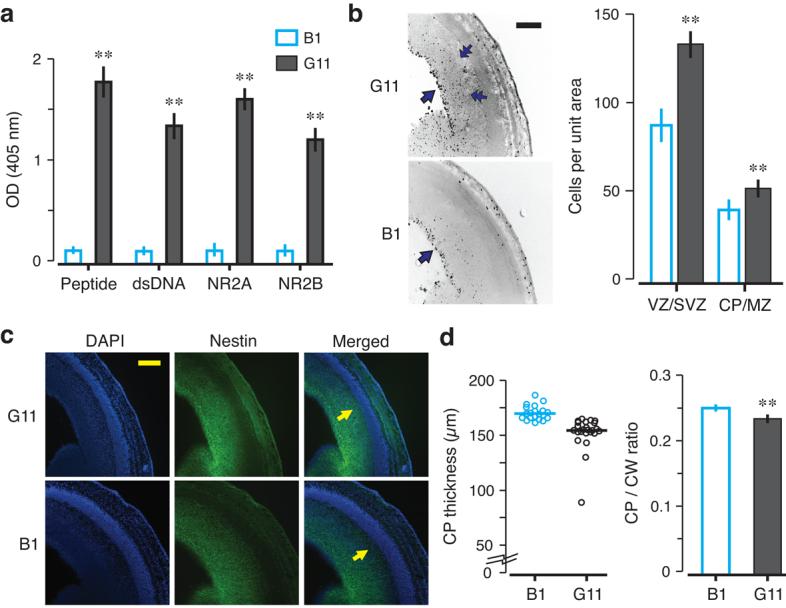
Monoclonal Abs (G11 and B1) were intravenously administered to pregnant dams (150 μg per dam), at E12. Embryos were harvested at E15. Coronal sections of fetal brains were stained with PH3, DAPI and nestin. (a) G11 and B1 were tested for peptide, dsDNA, NR2A and NR2B reactivity by ELISA. G11 shows robust reactivity with each antigen, while B1 shows negligible reactivity (mean ± s.d., ** P < 0.0001, Mann-Whitney U test). (b) PH3 staining shows that the VZ (single arrows) harbors most PH3+ cells. Similar to R4A-treated fetal cortex, PH3+ cells are found outside of the VZ in G11-exposed neocortex (double arrows). The graph displays the number of PH3+ cells per unit area (mean ± s.d., ** P < 0.0001, Mann-Whitney U test). (c) Coronal sections stained with DAPI and nestin were used to visualize the CP (arrows in merged sections). G11-exposed brains show CP thinning. (d) CP thickness is significantly decreased in G11-exposed neocortex when compared to B1 neocortex (P < 0.0001, Mann-Whitney U test). CP to CW ratio demonstrates that passive transfer of G11 from maternal circulation to fetal circulation causes disruption of the proper organization of the CP (mean ± s.e.m., ** P < 0.0001, Mann-Whitney U test). For each assay, 3 fetal brains per dam were analyzed; n = 2 dams for each group. Scale bar = 250 μm.
To assess a functional consequence of Aab exposure, we examined the negative geotaxis reflex which we could quantify reliably in very young pups23,24. MPH mice from dams with high NMDAR-specific AAb titers were exposed to ∼10-fold higher titer of NMDAR-specific AAbs in utero than MPL mice from dams with low AAb titers (Fig. 1a). MPH pups were delayed in reflex performance (Fig. 4a). By postnatal day 16 (P16), MC pups performed at adult level, MPL pups were somewhat sluggish, and MPH pups were significantly delayed. Late emergence of negative geotaxis correlated with levels of maternal NMDAR-specific AAbs to which the pups were exposed. Significantly, delayed acquisition of neonatal reflexes can presage cognitive abnormalities in adults25.
Figure 4. Congenital impairments in neonatal reflexes and cognitive tasks.
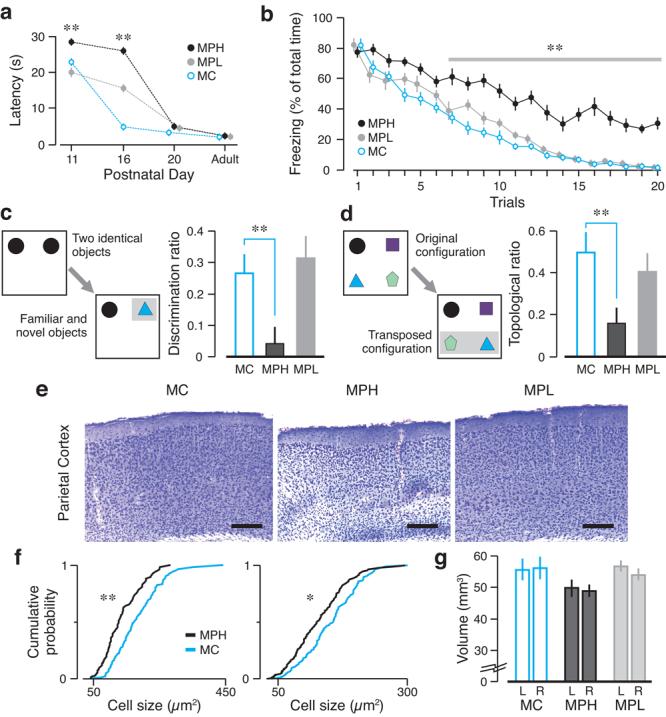
(a) Offspring born to MPH (n = 19), MPL (n = 14) and MC (n = 15) dams were examined in the negative geotaxis reflex. MPH offspring show significant delay in reflex acquisition (mean ± s.e.m., ** P < 0.001, t test MPH vs. MC). (b) The extinction task quantifies the ability to erase fearful responses to stimuli previously associated with foot-shocks. Adult MC (n = 10) and MPL (n = 8) groups show negligible freezing after 20 trials, whereas MPH (n = 10) mice display significantly higher freezing (means ± s.e.m., ** P < 0.001, ANOVA with trials as repeated measure). (c) The novel object recognition task (left diagram) shows that MPH mice are impaired (mean discrimination ratio ± s.e.m., ** P = 0.001, Mann-Whitney U test), whereas MPL mice behave normally. (d) The topological task (left diagram) shows that MPH mice fail to recognize novel configurations (mean topological ratio ± s.e.m., * P = 0.0015, Mann-Whitney U test). (e) Coronal sections of parietal cortex (cresyl violet) of adult mice show disorganization and thinning of cortical layers in MPH mice. Scale bar = 250 μm. (f) Cumulative probability plots for neuronal size are abnormal for MPH animals in the sensorimotor (left) and parietal (right) regions (* P < 0.05, ** P < 0.001, Kolmogorov-Smirnov test). (g) Regional volumes (mean ± s.d.) of adult neocortex show significant decrease in cortical volume in MPH mice (P < 0.0001, Kruskal-Wallis test).
Since NMDAR-specific AAbs disrupted neocortical development, we decided to test adult offspring in behavioral tasks that depended on proper neocortical processing. We used a fear extinction task26, subserved by the infralimbic and prelimbic areas of the prefrontal cortex, to measure behavioral flexibility. Initially, mice received five aversive footshocks that were paired with a tone (conditional stimulus, CS). All mice readily learned to associate the CS with the footshock, as measured by their increased freezing response. Fear extinction was tested 24 h later. MPH mice continued to freeze during CS-alone presentations, whereas MC and MPL mice did not freeze by the end of treatment (Fig. 4b, mean percent freezing on trial 20 ± s.e.m., MPH, 30.8±4, MPL, 1.9±0.4, MC, 1.6±0.3, n = 8–10, P = 0.0001, Mann-Whitney U test MPH vs. MC). A novel object recognition task27, depending specifically on the rhinal cortex, measured the preference of mice for new objects27. MPH mice failed to explore novel objects preferentially (Fig. 4c, mean discrimination ratios ± s.e.m., MPH, 0.04±0.05, MPL, 0.31±0.07, MC, 0.26±0.06, n = 8–10: P = 0.001, Mann-Whitney U test on MPH vs. MC). A topological task28, depending specifically on the parietal cortex, assessed the spontaneous exploration of transposed items. MPH mice did not show a preference for novel configurations, whereas MC and MPL mice behaved normally (Fig. 4d, mean topological ratios ± s.e.m., MPH, 0.16±0.07, MPL, 0.4±0.09, MC, 0.5±0.09, n = 8–10, P = 0.015, Mann-Whitney U test on MPH vs. MC). Thus, MPH mice were substantially altered in neocortically-based behaviors. MPL mice, despite an initial lag in reflex acquisition, displayed no long-term cognitive or behavioral sequelae. A neurological screen23 revealed no differences among groups (Supplementary Table 1). Additionally, all groups performed comparably on tasks that depended on the integrity of hippocampus, amygdala and striatum (Supplementary Fig. 5). Adult MPH mice showed disorganization of the neocortical architecture (Fig. 4e). MPH brains exhibited significant decreases in neocortical neuron size (Fig. 4f) and significant volume decreases in the neocortex (Fig. 4g). There were no detectable histological abnormalities and volume differences in the hippocampus and the amygdala in MPH mice (Supplementary Fig. 6).
Our study has clearly established that maternal Abs are transferred to the fetus during pregnancy and, in a dose-dependent manner, NMDAR-specific AAbs cause congenital brain injuries, resulting in long-term cognitive deficits. Fetal brain abnormalities translate into delayed neonatal reflex acquisition and impaired performance of neocortical-dependent tasks in adult offspring. Given that the BBB is not fully formed in utero, we show that pathogenic AAbs in maternal circulation represent a risk factor for the developing brain, even in the absence of brain disease in the mother.
The enhanced neocortical apoptosis of fetuses from dams harboring high titered NMDAR-specific AAbs during gestation suggests that maternal AAbs elicit NMDAR-mediated death of fetal neurons. While it is possible that maternal AAbs destroy fetal glial cells (NMDARs are expressed in astrocytes and oligodendrocytes29,30), we previously showed that NMDAR-specific AAbs did not trigger glial apoptosis6. Moreover, since neurogenesis precedes gliogenesis in brain development, it seems likely that fetal pathology, observed as early as E15, is mediated primarily by neuronal NMDARs.
NMDAR-specific AAbs disrupt the developmental program of the neocortex as evidenced by the failure to form a normal CP and the increase in mitotic cells distant from the VZ in affected fetuses. NMDARs help govern neuronal migration in the developing brain31. It is plausible, therefore, that the cortical abnormalities are related to altered migration of precursor neocortical neurons.
In this model, NMDAR-specific AAbs present in gestating dams cross-react with the NR2A and the NR2B subunits of the NMDAR. We think the observed phenotype in the fetal neocortex could be due to maternal AAbs targeting NR2B-containing NMDARs, as NR2B is the predominant NR2 subunit in the murine fetal cortex32. Unlike mouse NR2A expression, which appears to be strictly postnatal, the human NR2A subunit has been detected in fetal neocortex33. This, as well as potential differences in the NMDAR-associated signaling cascades, may be important distinctions for understanding species-specific responses to NMDAR-specific AAbs.
Previous reports of neurodevelopmental disorders with prenatal origin have implicated maternal serum factors34,35. Mice exposed in utero to human serum from a mother with both an autistic and a language-impaired child displayed abnormal exploratory behavior and cerebellar dysfunction35. Although this suggested that maternally derived antibodies could influence behavioral outcomes in offspring, the serum had no definitive neurotoxic properties and was obtained several years after the child's birth. Another study shows that IgG purified from serum of mothers with an autistic child, when transferred to pregnant monkeys, caused stereotypies akin to those exhibited by autistic children36.
Our study suggests that it would be quite informative to examine whether NMDAR-specific AAbs are associated with learning disorders in children born from lupus mothers. It is particularly interesting that histological studies of brains from individuals with learning disorders have demonstrated smaller-sized neurons in the neocortex and potential migration defects37,38, both of which are present in our model. Most importantly, this study suggests a powerful approach for examining how brain injury mediated by maternal AAbs might underlie multiple congenital, neurodevelopmental disorders. The model suggests that other AAbs from women with or without lupus could be explored for potential neurotoxic effects on the fetal brain.
METHODS
Animals and immunization
BALB/c female mice (6–8 week old) were obtained from Jackson Laboratory. Animal use conformed to institutional guidelines of Weill Cornell Medical College and the Feinstein Institute for Medical Research. Mice were immunized with multi-antigenic peptide coupled to DWEYSVWLSN (MP, 100 μg), control polylysine core (MC, 100 μg), scrambled WSDYEVWLSN (MS, 100 μg), or AAAAAVWLSN (MA, 100 μg, AnaSpec) as previously described15. ELISAs for Ab reactivity were performed as described15. We used mouse monoclonal AAb R4A or control IgG2b (200 μg, Southern Biotech), human monoclonal AAb G11, human control AAb B1 or human anti-DNA Ab D9 (150 μg), obtained by isolating peptide-reactive or non-reactive B cells and expressing the Ig heavy and light chain genes in 293 cells39,40. Abs were intravenously administered to gestating dams at E12. The D form of DWEYSVWLSN peptide (200 μg) or irrelevant peptide NEMQSSRLRE was given i.p. to mice at the time of intravenous R4A administration, and again 24 h and 48 h later. Eu-labeled monoclonal Abs (Perkin Elmer) were intravenously administered (100 μg) to gestating dams at E14. Fetal brains exposed to Eu-R4A were stained with Ab to CD-31 to identify blood vessels.
Immuno-histology of fetal brains
For TUNEL assays, coronal sections (20 μm thick) of E15 brains were processed according to the manufacturer (R&D Systems) and counterstained with methyl green. For PH3 assays of mitotic cells, frozen brain sections (20 μm) were blocked with 10% normal goat serum and incubated overnight at 4°C with PH3 Ab (1:1000; Chemicon, Millipore, Billerica, MA) followed by Red X goat anti-rabbit IgG (Jackson Immuno Research Laboratories). Measurements of the CP and the cerebral width were performed on nestin (1:200; Chemicon) and DAPI (1:1000; Invitrogen, Carlsbad, CA) stained E15 coronal sections, with OpenLab software (Improvision, PerkinElmer). Quantification of PH3+ and TUNEL+ cells was performed using automated image analysis programs on an Axio-Image microscope (Zeiss, Thornwood,NY). Anatomic regions were identified using within section coordinates and positive cells were counted in comparable areas of interest.
Behavioral assessment
Negative Geotaxis was measured23 in offspring born from MPH (n = 19), MPL (n = 14) and MC (n = 15) dams at ages P11, P15 or P16 and P20. Mice were placed with their heads facing downward on an incline (45°) and the time to turn 180° and climb the incline was measured (maximum 30 s, if the animal did not reach the top of the incline in the allotted time, a score of 30 s was assigned). Each mouse was given three trials and the mean latency was calculated. Adult male offspring from MPH (n = 10), MPL (n = 8) and MC (n = 10) groups underwent a behavioral battery15,16 (described in Supplementary Fig. 5). We used a blinded format and randomized the order in which the cohorts of animals were tested in different tasks. Tasks that depend on the integrity of neocortical regions are described below. The novel object recognition task27 consisted of three phases. In the first (sample), mice explored two identical objects for 5 min. In the second (delay), mice stayed in their home cage for 10 min. Test objects replaced sample objects, so that one was identical to those in the sample phase (familiar object) and the other was different (novel object). For the final phase (choice), mice inspected the test objects for 5 min. Object exploration was scored automatically with the PC-based AnyMaze software (Stoelting). We defined A3 as the exploration time for the familiar object, and B1 as the exploration time for the novel object, and measured the discrimination ratio, (B1−A3) / (B1+A3). The topological task28 consisted of five sessions (5 min) with varying intersession intervals (3 min, 10 min, 3 min and 10 min; sequentially). Four different objects were arranged in a square. In sessions 1 and 2, mice were familiarized to this arrangement. Before session 3, two objects were transposed, creating a novel configuration II. Animals were expected to explore displaced objects more avidly during session 3 and 4. Before Session 5, the two objects that had remained unchanged were switched, thus creating configuration III. Session 5 permitted exploration of another novel arrangement. Object exploration was scored automatically with AnyMaze. We defined D1 as the sum of the times exploring the displaced objects in configurations II and III (sessions 3 and 5) and D2 as the sum of the times exploring the non-displaced objects (sessions 3 and 5). Topological processing was measured with the ratio, (D1−D2) / (D1+D2). The fear extinction task26 initially required animals to associate the conditioned stimulus (CS, 20-s tone, 5 kHz, 80 dB) with the unconditioned stimulus (US, foot-shock, 1 s, 0.6 mA), as previously described16. Extinction learning was tested 24 h later by exposing mice to 20 trials of CS alone in a novel chamber, with an intertrial interval of 120 s. To determine fear extinction, we measured the amount of freezing during each tone, using FreezeFrame software (Actimetrics).
Adult Histology
Eight months after birth, mice that had been subjected to behavioral studies were anesthetized and perfused. The anterior striatum, and the anterior and posterior commissure were the internal landmarks to block and section the tissue in order to determine the volume of neocortical areas, hippocampus and amygdala. Tissue was stained with cresyl violet and mouse anti-NeuN (1:1000; Chemicon). Coronal sections were sampled every fourth 40-μm slice. Internal cortical coordinates were the interhemispheric fissure and the anterior corpus callosum; for hippocampus and amygdala (basolateral and lateral nuclei), the coronal perimeters were captured on video screen by Ibas software (Zeiss) from the dorsal third ventricle to the posterior commissure. For neuron size determinations, video capture of neurons was performed using an optical dissector randomly sampling each region of interest.
Statistical analysis
Statistical comparisons were performed with Kruskal-Wallis and Fisher's analysis of variance, Student's t test (unpaired, two-sided), Mann-Whitney U test, and Kolmogorov-Smirnov non-parametric test. We used the software packages SPSS (SPSS Inc.), Statistica (StatSoft) and Origin (OriginLab) for our analyses. P < 0.05 was determined as significant.
Supplementary Material
ACKNOWLEDGEMENTS
The work was supported by grants from the US National Institutes of Health (BD, PTH and BTV) and the Alliance for Lupus Research (BTV). We are grateful to L. DeGiorgio, S. Loncar, T. Huerta and R. Berlin for technical assistance; M. Scharff, E. Chang and T. Faust for suggestions on the manuscript; and S. Jones for help in preparing the manuscript.
REFERENCES
- 1.Lahita RG. Systemic lupus erythematosus: learning disability in the male offspring of female patients and relationship to laterality. Psychoneuroendocrinology. 1988;13:385–396. doi: 10.1016/0306-4530(88)90045-5. [DOI] [PubMed] [Google Scholar]
- 2.McAllister DL, et al. The influence of systemic lupus erythematosus on fetal development: cognitive, behavioral, and health trends. J Int Neuropsychol Soc. 1997;3:370–376. [PubMed] [Google Scholar]
- 3.Neri F, et al. Neuropsychological development of children born to patients with systemic lupus erythematosus. Lupus. 2004;13:805–811. doi: 10.1191/0961203304lu2018oa. [DOI] [PubMed] [Google Scholar]
- 4.Ross G, Sammaritano L, Nass R, Lockshin M. Effects of mothers' autoimmune disease during pregnancy on learning disabilities and hand preference in their children. Arch Pediatr Adolesc Med. 2003;157:397–402. doi: 10.1001/archpedi.157.4.397. [DOI] [PubMed] [Google Scholar]
- 5.Tincani A, et al. Impact of in utero environment on the offspring of lupus patients. Lupus. 2006;15:801–807. doi: 10.1177/0961203306071005. [DOI] [PubMed] [Google Scholar]
- 6.DeGiorgio LA, et al. A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nat Med. 2001;7:1189–1193. doi: 10.1038/nm1101-1189. [DOI] [PubMed] [Google Scholar]
- 7.Kowal C, et al. Human lupus autoantibodies against NMDA receptors mediate cognitive impairment. Proc Natl Acad Sci USA. 2006;103:19854–19859. doi: 10.1073/pnas.0608397104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoshio T, Onda K, Nara H, Minota S. Association of IgG anti-NR2 glutamate receptor antibodies in cerebrospinal fluid with neuropsychiatric systemic lupus erythematosus. Arthritis Rheum. 2006;54:675–678. doi: 10.1002/art.21547. [DOI] [PubMed] [Google Scholar]
- 9.Omdal R, et al. Neuropsychiatric disturbances in SLE are associated with antibodies against NMDA receptors. Eur J Neurol. 2005;12:392–398. doi: 10.1111/j.1468-1331.2004.00976.x. [DOI] [PubMed] [Google Scholar]
- 10.Lapteva L, et al. Anti-N-methyl-D-aspartate receptor antibodies, cognitive dysfunction, and depression in systemic lupus erythematosus. Arthritis Rheum. 2006;54:2505–2514. doi: 10.1002/art.22031. [DOI] [PubMed] [Google Scholar]
- 11.Husebye ES, et al. Autoantibodies to a NR2A peptide of the glutamate/NMDA receptor in sera of patients with systemic lupus erythematosus. Ann Rheum Dis. 2005;64:1210–1213. doi: 10.1136/ard.2004.029280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steup-Beekman GM, Steens SC, van Buchem MA, Huizinga TW. Anti-NMDA receptor autoantibodies in patients with systemic lupus erythematosus and their first-degree relatives. Lupus. 2007;16:329–334. doi: 10.1177/0961203307078224. [DOI] [PubMed] [Google Scholar]
- 13.Hanly JG. New insights into central nervous system lupus: a clinical perspective. Curr Rheumatol Rep. 2007;9:116–124. doi: 10.1007/s11926-007-0005-2. [DOI] [PubMed] [Google Scholar]
- 14.Loyo HF, et al. Serum and cerebrospinal fluid autoantibodies in patients with neuropsychiatric lupus erythematosus. Implications for diagnosis and pathogenesis. PloS Med. doi: 10.1371/journal.pone.0003347. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kowal C, et al. Cognition and immunity; antibody impairs memory. Immunity. 2004;21:179–188. doi: 10.1016/j.immuni.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 16.Huerta PT, Kowal C, DeGiorgio LA, Volpe BT, Diamond B. Immunity and behavior: antibodies alter emotion. Proc Natl Acad Sci USA. 2006;103:678–683. doi: 10.1073/pnas.0510055103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morphis LG, Gitlin D. Maturation of the maternofoetal transport system for human gamma-globulin in the mouse. Nature. 1970;228:573. doi: 10.1038/228573a0. [DOI] [PubMed] [Google Scholar]
- 18.Buyon JP, Clancy RM. Neonatal lupus syndromes. Curr Opin Rheumatol. 2003;15:535–541. doi: 10.1097/00002281-200309000-00003. [DOI] [PubMed] [Google Scholar]
- 19.Putterman C, Diamond B. Immunization with a peptide surrogate for double-stranded DNA (dsDNA) induces autoantibody production and renal immunoglobulin deposition. J Exp Med. 1998;188:29–38. doi: 10.1084/jem.188.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanashima C, et al. Brain factor-1 controls the proliferation and differentiation of neocortical progenitor cells through independent mechanisms. J Neurosci. 2002;22:6526–6536. doi: 10.1523/JNEUROSCI.22-15-06526.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kriegstein AR, Noctor SC. Patterns of neuronal migration in the embryonic cortex. Trends Neurosci. 2004;27:392–399. doi: 10.1016/j.tins.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 22.Zhang J, et al. Identification of DNA-reactive B cells in patients with systemic lupus erythematosus. J Immunol Methods. 2008;338:79–84. doi: 10.1016/j.jim.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rafael JA, Nitta Y, Peters J, Davies KE. Testing of SHIRPA, a mouse phenotypic assessment protocol, on Dmd(mdx) and Dmd(mdx3cv) dystrophin-deficient mice. Mamm Genome. 2000;11:725–728. doi: 10.1007/s003350010149. [DOI] [PubMed] [Google Scholar]
- 24.Ten VS, Bradley-Moore M, Gingrich JA, Stark RI, Pinsky DJ. Brain injury and neurofunctional deficit in neonatal mice with hypoxic-ischemic encephalopathy. Behav Brain Res. 2003;145:209–219. doi: 10.1016/s0166-4328(03)00146-3. [DOI] [PubMed] [Google Scholar]
- 25.Lubics A, et al. Neurological reflexes and early motor behavior in rats subjected to neonatal hypoxicischemic injury. Behav Brain Res. 2005;157:157–165. doi: 10.1016/j.bbr.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 26.Quirk GJ, Garcia R, González-Lima F. Prefrontal mechanisms in extinction of conditioned fear. Biol Psychiatry. 2006;15:337–343. doi: 10.1016/j.biopsych.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 27.Barker GR, et al. The different effects on recognition memory of perirhinal kainate and NMDA glutamate receptor antagonism: implications for underlying plasticity mechanisms. J Neurosci. 2006;29:3561–3566. doi: 10.1523/JNEUROSCI.3154-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goodrich-Hunsaker NJ, Hunsaker MR, Kesner RP. Dissociating the role of the parietal cortex and dorsal hippocampus for spatial information processing. Behav Neurosci. 2005;119:1307–1315. doi: 10.1037/0735-7044.119.5.1307. [DOI] [PubMed] [Google Scholar]
- 29.Verkhratsky A, Kirchhoff F. NMDA receptors in glia. Neuroscientist. 2007;13:28–37. doi: 10.1177/1073858406294270. [DOI] [PubMed] [Google Scholar]
- 30.Salter MG, Fern R. NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature. 2005;438:1167–1171. doi: 10.1038/nature04301. [DOI] [PubMed] [Google Scholar]
- 31.Manent JB, et al. A noncanonical release of GABA and glutamate modulates neuronal migration. J Neurosci. 2005;25:4755–4765. doi: 10.1523/JNEUROSCI.0553-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Zundert B, Yoshii A, Constantine-Paton M. Receptor compartmentalization and trafficking at glutamate synapses: a developmental proposal. Trends Neurosci. 2004;27:428–437. doi: 10.1016/j.tins.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 33.Ritter LM, Unis AS, Meador-Woodruff JH. Ontogeny of ionotropic glutamate receptor expression in human fetal brain. Brain Res Dev Brain Res. 2001;127:123–133. doi: 10.1016/s0165-3806(01)00126-2. [DOI] [PubMed] [Google Scholar]
- 34.Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci. 2003;23:297–302. doi: 10.1523/JNEUROSCI.23-01-00297.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dalton P, et al. Maternal neuronal antibodies associated with autism and a language disorder. Ann Neurol. 2003;53:533–7. doi: 10.1002/ana.10557. [DOI] [PubMed] [Google Scholar]
- 36.Martin LA, et al. Stereotypies and hyperactivity in rhesus monkeys exposed to IgG from mothers of children with autism. Brain Behav Immun. 2008;22:806–816. doi: 10.1016/j.bbi.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Galaburda AM, Sherman GF, Rosen GD, Aboitiz F, Geschwind N. Developmental dyslexia: four consecutive patients with cortical anomalies. Ann Neurol. 1985;18:222–233. doi: 10.1002/ana.410180210. [DOI] [PubMed] [Google Scholar]
- 38.Walsh CA. Genetic malformations of the human cerebral cortex. Neuron. 1999;23:19–29. doi: 10.1016/s0896-6273(00)80749-7. [DOI] [PubMed] [Google Scholar]
- 39.Newman J, Rice JS, Wang C, Harris SL, Diamond B. Identification of an antigen-specific B cell population. J Immunol Methods. 2003;272:177–187. doi: 10.1016/s0022-1759(02)00499-4. [DOI] [PubMed] [Google Scholar]
- 40.Wardemann H, et al. Predominant autoantibody production by early human B cell precursors. Science. 2003;301:1374–1377. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.