

Abstract
The Fas/APO-1-receptor associated cysteine protease Mch5 (MACH/FLICE) is believed to be the enzyme responsible for activating a protease cascade after Fas-receptor ligation, leading to cell death. The Fas-apoptotic pathway is potently inhibited by the cowpox serpin CrmA, suggesting that Mch5 could be the target of this serpin. Bacterial expression of proMch5 generated a mature enzyme composed of two subunits, which are derived from the precursor proenzyme by processing at Asp-227, Asp-233, Asp-391, and Asp-401. We demonstrate that recombinant Mch5 is able to process/activate all known ICE/Ced-3-like cysteine proteases and is potently inhibited by CrmA. This contrasts with the observation that Mch4, the second FADD-related cysteine protease that is also able to process/activate all known ICE/Ced-3-like cysteine proteases, is poorly inhibited by CrmA. These data suggest that Mch5 is the most upstream protease that receives the activation signal from the Fas-receptor to initiate the apoptotic protease cascade that leads to activation of ICE-like proteases (TX, ICE, and ICE-relIII), Ced-3-like proteases (CPP32, Mch2, Mch3, Mch4, and Mch6), and the ICH-1 protease. On the other hand, Mch4 could be a second upstream protease that is responsible for activation of the same protease cascade in CrmA-insensitive apoptotic pathways.
Transduction of the apoptotic signal and execution of apoptosis in mammalian cells require the coordinated action of several aspartate-specific cysteine proteases (ASCPs) (1–4). So far, there are 10 reported human ASCPs that can be divided into three subfamilies based on their homology to the human prototype interleukin 1β converting enzyme (ICE) and the nematode prototype CED-3 (4–6). The ICE-like subfamily includes ICE, TX (ICH-2/ICE-relII), and ICE-relIII. The CED-3-like subfamily includes CPP32 (Yama/Apopain), Mch2, Mch3 (ICE-lap3/CMH-1), Mch4, Mch5 (MACH/FLICE), and Mch6 (ICE-lap6). The NEDD2/ICH-1 subfamily contains the human ICH-1 and its mouse counterpart NEDD2. All these enzymes are initially synthesized as single-chain inactive proenzymes. The proenzymes require cleavage after aspartate residues present in conserved processing sites C-terminal to the catalytic cysteine residue to obtain the active protease. Under certain conditions, such as when the proenzymes are overexpressed, this process can occur by autocatalysis. However, under physiological conditions, it is more complex and may involve heterotypic protein–protein interactions and a cascade of ASCPs.
Clues to the mechanism of activation of ASCPs in apoptosis were recently provided by the discovery of two human ASCPs, which contain prodomains highly related to the death effector domain of the Fas/Apo-1-receptor associated protein FADD/Mort1 (5, 7, 8). One of these ASCPs (Mch5/MACH/FLICE) was found to be physically associated with FADD (7, 8). Upon Fas-ligation, Fas binds FADD, which then, through its ability to interact with the prodomain of proMch5, could trigger autocatalytic activation of the enzyme, possibly by bringing together several molecules of proMch5 (7). Because proMch4 also has a general structure similar to proMch5 (5), it is possible that its FADD-like prodomain serves as an adaptor for homotypic and heterotypic protein–protein interactions. However, there is no direct evidence that proMch4 is associated with the Fas/Apo-1-receptor complex. proMch4 may participate in as yet unidentified apoptotic signal-transducing complex distinct from the Fas/Apo-1-receptor complex.
Several lines of evidence suggest the existence of at least two distinct apoptotic pathways. For example, overexpression of the cowpox virus CrmA, inhibits Fas- and tumor necrosis factor (TNF)-induced apoptosis (9–11) but does not inhibit apoptosis induced by DNA-damaging agents and staurosporine (refs. 12 and 13; unpublished observations). Although there is conflicting evidence on the effects of bcl2 overexpression on Fas-induced apoptosis (13, 14), there is strong evidence that bcl-2 family members completely inhibit the latter (12, 13, 15). Interestingly, CPP32 activation was observed in all these cases (12–14). In addition, Mch2 and Mch3 activation was observed in Fas- and staurosporine-induced apoptosis (13, 16). Since CPP32, Mch2, and Mch3 are poorly inhibited by CrmA (17), they may be activated by upstream proteases with different sensitivity to CrmA.
To study the functions of the newly discovered apoptotic ASCPs Mch4 and Mch5, the ASCPs were overexpressed and purified, and their subunit structure was determined. We provide clear biochemical evidence that both proteases are capable of processing/activating each other and all the known ASCPs. However, the proteases differ in their sensitivity to CrmA. We propose that Mch5 mediates the CrmA-sensitive apoptotic pathways such as the Fas and TNF pathways, whereas Mch4 mediates the CrmA-insensitive apoptotic pathways, such as the DNA-damaging agents and staurosporine pathways.
MATERIALS AND METHODS
Expression of Mch4 and Mch5 in Bacteria and Assay of Enzyme Activity.
Human proMch4 and proMch5 (5) lacking the two N-terminal FADD-like domains were subcloned in the bacterial expression vector pET21b in frame with an N-terminal T7 tag and a C-terminal His6 tag. The proteins were expressed in BL21(DE3) bacteria and assayed as described recently (6, 17, 18). The recombinant proteases were purified on a Ni2+-affinity resin and used for microsequencing and the gel cleavage assays described below.
Bacterial Expression and Purification of CrmA.
CrmA cDNA was subcloned in frame into the EcoRI site of the bacterial expression vector pGEX-2T (Pharmacia). The glutathione S-transferase–CrmA fusion protein was purified from the bacterial extracts on glutathione-Sepharose and then used in the enzymatic assays.
In Vitro Transcription/Translation and Cleavage Assays.
Wild-type and mutated cDNAs of ASCPs were transcribed in vitro and translated in the presence of [35S]methionine using Promega’s coupled transcription/translation TNT kit according to the manufacturer’s recommendations. Two microliters of the translation reactions was incubated with purified Mch4 or Mch5 in ICE buffer (25 mM Hepes/1 mM EDTA/5 mM DTT/0.1% CHAPS, pH 7.5), in a final volume of 10 μl. The reaction was incubated at 37°C and then analyzed by Tricine-SDS/PAGE and autoradiography.
RESULTS AND DISCUSSION
Expression, Purification, and Microsequencing of Mch4 and Mch5.
The Fas/APO-1-receptor associated cysteine protease Mch5 and its homolog Mch4 are the most upstream proteases in Fas-apoptotic and probably other apoptotic pathways (5, 7, 8). Therefore, they must undergo autocatalytic processing to the active enzyme heterodimer (large subunit/small subunit) after they receive the apoptotic signal. To determine the primary structure and the exact autocatalytic processing sites in proMch4 and proMch5, they were expressed in bacteria. This is because bacteria do not contain any ASCP activity and Cys → Ala active site mutant ASCPs do not autoprocess in bacteria. Expression of full-length proMch4 or proMch5 generated mainly insoluble proenzymes in bacterial occlusion bodies and very small amount of mature enzymes (data not shown). However, expression of truncated proMch4 or proMch5 (Fig. 1) lacking most of the FADD-like prodomain, and containing N-terminal T7 tag and C-terminal His6 tag, produced soluble mature enzymes. The mature and active enzymes were purified, and the N termini of their subunits were sequenced. As shown in Fig. 1A, purified mature Mch5 migrates in SDS gel as two bands of apparent molecular masses of 18 and 13 kDa. The N terminus of the 13-kDa band starts with L402, indicating that processing occurred after Asp-401 of proMch5. The calculated molecular mass of this peptide excluding the C-terminal His6 tag is 11 kDa. In addition to processing at Asp-401, processing was also observed at Asp-391 (see below). This results in removal of a 10-aa peptide in between the two subunits. N-terminal amino acid sequencing of the p18 subunit revealed that the subunit is a mixture of two polypeptides. One polypeptide starts with Ser-228 and the other with Ser-234, indicating that processing occurred after Asp-227 and Asp-233 of proMch5. However, in some batches of purified Mch5, we observed a small amount of nonmature large subunit where the prodomain was still attached (data not shown). This observation confirms earlier observations that the prodomain is removed last by an autocatalytic activity of the active enzyme (5, 19). Processing at Asp-227 and Asp-233 removes the 28-kDa FADD-like prodomain. This prodomain has been found in a complex with FADD after processing of proMch5 (proFLICE) in mammalian cells (8). Based on these data, proMch5 can autoprocess after Asp-227, Asp-233, Asp-391, and Asp-401 to generate the two subunits (p18, large subunit, and p11, small subunit) of mature Mch5 enzyme (Fig. 1B).
Figure 1.
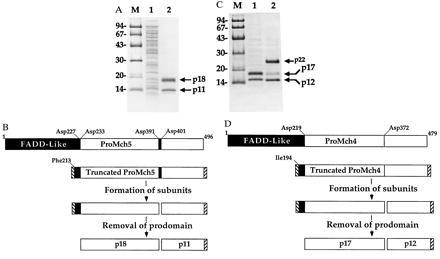
Subunit structure of mature Mch4 and Mch5. Truncated proMch5 (A) and proMch4 (C) lacking most of the FADD-like prodomain (see schematics B and D) were expressed in Escherichia coli, purified to homogeneity, analyzed by SDS/PAGE and Coomassie staining, and then microsequenced. (A) Lane 1, Total bacterial lysate; lane 2, Ni2+-affinity purified mature Mch5 enzyme. The N-terminal amino acid sequence of p11 is LSSPQTRYIPDEADFLLGMA, and the N-terminal amino acid sequences of p18 are SPREQDSESQTLDKVYQMKS and SESQTLD KVYQMKS. (B) A schematic diagram illustrating autoprocessing of proMch5. ProMch5 is autocatalytically processed in two steps to generate the mature p11/p18 enzyme complex. The first step involves processing at Asp-391 and Asp-401, which generates the p11 subunit and nonmature large subunit. This form is enzymatically active and can autoprocess the nonmature large subunit to the mature p18 species by cleavage at Asp-227 and Asp-233 to remove the FADD-like prodomain. (C) Lane 1, Ni2+-affinity purified mature Mch4 obtained after expression of truncated proMch4 (V220-I479); lane 2, Ni2+-affinity purified Mch4 obtained after expression of truncated proMch4 (I194-I479). The N-terminal amino acid sequence of p12 is ALNPEQAPTSLQDSIPAEAD, and the N-terminal amino acid sequence of p17 is VKTFLEALPRA. The N-terminal amino acid sequence of p23 is ASMTGGQQMGRDPIQIVTPP, which contains both the T7 tag (underlined) and the proMch4 sequence. (D) A schematic diagram illustrating autoprocessing of proMch4. Like proMch5, proMch4 undergoes autocatalytic processing in two steps to generate the mature p12/p17 enzyme complex. The first step involves processing at Asp-372, which generates the p12 subunit and nonmature large subunit (in this case p23). The nonmature large subunit is autocatalytically processed to the mature p17 species by cleavage at Asp-219 to remove the FADD-like prodomain. Hatched boxes at the N and C termini of truncated proMch4 or proMch5 represent the T7 and His6 tags, respectively. Lanes M in A and C contain molecular weight markers.
Expression of a truncated proMch4 (amino acids 194–479) generated an active Mch4 enzyme that migrates in SDS gel as two major bands of apparent molecular masses of 23 and 15 kDa and a minor 17-kDa band (Fig. 1C, lane 2). The N terminus of the 15-kDa band starts with Ala-373, indicating that processing occurred after Asp-372 of proMch4. The calculated molecular mass of this polypeptide excluding the C-terminal His6 tag is 12 kDa. This confirms our recent data using site directed mutagenesis, which showed that granzyme B processes proMch4 after Asp-372 to generate the p12 subunit (5). Thus granzyme B cleavage and the autocatalytic processing occur at the same site. No further processing between the two subunits was observed, indicating that proMch4, like proCPP32, is processed at a single site between the two subunits. N-terminal amino acid sequencing of the 23-kDa band revealed that it still has the N-terminal T7 tag, indicating that it was not processed at the N terminus. However, the 17-kDa band starts with Val-220, indicating that it is derived from the 23-kDa band by processing after Asp-219. Expression of proMch4 starting with Val-220 generated an active enzyme that was able to autoprocess to the p17 and p12 subunits (Fig. 1C, lane 1), suggesting that p17 is the minimal size of the large subunit of Mch4. Based on these, data proMch4 can autoprocess after Asp-219 and Asp-372 to generate the two subunits (p17, large subunit, and p12, small subunit) of mature Mch4 enzyme (Fig. 1D). This also removes the FADD-like prodomain in a manner similar to that observed with proMch5.
Autocatalytic Processing of proMch5 at Either Asp-391 or Asp-401 Is Sufficient to Activate the Enzyme.
ProASCPs require cleavage at a site 10–16 amino acids C-terminal to the active site pentapeptide (QACXG, X = R, Q, or G) to generate the two subunits that form the active enzyme heterocomplex. The proenzymes of Mch2, Mch5, Mch6, ICE, TX, ICE-relIII, and ICH-1 have two cleavage sites between their two subunits, whereas proenzymes of CPP32, Mch3, and Mch4 have one site (6). Cleavage and removal of the prodomain is not necessary for enzymatic activity and normally occurs after generation of the two subunits by an autocatalytic activity of the activated enzyme itself (refs. 5 and 19; also see Fig. 1C, lane 2). For example, we observed that mutant CPP32 with a D to A mutation in its prodomain cannot cleave its prodomain; however, it is as active as the wild-type enzyme (5).
To determine whether Mch5 can cleave its proenzyme at Asp-391 and Asp-401 in vitro, 35S-labeled wild-type and mutated proMch5 lacking the FADD-like prodomain were incubated with recombinant active Mch5 enzyme. As expected, no processing was observed when both Asp-391 and Asp-401 were mutated (Fig. 2A, lane 8). However, when only Asp-391 was mutated, proMch5 was cleaved at Asp-401 to generate the small subunit (p11) (Fig. 2A, lane 4). Vice versa, when only Asp-401 was mutated, proMch5 was cleaved at Asp-391 to generate a longer small subunit (p13 instead of p11) (Fig. 2A, lane 6). The wild-type proMch5 was processed at both Asp-391 and Asp-401, as evident from the presence of the p11 and p13 species (Fig. 2A, lane 2). Equivalent shifting in the size of the large subunit was observed with each mutation (Fig. 2A, lanes 4 and 6). This confirms that Asp-391 and Asp-401 are the exact autoprocessing sites between the two subunits of Mch5.
Figure 2.
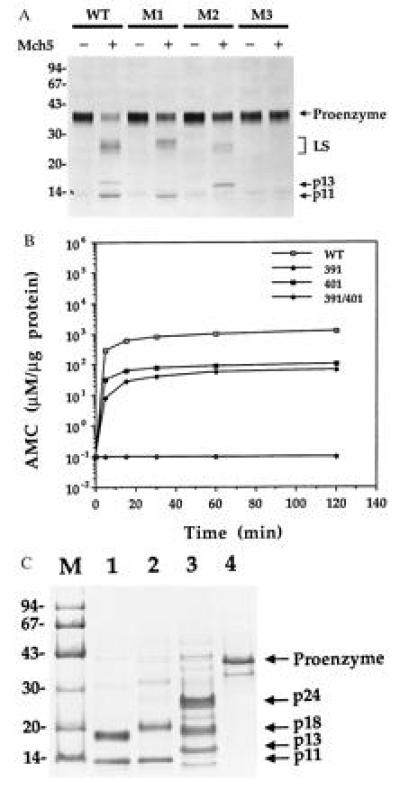
Processing and enzymatic activity of wild-type and mutated proMch5. (A) In vitro processing of proMch5 by mature Mch5 enzyme. 35S-labeled truncated wild-type (WT), Asp-391 mutant (M1), Asp-401 mutant (M2), or Asp-391/Asp-401 double mutant (M3) proMch5 was incubated with (+) or without (−) pure mature Mch5 (200 ng per reaction) for 2 h at 37°C. The reaction products were then analyzed by Tricine-SDS/PAGE and autoradiography. LS, large subunit. (B) Enzymatic activity of wild-type and mutant Mch5 enzymes. Pure wild-type (WT), Asp-391 mutant (391), Asp-401 mutant (401), or Asp-391/Asp-401 double mutant (391/401) Mch5 enzymes was incubated with the peptide substrate DEVD–7-amino-4-methylcoumarin (AMC) (50 μM final) for the indicated times at 37°C. The release of AMC was determined by spectrofluorometry and expressed in micromolar per microgram protein. (C) SDS/PAGE analysis of Ni2+-affinity purified wild-type and mutant Mch5 enzymes. Lanes 1–4 are wild-type, Asp-391 mutant, Asp-401 mutant, and Asp-391/Asp-401 double mutant Mch5 enzymes, respectively. The band seen below the p11 band in lane 3 is a contaminant bacterial protein seen occasionally in some batches of purified Mch5.
To determine whether mutant proMch5 can autoprocess in bacteria to generate active enzymes, they were expressed in E. coli and purified on Ni2+-affinity resin, and their activity was determined with the DEVD-AMC peptide substrate. Their subunit structure was also analyzed by SDS/PAGE and Coomassie staining. As shown in Fig. 2B, except for the double mutant Asp-391/Asp-401, both single mutants were active and able to cleave the DEVD-AMC substrate, although less efficiently than the wild-type enzyme. The Asp-391 and Asp-401 mutant enzymes were about 40- and 10-fold less active than the wild-type enzyme, respectively. This suggests that processing at both sites is necessary for complete activity. Electrophoretic analysis revealed that, except for the double mutant Asp-391/Asp-401 (Fig. 2C, lane 4), both single mutants were also able to autoprocess to the small/large subunit complex in E. coli (Fig. 2C, lanes 2 and 3). Complete autoprocessing was observed in the wild-type and Asp-391 mutant enzymes (p18 and p11 subunits in WT, Fig. 2C, lane 1; p20 and p11 in Asp-391 mutant, lane 2). The large subunit of Asp-391 mutant was longer than that of the wild type, because there was no processing at Asp-391. The Asp-401 mutant had a longer small subunit (p13 instead of p11), because there was no processing at Asp-401. Unlike the wild type or the Asp-391 mutant, the Asp-401 mutant was less efficient in processing its propeptide at Asp-233 (Fig. 2C, lane 3). Nevertheless, an active enzyme was generated. These data establish that a single cleavage site between the large and small subunits of ASCPs is sufficient for autoprocessing and generation of an active enzyme. The two subunits must be physically separated from each other before an active enzyme can be formed. This is evident from the absence of any measurable activity when the double mutant Asp-391/Asp-401 was expressed in E. coli (Fig. 2B). We still do not know why some proASCPs contain two processing sites, and others only one, between their subunits. This could be a regulatory mechanism to control activation of ASCPs. An enzyme with two cleavage sites is expected to be more susceptible to activation than an enzyme with a single cleavage site.
Mch4 and Mch5 Can Process and Activate All Known ASCPs.
Recently, we demonstrated that mature Mch4 can activate the proenzymes of the apoptotic mediators CPP32 and Mch3 (ref. 5; also see Fig. 4C, lanes 2 and 6). A time-course analysis of the activity of Mch5 toward proCPP32 and proMch3 revealed that Mch5 can also efficiently process these two proenzymes (Fig. 3). Mch5 cleaved proCPP32 to generate the p20, p19, and p12 subunits within 5 min of incubation (Fig. 3A, lane 2). The p20 was then converted by an autocatalytic activity of activated CPP32 (5) to the p19 subunit. As recently observed with granzyme B (5), the conversion of p20 to p19 was inhibited by mutation of Asp-9 of proCPP32 (Fig. 3A, lane 7). Similarly, proMch3 was processed to its individual subunits p20 and p12 in less than 15 min (Fig. 3B). Therefore, like Mch4, Mch5 is able to activate CPP32 and Mch3.
Figure 4.
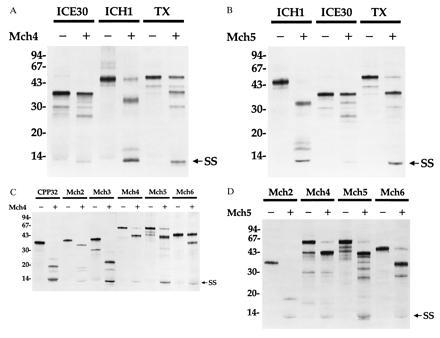
Processing of proASCPs by mature Mch4 or Mch5. 35S-labeled proASCPs were incubated with (+) or without (−) purified equivalent amounts (200 ng per reaction) of Mch4 (A and C) or Mch5 (B and D) for 1 h at 37°C. The reaction products were then analyzed by Tricine-SDS/PAGE and autoradiography. (A and B) ICE-like (ICE30 and TX) and NEDD2-like (ICH1) proASCPs. (C and D) CED-3-like (CPP32, Mch2, Mch3, Mch4, Mch5, and Mch6) proASCPs. SS, small subunit.
Figure 3.
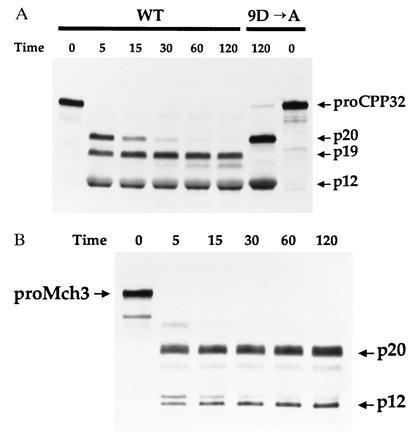
Processing of proCPP32 and proMch3 by mature Mch5. 35S-labeled wild-type (WT) or Asp-9 mutant (9D → A) proCPP32 (A) or proMch3 (B) were incubated with pure mature Mch5 (40 ng) for the indicated times at 37°C. The reaction products were then analyzed by Tricine-SDS/PAGE and autoradiography.
Recently, it was demonstrated that a rise in ICE-like activity, using YVAD as a substrate, precedes the appearance of CPP32-like activity using DEVD as a substrate in Fas-induced apoptosis (20). Because Mch5 does not cleave YVAD efficiently (ref. 7 and unpublished data), it is possible that Mch5 could activate the ICE-like proteases to generate the ICE activity observed in Fas-induced apoptosis. Incubation of Mch4 or Mch5 with proTX, proICE30, or proICH-1 resulted in cleavage of these substrates between their subunits to generate the small subunit (p10) and several intermediate products of the large subunit with or without the prodomain (Fig. 4 A and B, lanes 2, 4, and 6). ICE-relIII has cleavage sites similar to those of ICE and TX between its two subunits, and it is expected to be cleaved by Mch4 or Mch5. Because mature TX can also activate proICE, activation of TX by Mch4 or Mch5 could lead to further activation of proICE. This could explain the observed rise in ICE-like activity in apoptosis.
Mch4 and Mch5 can also process their proenzymes, each other, and the remaining mammalian CED-3-like ASCPs, Mch2 and Mch6 (Fig. 4 C and D). Taken together, these data establish that Mch4 and Mch5 have broad specificity toward proASCPs similar to the serine protease granzyme B. Therefore, all ASCPs are potential targets of mature Mch4 or Mch5 in apoptosis. This should be expected, because Mch4 and Mch5 are the most upstream proteases that first receive the apoptotic signals. To be efficient apoptotic inducers like granzyme B, they should be able to activate all the downstream apoptotic effectors.
CrmA Is a Potent Inhibitor of Mch5 but Not Mch4.
The cowpox virus gene product CrmA has been shown to inhibit apoptosis in several model systems, including Fas/Apo-1-, TNF-, and growth factor withdrawal-induced apoptosis (9–11, 21). The ability of CrmA to potently inhibit ICE suggested that ICE is a critical component in these model systems. However, the absence of a significant effect on Fas- or TNF-induced apoptosis in ICE-deficient mice (22, 23) suggests that a second member of the ASCP family might be the critical target of CrmA inhibition. Because Mch5 and possibly Mch4 are associated with the Fas/Apo-1 receptor and able to activate all ASCP family members, they are most likely to be the target of CrmA inhibition.
To determine the effect of CrmA on Mch4 and Mch5, purified recombinant CrmA was incubated with purified Mch4 or Mch5 and the activity of the two enzymes toward 35S-labeled proCPP32 was determined by quantitative analysis of the cleavage products (Fig. 5). Interestingly, Mch5 was potently inhibited by CrmA (IC50 ≈ 2 nM). On the other hand, CrmA had very little effect on Mch4 activity under the same conditions. The concentration of CrmA that would produce 50% inhibition of Mch4 was calculated to be 2–3 μM, which is 1000- to 1500-fold more than that required to inhibit 50% of Mch5 activity. These results clearly establish that mature Mch5 is a CrmA-inhibitable enzyme and suggest that it is the target of CrmA inhibition in Fas/Apo-1-, TNF-, and growth factor withdrawal-induced apoptosis (Fig. 6). Mch4, which is poorly inhibited by CrmA, could be involved in the CrmA-insensitive apoptotic pathways such as apoptosis induced by DNA-damaging agents and staurosporine (Fig. 6).
Figure 5.
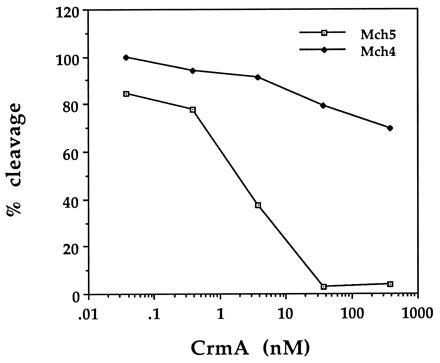
Effect of CrmA on the enzymatic activity of Mch4 and Mch5. Purified 35S-labeled proCPP32 was incubated with equivalent amounts of pure mature Mch4 or Mch5 (20 ng per reaction) in the presence of increasing concentrations of CrmA for 45 min at 37°C. The reaction products were then analyzed by Tricine-SDS/PAGE, autoradiography, and densitometric scanning. The extent of cleavage was determined from the intensity of the cleavage products relative to the total input and expressed as percentage of cleavage.
Figure 6.
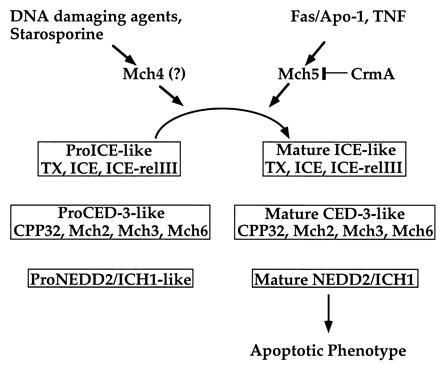
A schematic diagram illustrating the molecular ordering of two potential apoptotic pathways. See text for details.
In conclusion, we have determined the exact subunit structure of the upstream apoptotic ASCPs Mch4 and Mch5. These two proteins are similar in structure (both contain FADD-like death effector domains) and activity (both can process all ASCPs), but they are different in their sensitivity to CrmA. Although there is compelling circumstantial evidence for the involvement of Mch5 in the Fas pathway (7, 8), this cannot be said for Mch4. However, in vitro biochemical evidence supports the idea that Mch4 and Mch5 could mediate two distinct apoptotic pathways (Fig. 6). The FADD-like domains may act as adaptors to allow interactions with different proximal apoptotic effectors like FADD. For example, the Fas signal transduction pathway could recruit Mch5 through interaction with FADD. In a similar fashion, the CrmA-insensitive pathway could recruit Mch4 through interaction with a FADD-like protein. These protein–protein interactions result in activation of Mch4 or Mch5, which leads to activation of the entire ASCP complement (Fig. 6). Because all the downstream ASCPs can potentially be activated once Mch4 or Mch5 are activated, it is clearly difficult to order them in a cascade fashion. These proteases have the ability to also interact with each other once they are activated to generate protease amplification cycles (6). For example, we have shown recently that CPP32 can process/activate Mch2, Mch3, Mch6, and NEDD2 (6, 17, 24). Mch2 can also activate CPP32 (6). These interactions between downstream proteases could serve to further amplify the initial activation signal from Mch4 or Mch5.
Acknowledgments
This work was supported by Research Grants AG 13487 and AI 35035 from the National Institutes of Health and in part by Research Grant from IDUN Pharmaceuticals.
Footnotes
Abbreviations: ASCP, aspartate-specific cysteine protease; ICE, interleukin 1β converting enzyme; TNF, tumor necrosis factor; AMC, 7-amino-4-methylcoumarin.
References
- 1.Martin S J, Green D R. Cell. 1995;82:349–352. doi: 10.1016/0092-8674(95)90422-0. [DOI] [PubMed] [Google Scholar]
- 2.Henkart P A. Immunity. 1996;4:195–201. doi: 10.1016/s1074-7613(00)80428-8. [DOI] [PubMed] [Google Scholar]
- 3.Earnshaw W C. Curr Opin Cell Biol. 1995;7:337–343. doi: 10.1016/0955-0674(95)80088-3. [DOI] [PubMed] [Google Scholar]
- 4.Alnemri, E. S. (1996) J. Cell. Biochem., in press. [DOI] [PubMed]
- 5.Fernandes-Alnemri T, Armstrong R, Krebs J, Srinivasula S M, Wang L, Bullrich F, Fritz L, Trapani J A, Tomaselli K J, Litwack G, Alnemri E S. Proc Natl Acad Sci USA. 1996;93:7464–7469. doi: 10.1073/pnas.93.15.7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Srinivasula S M, Fernandes-Alnemri T, Zangrilli J, Robertson N, Armstrong R C, Wang L, Trapani J A, Tomaselli K J, Litwack G, Alnemri E S. J Biol Chem. 1996;271:27099–27106. doi: 10.1074/jbc.271.43.27099. [DOI] [PubMed] [Google Scholar]
- 7.Boldin M P, Goncharov T M, Goltsev Y V, Wallach D. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- 8.Muzio M, Chinnaiyan A M, Kischkel F C, O’Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz J D, Zhang M, Gentz R, Mann M, Krammer P H, Peter M E, Dixit V M. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- 9.Los M, Van de Craen M, Penning L C, Westendorp M, Baeuerle P A, Droge W, Krammer P H, Fiers W, Schulze-Osthoff K. Nature (London) 1995;375:81–83. doi: 10.1038/375081a0. [DOI] [PubMed] [Google Scholar]
- 10.Enari M, Hug H, Nagata S. Nature (London) 1995;375:78–81. doi: 10.1038/375078a0. [DOI] [PubMed] [Google Scholar]
- 11.Tewari M, Dixit V M. J Biol Chem. 1995;270:3255–3260. doi: 10.1074/jbc.270.7.3255. [DOI] [PubMed] [Google Scholar]
- 12.Datta R, Banach D, Hiromi K, Talanian R V, Alnemri E S, Wong W W, Kufe D W. Blood. 1996;88:1936–1943. [PubMed] [Google Scholar]
- 13.Chinnaiyan A M, Orth K, O’Rourke K, Hangjun D, Poirier G G, Dixit V M. J Biol Chem. 1996;271:4573–4576. doi: 10.1074/jbc.271.9.4573. [DOI] [PubMed] [Google Scholar]
- 14.Armstrong R C, Aja T, Xiang J, Gaur S, Krebs J F, Hoang K, Bai X, Korsmeyer S J, Karanewsky D S, Fritz L C, Tomaselli K J. J Biol Chem. 1996;271:16850–16855. doi: 10.1074/jbc.271.28.16850. [DOI] [PubMed] [Google Scholar]
- 15.Chao D T, Linette G P, Boise L H, White L S, Thompson C B, Korsmeyer S J. J Exp Med. 1995;182:821–828. doi: 10.1084/jem.182.3.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orth K, Chinnaiyan A M, Garg M, Froelich C J, Dixit V M. J Biol Chem. 1996;271:16443–16446. [PubMed] [Google Scholar]
- 17.Fernandes-Alnemri T, Takahashi A, Armstrong R, Krebs J, Fritz L, Tomaselli K J, Wang L, Yu Z, Croce C M, Earnshaw W C, Litwack G, Alnemri E S. Cancer Res. 1995;55:6045–6052. [PubMed] [Google Scholar]
- 18.Fernandes-Alnemri T, Litwack G, Alnemri E S. Cancer Res. 1995;55:2737–2742. [PubMed] [Google Scholar]
- 19.Ramage P, Cheneval D, Chvei M, Graff P, Hemmig R, Heng R, Kocher H P, Mackenzie A, Memmert K, Revesz L, Wishart W. J Biol Chem. 1995;270:9378–9383. doi: 10.1074/jbc.270.16.9378. [DOI] [PubMed] [Google Scholar]
- 20.Enari M, Talanian R V, Wong W W, Nagata S. Nature (London) 1996;380:723–726. doi: 10.1038/380723a0. [DOI] [PubMed] [Google Scholar]
- 21.Gagliardini V, Fernandez P A, Lee R K, Drexler H C, Rotello R J, Fishman M C, Yuan J. Science. 1994;263:826–828. doi: 10.1126/science.8303301. [DOI] [PubMed] [Google Scholar]
- 22.Kuida K, Lippke J A, Ku G, Harding M W, Livingston D J, Su M S-S, Flavell R A. Science. 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- 23.Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M, Rodman L, Salfeld J, Towne E, Tracey D, Wardwell S, Wei F-Y, Wong W, Kamen R, Seshadri T. Cell. 1995;80:401–411. doi: 10.1016/0092-8674(95)90490-5. [DOI] [PubMed] [Google Scholar]
- 24.Harvey N L, Trapani J A, Fernandes-Alnemri T, Litwack G, Alnemri E S, Kumar S. Genes Cells. 1996;1:673–686. doi: 10.1046/j.1365-2443.1996.00255.x. [DOI] [PubMed] [Google Scholar]