

Abstract
Peripheral inflammation and edema are often accompanied by primary and secondary hyperalgesia which are mediated by both peripheral and central mechanisms. The role of cyclooxygenase-2 (COX-2)-mediated prostanoid production in hyperalgesia is a topic of substantial current interest. We have established a murine foot-pad inflammation model in which both pharmacologic and genetic tools can be used to characterize the role of COX-2 in hyperalgesia. Zymosan, an extract from yeast, injected into the plantar surface of the hind paw induces an edema response and an increase in COX-2 expression in the hindpaw, spinal cord and brain. Zymosan-induced primary hyperalgesia, measured as a decrease in hindpaw withdrawal latency in response to a thermal stimulus, is long-lasting and is not inhibited by pre-treatment with the systemic COX-2 selective inhibitor, parecoxib (20 mg/kg). In contrast, the central component of hyperalgesia, measured as a reduction in tail flick latency in response to heat, is reduced by parecoxib. Zymosan-induced primary hyperalgesia in Cox-2−/− mice is similar to that of their Cox-2+/+ littermate controls. However, the central component of hyperalgesia is substantially reduced in Cox-2−/− versus Cox-2+/+ mice, and returns to baseline values much more rapidly. Thus pharmacological data suggest, and genetic experiments confirm, (i) that primary hyperalgesia in response to zymosan inflammation in the mouse paw is not mediated by COX-2 function and (ii) that COX-2 function plays a major role in the central component of hyperalgesia in this model of inflammation.
Introduction
Cyclooxygenases (COX) are the key enzymes in the synthesis of prostaglandins, prostacyclins and thromboxanes. The cyclooxygenases convert free arachidonic acid, following its release from membrane phospholipids by phospholipases, to prostaglandin H2 (PGH2), the common precursor for all prostanoids. There are two cyclooxygenases; cyclooxygenase-1 (COX-1) is generally constitutively expressed. In contrast, cyclooxygenase-2 (COX-2), an inducible COX isoform, is expressed in inflammatory cells and tissues in response to cellular activation by endotoxin, cytokines, mitogens and other stimuli [1]. Non-steroidal anti-inflammatory drugs (NSAIDs), which inhibit both COX-1 and COX-2 enzymatic activity, and COX-2 selective inhibitors (coxibs) are widely used for the treatment of pain and inflammatory conditions [2].
Acute inflammatory responses are characterized by an increase in vascular permeability and cellular infiltration, leading to edema. After inflammation, allodynia and hyperalgesia occur, generally because of an increase in PGE2 in the inflamed tissue and in the spinal cord that is associated with an induction and activation of COX-2 [3–5]. The COX-2 enzyme is the major source of PGE2 in many inflammatory pain models; consequently COX-2-selective inhibitors are potent antihyperalgesic agents. In rat models of carrageenan, zymosan or formalin-evoked hyperalgesia, coxibs markedly reduce pain symptoms [6–8].
Zymosan, a yeast cell wall extract, is a phagocytic stimulant which induces inflammatory reactions that are accompanied by the synthesis of eicosanoids and cytokines [9,10]. Zymosan-induced paw inflammation in the rat is well characterized and is frequently used to study edema and hyperalgesia responses [11,12]. In contrast, the literature on similar mouse peripheral inflammation models is relatively scant [13,14], and detailed analysis of COX-2 gene activation and function in the hyperalgesic response to paw inflammation in mice is not well studied [15–17]. Because of the advantages of mouse genetics and the ability to make murine transgenic and knockout models, inflammation models in this species should facilitate our understanding of the role of COX-2 in inflammation and hyperalgesia.
We describe here the development of a murine foot-pad inflammation model to characterize the role of COX-2 in inflammatory responses and the accompanying hyperalgesia that follow intraplantar zymosan administration. By combining pharmacologic approach (using a selective COX-2 inhibitor) and a genetic manipulation (using COX-2 knockout mice), we have explored both the molecular response of the Cox-2 gene to this inflammatory stimulus and the functional role of COX-2 in peripheral and central components of hyperalgesia.
Materials and methods
Animals
Experiments were performed on C57BL/6 mice (8–10 weeks old, 20–30 g; Charles River, USA), Cox-2−/− knockout mice (C57BL/6 and 129 background) [18] and their Cox-2+/+ littermate controls. All mice were housed under standard laboratory conditions and kept under a 12/12 hour light/dark cycle. Mice were fed a standard NIH-31 modified 6% mouse/rat pellet diet 7013 (Teklad, Madison, WI) and water ad libitum. Behavioral studies were carried out between 0900 and 1800 h. All experimental protocols have been approved by the Animal Research Committee, University of California, Los Angeles, USA.
Zymosan-induced hindpaw inflammation
To induce inflammation, mice received intraplantar injections of zymosan (Sigma, St. Louis, MO) in 30 μl of phosphate buffered saline (PBS) in the right (ipsilateral) hindpaw and 30 μl of PBS in the left (contralateral) hindpaw. Hindpaw edema was measured with a calibrated dental caliper with 1 mm gradations, and is expressed as percent change in dorso-ventral hindpaw thickness relative to baseline values observed before intraplantar injection.
Measurement of thermal hyperalgesia
A modified Hargreaves apparatus (model #390, IITC instruments, Woodland Hills, CA) was used to assess thermal hyperalgesia by application of a focused radiant heat source to the hindpaw [19]. Prior to testing, the mice were allowed to acclimate for 30 minutes to the testing environment; a 10×10×20 cm translucent plastic box with a 3 mm glass bottom (preheated to 30 °C) through which the thermal stimulus was applied. Each mouse was tested three times, to obtain consistent baseline paw withdrawal latencies (PWL). The stimulus intensity was adjusted to yield a baseline hindpaw withdrawal latency of approximately 7–9 seconds for naïve mice, with a cutoff set at 20 seconds to avoid tissue damage. Three responses were measured alternately in both ipsilateral and contralateral hindpaws, with a 5–10 minute interval for each experimental point. After two days of baseline testing, thermal hyperalgesia was assessed at times indicated following intraplantar zymosan injection.
Measurement of tail-flick latency
A modified Hargreaves apparatus was also used to measure tail-flick latency (TFL). Radiant heat was directed toward the proximal part of the tail and the TFL was observed and timed with a photo cell counter. The intensity of the radiant heat was adjusted for a baseline TFL of approximately 5–7 seconds for naïve mice, with a 15 seconds cutoff set to avoid tissue damage. Control mice for TFL studies were injected with PBS (30 μl) in the ipsilateral hindpaw.
RT-PCR for COX-2
Paw tissues were homogenized in Trizol Reagent (Invitrogen, Carlsbad, CA). RNA was extracted from homogenate supernatants according to the manufacturer’s instructions. RNA concentrations were determined spectrophotometrically. Single stranded cDNA was synthesized from 1 μg of total RNA, using oligo dT primer and AMV reverse transcriptase (Takara RNA PCR Kit, Takara Bio Inc., Otsu, Japan). PCR was performed using Takara Taq. The oligonucleotide primers used for PCR analysis were: COX-2 (forward), 5′-CAAGCAGTGGCAAAGGCCTCCA-3′; COX-2 (reverse), 5′-GGCACTTGCATTGATGGTGGCT-3′; GAPDH (glyceraldehyde-3-phosphatedehydrogenase, forward), 5′-CATGGAGAAGGCCGGGGCCC-3′; GAPDH (reverse), 5′-GACGGACACATTGGGGGTAGG-3′. PCR was performed for 45 cycles. Cycling conditions were 94° for 30 seconds 60° for 30 seconds and 72° for 1.5 minutes. PCR products were analyzed using 1.5% agarose gel electrophoresis; DNA was visualized by staining with ethidium bromide. RT-PCR product sizes for COX-2 and GAPDH were 459 and 415 nucleotides respectively.
Western blot for COX-2
Tissue samples were lysed and homogenized in RIPA buffer (1% NP-40, 0.5% sodium deoxycholate and 0.1% SDS in 1× PBS) containing a proteinase inhibitor cocktail (Roche Diagnostics, Indianapolis, IN). After centrifugation, supernatant proteins (100 μg) were separated in 10% SDS polyacrylamide gels and transferred to nitrocellulose membranes. The membranes were blocked with 5% skim milk and incubated with polyclonal antibodies against COX-2 (1000-fold dilution, Cayman Chemical, Ann Arbor, MI) or 14-3-3 (500-fold dilution, Santa Cruz Biotechnology Inc, Santa Cruz, CA), used as a loading control. The ECL detection system (Amersham Biosciences, Piscataway, NJ) was used to detect the signals on X-ray film. The film was scanned and analyzed for quantification using NIH image software.
Immunostaining for COX-2
Hindpaws were fixed with 10% formalin and embedded in paraffin, and sections (4 μm) were cut. Some sections were stained with hematoxylin & eosin, while others were processed for COX-2 immunostaining. The sections were deparaffinized with xylene, and antigen retrieval was performed with citrate buffer [20], then blocked with 5% normal goat serum in PBS and incubated with rabbit anti-COX-2 (100-fold dilution, overnight at 4°C, Cayman Chemical) in PBS with 5% normal goat serum. The slides were then washed three times at room temperature with PBS; each wash was 5 min. The sections were incubated with biotinylated goat anti-rabbit IgG (300-fold dilution, at room temperature, Vector Labs, Burlingame, CA). After washing in PBS, sections were processed with the ABC kit (Vector labs), according to the manufacturer’s instructions. Slides were examined with a BX60 microscope (Olympus, Melville, NY). Images were captured with a Macrofire 599831 camera and processed with Pictureframe software (Optronics, Goleta, CA).
Reagents
Zymosan A was from Sigma, USA. Parecoxib sodium was gift from Panacea Biotec Ltd. (New Delhi, India). Parecoxib was dissolved in PBS, and administered intraperitoneally (10 ml/kg of body weight) 0.5 hr prior to zymosan injection.
Statistical analysis
Results are presented as means ± S.E.M. Significant differences were determined by unpaired Student’s t-test. p<0.01 was considered statistically significant.
Results
Paw inflammation and COX-2 expression following intraplantar zymosan administration in C57BL/6 mice
We first determined the dose-response relationship for zymosan concentration and hindpaw inflammation in C57BL/6 mice. Intraplantar injection of zymosan (0.5, 1.0 or 2.0% w/v) produces pronounced, dose-dependent edema (Fig. 1A). This inflammatory response is time-dependent and long-lasting; up to 14 days following zymosan administration. Injection of saline into the contralateral hindpaw does not result in significant edema (data not shown).
Fig. 1.
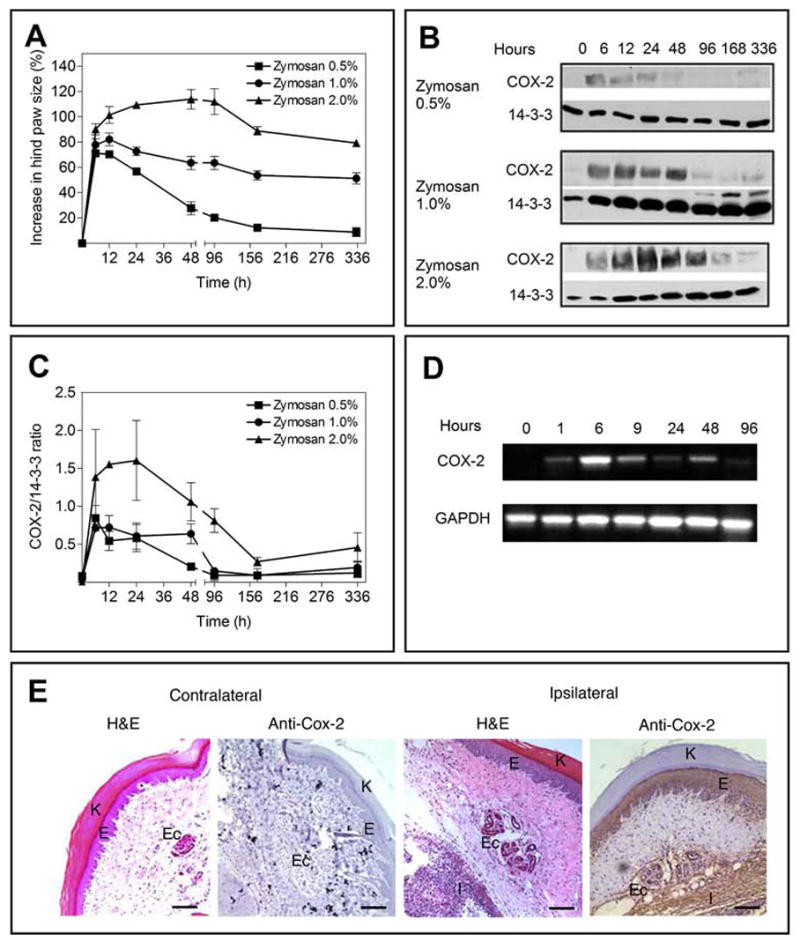
(A) Zymosan (0.5, 1.0 and 2% w/v) induces paw edema following intraplantar injection in C57BL/6 mice. Zymosan suspensions (30 μl) were injected into the right (ipsilateral) hindpaw and PBS was injected into the left (contralateral) hindpaw. Paw thickness was measured at the times shown following zymosan injection. Data are expressed as percent increase in paw edema and are presented as means ± SEM for 6 mice per group. (B) Western blots were performed on homogenates prepared from mouse hindpaws at the times shown following intraplantar zymosan injection (30 μl). 100 μg of total protein was loaded in each well. Antibody to 14-3-3 protein was used as a loading control. (C) Quantification of the data from panel B. Data are expressed as the ratios of COX-2/14-3-3 intensities from optical density analyses, and are presented as means ± SEM for three mice at each time point. (D) RT-PCR for COX-2 mRNA was performed on homogenates prepared from mouse hindpaws at the times shown following intraplantar zymosan A injection (2%, 30 μl). GAPDH mRNA was analyzed as a control. (E) Histological appearance of mouse hindpaws 24 hrs following intraplantar zymosan (2%, 30 μl) and contralateral PBS (30 μl) administration. H&E staining and COX-2 immunostaining (brown color) are shown for the contralateral paw (left two panels) and the ipsilateral paw (right two panels). K; keratin layer, E; epidermal layer, Ec; eccrine tubular glands, I; infiltrating inflammatory cells. Scale bar: 100 μm.
To determine the time course and the location of COX-2 expression in response to zymosan injection we performed Western blotting, RT-PCR and immunohistological studies. COX-2 protein and mRNA are not detectable in saline-injected hindpaws. A zymosan (2%) injection that induces maximal edema induces COX-2 protein and mRNA accumulation in the hindpaw, peaking at 12--24 hours and 6 hours respectively, and subsequently returning to baseline values (Figs. 1B, 1C and 1D). Zymosan injected paws exhibit massive accumulation of degenerating neutrophils within the space between the muscles of the digits, and in the muscles and tendons of the primary palmar pad (Fig. 1E). Additionally there is a severe generalized subcutaneous edema with moderate to severe infiltration of neutrophils, fewer macrophages and minimal numbers of lymphocytes. In contrast, the cell architecture of contralateral, saline-injected paws remains normal. Immunohistological staining with anti-COX-2 antibody confirms the local accumulation of COX-2 protein in epidermal cells and infiltrating inflammatory cells of the ipsilateral, zymosan-injected paw.
Hyperalgesia in response to intraplantar zymosan administration
To determine the hyperalgesic response in mice following inflammation resulting from intraplantar zymosan injection, we used the Hargreaves thermal hyperalgesia test [19]. Prior to intraplantar zymosan injection, there are no significant differences in baseline hindpaw withdrawal latencies (HWL) for the right and left (ipsilateral and contralateral) hindpaws of C57BL/6 mice (data for contralateral hindpaws are not shown). At all tested zymosan doses, thermal hyperalgesia is observed in the ipsilateral, zymosan-injected hindpaws. The hyperalgesic response occurs within 6 hrs of zymosan administration, and is long-lasting. Doses of 1% and 2% induce similar hyperalgesia, suggesting that a maximal hyperalgesic response to inflammation occurs at these concentrations (Fig. 2A). No hyperalgesia occurs in the contralateral, saline-injected paw (data not shown).
Fig. 2.

(A) Zymosan-induced thermal hyperalgesia in C57BL/6 mice. Zymosan suspensions (0.5, 1.0 and 2%; 30 μl) were injected into the right (ipsilateral) hindpaw and PBS was injected as a control treatment into the left (contralateral) hindpaw. Hindpaw (HWL) withdrawal latency data are expressed as time (in seconds) to paw withdrawal, and are presented as means ± SEM for 4–6 mice per group. *p<0.01, ipsilateral HWL compared to contralateral HWL (not shown) at each time point. (B) Tail-flick latency (TFL) following zymosan-induced paw inflammation, for the mice described in Panel A. TFL data are expressed as means ± SEM for 4–6 mice per group. *p<0.01 for each time point, compared to TFL prior to zymosan/saline injection.
To study central mechanisms of hyperalgesia following peripheral tissue injury, we used the tail-flick latency (TFL) test. In this test, reduced latency is attributable to central nervous system mechanisms since thermal activation of primary afferent fibers that innervate the tail does not directly activate primary afferents innervating the inflamed hindpaw [8]. All tested zymosan doses induce a significant reduction in TFL after intraplantar injection, indicating central sensitization (Fig. 2B). Zymosan doses of 1% and 2% induce similar reductions in TFL, suggesting a maximal centrally-mediated hyperalgesic response to the peripheral (hindpaw) inflammation is occurring at these doses. TFL remains unchanged throughout the study period in mice injected with saline in the ipsilateral paw (data not shown).
Zymosan-induced peripheral inflammation is accompanied by COX-2 induction in the spinal cord and brain
Since we observed a central component of hyperalgesia following intraplantar zymosan administration (Fig. 2B), we also examined COX-2 induction in the brain and spinal cord. Control mice were injected with saline in the intraplantar hindpaw region; experimental mice were injected with zymosan (2%). Brain and spinal cord tissue extracts were prepared 24 hrs following injection. Western blots demonstrate that COX-2 protein levels are elevated in the spinal cord and in brain in response to zymosan-induced peripheral inflammation (Fig. 3).
Fig. 3.
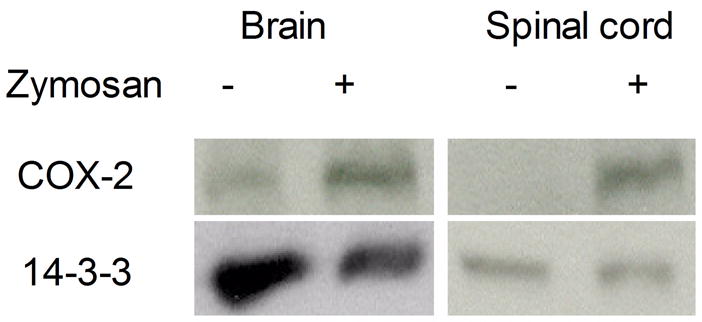
COX-2 protein accumulation in the brain and spinal cord following intraplantar zymosan administration in the paws of C57BL/6 mice. Western blots were performed on homogenates prepared from the brains and spinal cords of mice 24 hrs after intraplantar zymosan injection (2.0%; 30 μl). 100 μg of total protein was loaded in each well. Antibody to 14-3-3 protein was used as a loading control.
Selective COX-2 inhibition decreases the central component of hyperalgesia, but not primary hyperalgesia
Elevated COX-2 expression, both in the inflamed hindpaw and in the central nervous system following intraplantar zymosan injection, prompted us to examine whether blocking COX-2 activity with the selective COX-2 inhibitor parecoxib can abrogate local edema and either primary hyperalgesia (measured by the HWL test) and/or the central component of hyperalgesia (measured by the TFL test). A single administration of parecoxib (20 mg/kg, i.p.) [21] significantly ameliorated local edema (Fig. 4A) and the central component of hyperalgesia (Fig. 4C), but had no effect on primary hyperalgesia (Fig. 4B) in this experimental paradigm during the first 24 hrs after zymosan administration.
Fig. 4.

Effect of parecoxib on (A) hindpaw edema, (B) hindpaw withdrawal latency and (C) tail-flick latency following intraplantar zymosan administration in the ipsilateral hindpaw and PBS in the contralateral hindpaw. Parecoxib (20 mg/kg, i.p., 30 min prior to the zymosan challenge) or vehicle was administered. Data are presented as means ± SEM (6 mice per group). *p<0.01 compared to vehicle-treated group.
Deletion of the Cox-2 gene confirms a prominent role for COX-2 in centrally-mediated hyperalgesia
The pharmacological data for parecoxib (Fig. 4) suggest that COX-2 plays a role in both paw edema and in the centrally-mediated component of hyperalgesia. To validate this pharmacological response, we further measured edema and hyperalgesia induced by zymosan (2%) in C57BL/6/129 Cox-2−/− mice and their wild-type Cox-2+/+ littermates.
The hindpaw edema in C57BL/6 mice (Fig. 1) and C57BL/6/129 Cox-2+/+ mice (Fig. 5A) in response to zymosan (2%) intraplantar injection appears to be essentially the same. However, the inflammatory response in C57BL/6/129 Cox-2−/− mice following zymosan injection is significantly reduced, relative to that observed in Cox-2+/+ littermates (Fig. 5A), suggesting that COX-2 does contribute to zymosan-induced edema. The decrease of inflammatory response in the Cox-2−/− mouse (Fig. 5A) during the first 24 hours after zymosan administration appears similar to the anti-inflammatory effect of parecoxib pre-treatment in wild-type mice (Fig. 4A), confirming that COX-2 induction does contribute to zymosan-induced paw edema.
Fig. 5.

Comparison of (A) hindpaw edema, (B) hindpaw withdrawal latency and (C) tail-flick latency in Cox-2−/− and Cox-2+/+ C57BL6/129 mice, following intraplantar zymosan administration. (A) Intraplantar zymosan (2%, 30 μl) induced paw edema. Zymosan suspension (2%) was injected into the right (ipsilateral) hindpaw and PBS was injected as a control treatment into the left (contralateral) hindpaw. Data are presented as means ± SEM (5–6 mice per group). *p<0.01 for all values for Cox-2−/− mice when compared to the same time points for Cox-2+/+ mice. (B) Intraplantar zymosan (2%)-induced Hindpaw withdrawal latency (HWL) for Cox-2−/− and Cox-2+/+ mice. Data are presented as means ± SEM for 5–6 mice per group. *p<0.01 for ipsilateral HWLs of Cox-2−/− when compared to ipsilateral HWLs of Cox-2+/+ mice. (C) Intraplantar zymosan (2%)-induced tail-flick latency (TFL) in Cox-2−/− and Cox-2+/+ mice. Data are presented as means ± SEM (5–6 mice per group). *p<0.01 for all time points for Cox-2−/− mice TFL values, when compared to Cox-2+/+ mice. The inset in panel B shows Western blots for COX-2 and 14-3-3 proteins in the hindpaws of Cox-2−/− mice and Cox-2+/+ mice 20 hrs after zymosan injection. Note the complete absence of the COX-2 protein signal in the Cox-2−/− mouse.
Zymosan-induced primary hyperalgesia in C57BL/6 wild type mice (Fig. 2) and C57BL/6/129 Cox-2+/+ mice (Fig. 5B) are indistinguishable. Hindpaw withdrawal latency in naïve, uninjected C57BL/6/129 Cox-2−/− mice is slightly, but significantly longer (HWL = 10.94 ± 0.25 seconds) in comparison to their C57BL6/129 Cox-2+/+ littermates (HWL = 8.95 ± 0.31 seconds) in response to thermal stimuli (Fig. 5B). Zymosan produces profound, long-lasting hyperalgesia in Cox-2−/−mice. Despite a significant reduction in the degree of inflammation/edema in the hindpaws of Cox-2−/− mice receiving 2% zymosan relative to their Cox-2+/+ littermates (Fig. 5A), no difference in hindpaw withdrawal latency is observed in Cox-2−/− mice, when compared to wild-type littermates (Fig. 5B); primary hyperalgesia to thermal stimuli appears to be independent of Cox-2 gene expression in this murine experimental paradigm.
Baseline TFL is similar between naïve, uninjected C57BL/6 Cox-2+/+ mice (6.50 ± 0.20 seconds, Fig. 2B) and C57BL/6/129 Cox-2+/+ mice (6.86 ± 0.41 seconds, Fig. 5C). In contrast, naïve (untreated) Cox-2−/− mice have a significantly longer tolerance to tail warming (9.67 ± 0.24 seconds) than their Cox-2+/+ littermates (Fig. 5C). Intraplantar zymosan (2%) injection elicits a decrease in TFL of Cox-2−/− knockout mice, as it does in Cox-2+/+ mice. The absolute reduction in TFL in C57BL/6/129 Cox-2+/+ mice and Cox-2−/− mice is the same; 24 hr after zymosan injection TFL in both Cox-2+/+ mice and Cox-2−/− mice is reduced by 3.5 seconds. However, in the Cox-2+/+ mice this is a 56% reduction in TFL, while in Cox-2−/− mice, maximal TFL reduction is only 36% of the control value. Moreover, the centrally-mediated TFL hyperalgesic response fully recovers to baseline values in Cox-2−/− mice, but is persistent in Cox-2+/+ mice. Thus, the centrally-mediated component of hyperalgesia appears to be substantially diminished as a consequence of Cox-2 gene deletion. Western blot analysis confirms the absence of COX-2 protein expression in the hindpaws of the zymosan-injected Cox-2−/− mice (inset of Panel B, Fig. 5).
Discussion
Edema and hyperalgesia occurring in response to inflammatory stimuli are generally attributed at least in part to increased prostaglandins in the inflamed tissue, spinal cord and brain, as a result of inflammation-induced COX-2 expression [3,4,6,8,14,21–23]. Because of the advantages offered by mouse genetics and the ability to modify the genes of mice, development of well characterized, easily monitored inflammation models in the mouse should aid in our understanding of inflammatory hyperalgesia mechanisms.
Zymosan and carrageenan are potent inflammatory stimuli, often used to study inflammation-induced edema and hyperalgesia in rodents. Carrageenan–induced paw inflammation in mice and rats has been well studied, and commonly used in the development of non-steroidal anti-inflammatory drugs (NSAIDs) and coxibs [5,22,24]. In contrast, zymosan – while well studied in rat paw inflammation models – has not been as extensively utilized in murine footpad studies [15].
Intraplantar zymosan injection into the hindpaws of mice produces pronounced, dose- and time-dependent edema (Fig. 1). In contrast, zymosan-induced paw inflammation in rats is maximal at 0.5–1.0 hr post-injection and returns to baseline at 24–48 hrs [11,12]. The sustained edema we observe in the mouse paw in response to zymosan administration also differs from the biphasic response reported for carrageenan-induced edema in the mouse paw [17].
Histologically, the zymosan-injected mouse hindpaws exhibit severe generalized subcutaneous edema with moderate to severe infiltration of neutrophils, fewer macrophages and minimal numbers of lymphocytes. These histology results are also similar to the infiltration pattern observed in zymosan-induced inflammation in a murine air pouch model [10]. Immunohistochemical localization of COX-2 protein in the inflamed hindpaw following zymosan injection is similar to that observed in the rat paw following carrageenan-induced inflammation [25].
Parecoxib pre-treatment prior to zymosan injection produces a significant reduction in paw edema. The parecoxib concentration used in these experiments has been shown to be optimally effective in several analgesic assays and in a paw edema assay [26]. To determine unequivocally the role of COX-2 in paw edema in this model, we examined the response in mice unable to express COX-2. The edema response observed in response to zymosan injection is also reduced significantly in Cox-2−/− mice, relative to that observed in littermate Cox-2+/+ mice; moreover, the reduction in zymosan-induced hindpaw edema in Cox-2−/− mice over the first 24 hours is similar to that observed in parecoxib-treated wild-type mice (compare Fig. 5A and Fig. 4A). These data demonstrate that inflammation-induced COX-2 production does contribute to zymosan-induced paw edema. In contrast to our result with zymosan-induced edema in Cox-2−/− mice, Dinchuk et al. 1995 [27] reported no differences in carrageenan-induced edema between Cox-2−/− mice and wild-type littermate controls.
The localized primary hyperalgesia accompanying inflammation induced by intraplantar zymosan or carrageenan injection is classically monitored by a decrease in paw withdrawal latency in response to a thermal stimulus [19]. We observe long-lasting (at least 14 days; Fig. 2A) thermal hyperalgesia following intraplantar zymosan injection, in contrast to the more transient response previously reported in mice [13]. Doses of 1% and 2% zymosan induce similar hyperalgesia (Fig. 2A), suggesting a maximal primary hyperalgesic response to zymosan-induced inflammation in the mouse paw occurs at these concentrations.
Primary hyperalgesia in the hindpaw inflammation model is thought to result from release of pro-inflammatory mediators that include bradykinin, cytokines and prostaglandins [28]. Carrageenan-induced inflammation in the rat hindpaw is accompanied by increased tissue concentrations of PGE2 and PGI2 [6,29]. Systemic injections of monoclonal antibody to PGE2, which cannot cross the blood-brain barrier, and rofecoxib, a selective COX-2 inhibitor, reverse carrageenan-mediated hyperalgesia in this model [6,30] suggesting that COX-2-dependent prostaglandin production plays a major role in primary hyperalgesia. In contrast to these results for the rat hindpaw edema model, we observe no parecoxib-mediated reduction in paw withdrawal latency in mice following intraplantar zymosan injection (Fig. 4B). Consistent with this observation, Mazario et al. 2001 [14] reported that the selective COX-2 inhibitor rofecoxib is not effective in reversing hyperalgesia to mechanical stimulation in carrageenan-evoked paw inflammation in mice. Together, these data suggest potential species differences in the relative contribution of COX-2 to primary hyperalgesia in rodents.
To determine whether the pharmacologic data suggesting that COX-2 does not play a role in peripheral hyperalgesia (as measured by paw withdrawal latency) in mice following zymosan-induced paw inflammation are correct, we examined hindpaw withdrawal latency following intraplantar zymosan injection in Cox-2−/− mice. Zymosan (2.0%) produces profound, maximal, long-lasting primary hyperalgesia in Cox-2−/− mice (Fig. 5B). Despite a reduction in the edema response in the hindpaws of Cox-2−/− mice receiving 2% zymosan relative to their Cox-2+/+ littermates, unambiguous lack of COX-2 expression as a result of Cox-2 gene deletion has no effect on hindpaw withdrawal latency; zymosan-induced peripheral hyperalgesia is not mediated by COX-2 dependent pathways in this murine model.
We used the tail-flick latency test to evaluate the role of COX-2 in the central nervous system component of hyperalgesia during zymosan-induced hindpaw inflammation in the mouse. Zymosan concentrations of 1% and 2% induce similar reductions in TFL, suggesting a maximal centrally-mediated hyperalgesic response (Fig. 2B). As reported in other studies in mice [13,16], we observe elevated COX-2 levels in the brain and spinal cord in response to peripheral inflammation, as might be expected if COX-2 plays a role in central sensitization. Parecoxib administration to wild type mice significantly ameliorates central sensitization, measured as reduction in TFL following intraplantar 2.0% zymosan injection (Fig. 4C). Alternatively, mechanical stimuli to assess mechanical hyperalgesia and allodynia could be used to study central mechanisms of COX-2 involvement in this model of peripheral inflammation.
Cox-2−/− mice that have not been injected with zymosan have a significantly longer tolerance to tail warming than do their littermate controls (Fig. 5), suggesting that “constitutive” COX-2 expression plays a role in centrally mediated pain perception in the absence of any inflammation. Recently, Martin et al. 2007 [31] reported, using parecoxib inhibition of COX-2 enzyme activity, that constitutive COX-2 expression mediates central nociception in humans. Intraplantar zymosan (2%) injection elicits a reduction in tail flick latency in Cox-2−/− mice, as it does in Cox-2+/+ mice. However, centrally-mediated component of hyperalgesia persists for a much greater length of time in wild-type mice receiving 2% zymosan than it does in the Cox-2−/−mice. Further, at this high zymosan dose, deletion of the Cox-2 gene is more effective at ameliorating central sensitization than is parecoxib administration, suggesting an incomplete inhibition of COX-2 in the pharmacologic experiment. Both the pharmacologic data and the data obtained from the Cox-2−/− mouse suggest that COX-2 plays an important role in central sensitization after peripheral inflammation in the mouse, consistent with reported data for rat models of inflammation [4,8,32,33].
Conclusion
In conclusion, zymosan injection in the mouse paw induces edema, peripheral COX-2 expression, primary hyperalgesia, central nervous system COX-2 expression and central sensitization. Pharmacologic and genetic data demonstrate that COX-2 function plays a role in zymosan-induced edema and the centrally-mediated component of hyperalgesia in this experimental paradigm. However, despite local COX-2 induction in response to zymosan injection in the mouse paw, pharmacologic and genetic data demonstrate that lack of COX-2 function does not alter primary hyperalgesia. Murine models in which the COX-2 protein can be overexpressed in a targeted, cell type specific fashion [34] and, conversely, murine models in which the Cox-2 gene can be inactivated in a targeted, cell type specific fashion [35], have been developed recently. Consequently, it should now be possible to determine in which central nervous system cell types COX-2 function is required for the centrally-mediated component of hyperalgesia.
Acknowledgments
These studies were supported by National Cancer Institute Grants RO1 CA84572 and P50 CA863606 to HRH. We also thank Arthur Catapang for technical assistance, and Dr. Gregory W. Lawson, DLAM, UCLA for helpful discussions concerning histology studies.
Abbreviations
- NSAID
nonsteroidal anti-inflammatory drug
- COX
cyclooxygenase
- COX-1
cyclooxygenase-1
- COX-2
cyclooxygenase-2
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HPWL
hindpaw withdrawl latency
- PGE2
prostaglandin E2
- PGH2
prostaglandin H2
- PGI2
prostaglandin I2, TFL, tail flick latency
Footnotes
Primary Laboratory of Origin: Harvey R. Herschman
Competing interests
The author(s) declare that they have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular and molecular biology. Ann Rev Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- 2.Herschman HR, Talley JJ, DuBois R. Cyclooxygenase 2 (COX-2) as a target for therapy and noninvasive imaging. Mol Imag Biol. 2003;5:286–303. doi: 10.1016/j.mibio.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 3.Ebersberger A, Grubb BD, Willingale HL, Gardiner NJ, Nebe J, Schaible HG. The intraspinal release of prostaglandin E2 in a model of acute arthritis is accompanied by an upregulation of cyclooxygenase-2 in the spinal cord. Neuroscience. 1999;93:775–781. doi: 10.1016/s0306-4522(99)00164-5. [DOI] [PubMed] [Google Scholar]
- 4.Ibuki T, Matsumura K, Yamazaki Y, Nozaki T, Tanaka Y, Kobayashi S. Cyclooxygenase-2 is induced in the endothelial cells throughout the central nervous system during carrageenan-induced hind paw inflammation; its possible role in hyperalgesia. J Neurochem. 2003;86:318–328. doi: 10.1046/j.1471-4159.2003.01848.x. [DOI] [PubMed] [Google Scholar]
- 5.Guay J, Bateman K, Gordon R, Mancini J, Riendeau D. Carrageenan-induced paw edema in rat elicits a predominant prostaglandin E2 (PGE2) response in the central nervous system associated with the induction of microsomal PGE2 synthase-1. J Biol Chem. 2004;279:24866–24872. doi: 10.1074/jbc.M403106200. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y, Shaffer A, Portanova J, Seibert K, Isakson PC. Inhibition of COX-2 rapidly reverses inflammatory hyperalgesia and prostaglandin E2 production. J Pharmacol Exp Ther. 1997;283:1069–1075. [PubMed] [Google Scholar]
- 7.Niederberger E, Tegeder I, Vetter G, Schmidtko A, Schmidt H, Euchenhofer C, Brautigam L, Grosch S, Geisslinger G. Celecoxib loses its anti-inflammatory efficacy at high doses through activation of NF-kB. FASEB J. 2001;15:1622–1624. doi: 10.1096/fj.00-0716fje. [DOI] [PubMed] [Google Scholar]
- 8.Veiga APC, Duarte IDG, Avila MN, da Motta PG, Tatsuo MAKF, Francischi JN. Prevention by celecoxib of secondary hyperalgesia induced by formalin in rats. Life Sci. 2004;75:2807–2817. doi: 10.1016/j.lfs.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 9.Ferrandiz ML, Foster SJ. Tumor necrosis factor production in a rat air pouch model of inflammation: role of eicosanoids. Agents Action. 1991;32:289–294. doi: 10.1007/BF01980888. [DOI] [PubMed] [Google Scholar]
- 10.Posadas I, Terencio MC, Guillen I, Ferrandiz ML, Coloma J, Paya M, Alcaraz MJ. Co-regulation between cyclooxygenase-2 and inducible nitric oxide synthase expression in the time-course of murine inflammation. Naunyn-Schmied’s Arch Pharmacol. 2000;361:98–106. doi: 10.1007/s002109900150. [DOI] [PubMed] [Google Scholar]
- 11.Meller ST, Gebhart GF. Intraplantar zymosan as a reliable, quantifiable model of thermal and mechanical hyperalgesia in the rat. Eur J Pain. 1997;1:43–52. doi: 10.1016/s1090-3801(97)90052-5. [DOI] [PubMed] [Google Scholar]
- 12.Guhring H, Tegeder I, Lotsch J, Pahl A, Werner U, Reeh PW, Rehse K, Brune K, Geisslinger G. Role of nitric oxide in zymosan induced paw inflammation and thermal hyperalgesia. Inflamm Res. 2001;50:83–88. doi: 10.1007/s000110050728. [DOI] [PubMed] [Google Scholar]
- 13.Guhring H, Gorig M, Ates M, Coste O, Zeilhofer HU, Pahl A, Rehse K, Brune K. Suppressed injury induced rise in spinal prostaglandin E2 production and reduced early thermal hyperalgesia in iNOS deficient mice. J Neurosci. 2000;20:6714–6720. doi: 10.1523/JNEUROSCI.20-17-06714.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mazario J, Gaitan G, Herrero JF. Cyclooxygenase-1 vs cyclooxygenase-2 inhibitors in the induction of antinociception in rodent withdrawal reflexes. Neuropharmacol. 2001;40:937–946. doi: 10.1016/s0028-3908(01)00020-x. [DOI] [PubMed] [Google Scholar]
- 15.Tarayre JP, Delhon A, Aliaga M, Barbara M, Bruniquel F, Caillol V, Puech L, Consul N, Tisne-Versailles J. Pharmacological studies on zymosan inflammation in rats and mice: zymosan induced paw oedema in rats and mice. Pharmacol Res. 1989;21:375–384. doi: 10.1016/1043-6618(89)90155-2. [DOI] [PubMed] [Google Scholar]
- 16.Doi Y, Minami T, Nishizawa M, Mabuchi T, Mori H, Ito S. Central nociceptive role of prostacyclin (IP) receptor induced by peripheral inflammation. Neuroreport. 2002;13:93–96. doi: 10.1097/00001756-200201210-00022. [DOI] [PubMed] [Google Scholar]
- 17.Posadas I, Bucci M, Roviezzo F, Rossi A, Parente L, Sautebin L, Cirino G. Carrageenan-induced mouse paw edema is biphasic, age-weight dependent and displays differential nitric oxide cyclooxygenase–2 expression. Br J Pharmacol. 2004;142:331–338. doi: 10.1038/sj.bjp.0705650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishikawa T, Jain NK, Taketo MM, Herschman HR. Imaging cyclooxygenase-2 (Cox-2) gene expression in living animals with a luciferase knock-in reporter gene. Mol Imag Biol. 2006;8:171–187. doi: 10.1007/s11307-006-0034-7. [DOI] [PubMed] [Google Scholar]
- 19.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–78. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 20.Zha S, Gage WR, Sauvageot J, Saria EA, Putzi MJ, Ewing CM, Faith DA, Nelson WG, De Marzo AM, Isaacs WB. Cyclooxygenase-2 is up-regulated in proliferative inflammatory atrophy of the prostate, but not in prostate carcinoma. Cancer Res. 2001;61:8617–8623. [PubMed] [Google Scholar]
- 21.Satyanarayana PS, Jain NK, Singh S, Kulkarni SK. Effect of selective inhibition of cyclooxygenase-2 on lipopolysaccharide-induced hyperalgesia. Inflammopharmacology. 2004;12:57–68. doi: 10.1163/156856004773121374. [DOI] [PubMed] [Google Scholar]
- 22.Dirig DM, Isakson PC, Yaksh TL. Effect of COX-1 and COX-2 inhibition on induction and maintenance of carrageenan-evoked thermal hyperalgesia. J Pharmacol Exp Ther. 1998;285:1031–1038. [PubMed] [Google Scholar]
- 23.Samad TA, Moore KA, Saperstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–76. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- 24.Gierse JK, Zhang Y, Hood WF, Walker MC, Trigg JS, Maziasz TJ, Koboldt CM, Muhammad JL, Zweifel BS, Masferrer JL, Isakson PC, Seibert K. Valdecoxib: assessment of cyclooxygenase-2 potency and selectivity. J Pharmacol Exp Ther. 2005;312:1206–1212. doi: 10.1124/jpet.104.076877. [DOI] [PubMed] [Google Scholar]
- 25.Nantel F, Denis D, Gordon R, Northey A, Cirino M, Metters KM, Chan CC. Distribution and regulation of cyclooxygenase-2 in carrageenan-induced inflammation. Br J Pharmacol. 1999;128:853–859. doi: 10.1038/sj.bjp.0702866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pandi SV, Jain NK, Sing S, Kulkarni SK. Pharmacological profile of parecoxib: a novel potent injectable selective cyclooxygenase-2 inhibitor. Eur J Pharm. 2004;491:69–76. doi: 10.1016/j.ejphar.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 27.Dinchuk JE, Car BD, Focht RL, Johnston JL, Jaffee BD, Covington MB, Contel NR, Eng VM, Collins RJ, Czernlak PM, Gorry SA, Trazaskos JM. Renal abnormalities and an altered inflammatory responses in mice lacking cyclooxygenase II. Nature. 1995;378:406–409. doi: 10.1038/378406a0. [DOI] [PubMed] [Google Scholar]
- 28.Poole S, Cunha FQ, Selkirk S, Lorenzetti BB, Ferreria SH. Cytokine mediated inflammatory hyperalgesia limited by interleukins-10. Br J Pharmacol. 1995;115:684–688. doi: 10.1111/j.1476-5381.1995.tb14987.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Toriyabe M, Omote K, Kawamata T, Namiki A. Contribution of interaction between nitric oxide and cyclooxygenases to the production of prostaglandins in carrageenan-induced inflammation. Anesthesiology. 2004;101:983–990. doi: 10.1097/00000542-200410000-00025. [DOI] [PubMed] [Google Scholar]
- 30.Chan C-C, Boyce S, Brideau C, Charleson S, Cromlish W, Ethier D, Evans J, Ford-Hutchinson AW, Forrest MJ, et al. Rofecoxib [Vioxx, MK-0966; 4-(4′-Methylsulfonylphenyl)-3-phenyl-2-(5H)-furanone]: A Potent and Orally Active Cyclooxygenase-2 Inhibitor. Pharmacological and Biochemical Profiles. J Pharmacol Exp Ther. 1999;290:551–560. [PubMed] [Google Scholar]
- 31.Martin F, Fletcher D, Chauvin M, Bouhassira D. Constitutive cyclooxygenase-2 is involved in central nociceptive processes in humans. Anesthesiology. 2007;106:1013–1018. doi: 10.1097/01.anes.0000265162.39932.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seybold VS, Jia Y-P, Abrahams LG. Cyclooxygenase-2 contributes to central sensitization in rats with peripheral inflammation. Pain. 2003;105:47–55. doi: 10.1016/s0304-3959(03)00254-9. [DOI] [PubMed] [Google Scholar]
- 33.Ghilardi JR, Svensson CI, Rogers SD, Yaksh TL, Mantyh PW. Constitutive spinal cyclooxygenase-2 participates in the initiation of tissue injury-induced hyperalgesia. J Neurosci. 2004;24:2727–2732. doi: 10.1523/JNEUROSCI.5054-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamei KI, Ishikawa T, Herschman HR. A transgenic mouse for conditional, tissue-specific Cox-2 over-expression. Genesis. 2006;44:177–182. doi: 10.1002/dvg.20199. [DOI] [PubMed] [Google Scholar]
- 35.Ishikawa T, Herschman HR. Conditional knockout mouse for tissue-specific disruption of the cyclooxygenase-2 (Cox-2) gene. Genesis. 2006;44:143–149. doi: 10.1002/gene.20192. [DOI] [PubMed] [Google Scholar]