

Abstract
The nitric-oxide synthase (NOS; EC 1.14.13.39) reaction is formulated as a partially tetrahydrobiopterin (H4Bip)-dependent 5-electron oxidation of a terminal guanidino nitrogen of l-arginine (Arg) associated with stoichiometric consumption of dioxygen (O2) and 1.5 mol of NADPH to form l-citrulline (Cit) and nitric oxide (·NO). Analysis of NOS activity has relied largely on indirect methods such as quantification of nitrite/nitrate or the coproduct Cit; we therefore sought to directly quantify ·NO formation from purified NOS. However, by two independent methods, NOS did not yield detectable ·NO unless superoxide dismutase (SOD; EC 1.15.1.1) was present. In the presence of H4Bip, internal ·NO standards were only partially recovered and the dismutation of superoxide (O2⨪), which otherwise scavenges ·NO to yield ONOO−, was a plausible mechanism of action of SOD. Under these conditions, a reaction between NADPH and ONOO− resulted in considerable overestimation of enzymatic NADPH consumption. SOD lowered the NADPH:Cit stoichiometry to 0.8–1.1, suggesting either that additional reducing equivalents besides NADPH are required to explain Arg oxidation to ·NO or that ·NO was not primarily formed. The latter was supported by an additional set of experiments in the absence of H4Bip. Here, recovery of internal ·NO standards was unaffected. Thus, a second activity of SOD, the conversion of nitroxyl (NO−) to ·NO, was a more likely mechanism of action of SOD. Detection of NOS-derived nitrous oxide (N2O) and hydroxylamine (NH2OH), which cannot arise from ·NO decomposition, was consistent with formation of an ·NO precursor molecule such as NO−. When, in the presence of SOD, glutathione was added, S-nitrosoglutathione was detected. Our results indicate that ·NO is not the primary reaction product of NOS-catalyzed Arg turnover and an alternative reaction mechanism and stoichiometry have to be taken into account.
Keywords: NADPH, superoxide dismutase, hydroxylamine, nitroxyl, peroxynitrite
·NO is a widespread intra- and intercellular messenger and cytotoxin (1). Its biosynthesis is encompassed by enzymic oxidation of a terminal guanidino nitrogen of Arg (2, 3) yielding Cit as a coproduct (Eq. 1) and is catalyzed by the NO synthase enzyme family (NOS; EC 1.14.13.39; refs. 4 and 5).
 |
1 |
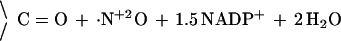 |
Electron transfer depends on interaction with the calcium-binding protein calmodulin (CaM; ref. 6), which, in the case of neuronal NOS (nNOS or NOS-I), requires elevated intracellular free calcium concentrations (>200 nM). Dioxygen (O2) serves as the cosubstrate, NADPH donates reducing equivalents (7), and glutathione (GSH) is required, at least in vitro, to protect essential protein thiols from oxidation (8). Furthermore, all NOS bind and require tetrahydrobiopterin (H4Bip; refs. 9 and 10), a redox-active cofactor of aromatic amino acid hydroxylases and an allosteric regulator. However, the biological function of H4Bip for NOS, in particular its potential redox role in the conversion of Arg, is unclear (11). The prevailing working hypothesis for the overall 5-electron transfer off the guanidino nitrogen of l-arginine (Arg) to yield ·NO (Eq. 1) does not involve H4Bip. It is based on the consumption of 1.5 mol of NADPH per mol of l-citrulline (Cit) as reported for murine and rat NOS (12, 13), a stoichiometry that is fully consistent with ·NO formation and serves as indirect evidence that the mechanism outlined in Eq. 1 and Fig. 1 is correct. However, for bacterial NOS (22) and another rat NOS preparation (23), a stoichiometry of 2 mol of NADPH per mol of Cit was found.
Figure 1.
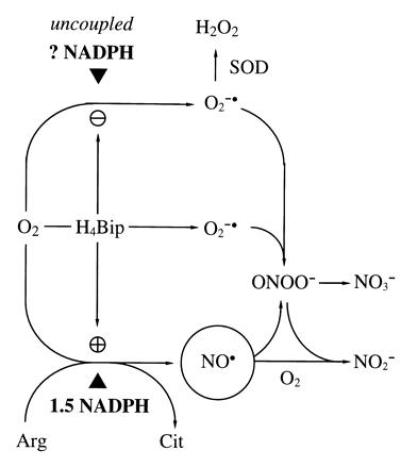
Prevailing hypothesis of the NOS reaction mechanism. O2 is reductively activated by NADPH and NOS to oxidize Arg (14) to the proximal NOS product, ·NO (circled). A fraction of total NADPH consumption and O2 activation is uncoupled from Arg turnover (15), more so in the absence of H4Bip (16), resulting in substantial O2⨪ formation (NADPH oxidase activity of NOS). O2⨪ limits detection of ·NO by a diffusion-limited reaction to ONOO−, which breaks down to NO3− (17, 18). Addition of H4Bip will further stimulate NOS activity and prevent uncoupling of NADPH consumption and O2 activation (16) but, due to its autoxidation in aerobic solutions, also provides an alternative source for O2⨪ (19, 20). Superoxide dismutase (SOD; EC 1.15.1.1) enables ·NO detection (19, 21), in the absence and presence of H4Bip, by dismutating O2⨪ to H2O2, which does not interfere with ·NO detection. In the absence of O2⨪, ·NO decays in a third-order reaction with O2 to yield NO2−. ▴ indicates required reducing equivalents—i.e., 1.5 mol of NADPH per mol of Cit. H4Bip is assumed to activate NOS allosterically but not to donate reducing equivalents.
It is a general shortcoming in NOS biochemistry that ·NO is rarely measured as such. The reason for this is the instability of ·NO in oxygenated solutions and, more importantly, its diffusion-limited reaction with superoxide (O2⨪) to yield peroxynitrite (ONOO−; refs. 24 and 25; see Fig. 1). The standard NOS activity assay features at least two in vitro sources for O2⨪ which differ depending on whether or not the incubation buffer contains H4Bip (Fig. 1): (i) in the absence of H4Bip, NOS reduces O2 at the expense of NADPH to form hydrogen peroxide (H2O2; ref. 26), possibly via intermediary O2⨪ (15); (ii) in the presence of H4Bip, this uncoupling is suppressed, but autoxidation of added H4Bip will yield H3Bip+· and O2⨪ (19, 20). Thus formation of O2⨪ is considered inherent into every NOS activity assay.
We therefore sought to directly quantify ·NO formation by purified NOS, using two different methods. Results, however, were negative even under conditions where internal ·NO standards were recovered. We instead identified two N-oxide species that could not be accounted for by ·NO formation and degradation. Since, furthermore, the stoichiometry of NADPH consumption—hitherto the strongest support in favor of Eq. 1—may have been overestimated by a secondary, nonenzymatic, reaction of NADPH, all our data are consistent with a different NOS reaction mechanism and product.
MATERIALS AND METHODS
Reagents and Enzymes.
HbO2 (27), ·NO (28), and ONOO− (29, 30) were prepared as described. Angeli’s salt (sodium trioxodinitrate, Na2N2O3; ref. 31) and S-nitrosoglutathione (GSNO; ref. 32) were dissolved fresh in argon-purged water. NOS-I (up to 428 nmol of Cit per min per mg of protein) was isolated from cerebellum (8) or extracted in crude form from Sf9 cells transfected with a recombinant baculovirus expressing human NOS-I (33). Other reagents were of the highest analytical grade available.
NOS Incubation.
NOS was incubated at either pH 7.5 (50 mM Hepes, for NADP+ formation) or pH 7.0 [50 mM Mops, for ·NO electrode; 50 mM triethanolamine (TEA)·HCl, in other assays] in the presence of 50 nM CaM, 10 μM H4Bip, ≥3 μM free calcium, 10–100 μM Arg, 0.1–1 mM NADPH, 5 μM FAD, and 5 μM FMN.
Guanylyl Cyclase Activation.
Soluble guanylyl cyclase activity was estimated by measurement of intracellular cGMP levels of RFL-6 detector cells (34) coincubated with NOS (35).
·NO Assays.
For electrochemical ·NO detection, a Celgard 2400 (Hoechst Celanese, Charlotte, NC) membrane was attached to a Universal Sensors 4000–2 oxygen electrode (New Orleans, LA) filled with degassed buffer (50 mM Na2HPO4/50 mM KH2PO4, pH 7.0/0.1 M NaCl, saturated with AgCl). For enzyme incubations, the electrode was inverted, a polypropylene cylinder was mounted on the outside of the electrode housing, and the resulting incubation chamber was filled with 0.3 ml of sample. The potential of the working electrode was set at +800 mV versus the Ag/AgCl counter electrode, using an electrochemical detector (model 400, EG & G, Princeton, NJ) or an operational amplifier (AD549 shielded preamplifying unit, AD515A FET-input electrometer; Analog Devices, Norwood, MA). Calibration [with authentic ·NO or spermine NONOate (SperNO)] and all measurements were performed at 23°C. The detection limit was 10 nM or 3 pmol of ·NO.
For chemiluminometric ·NO detection, a CLD 780 TR (eco Physics, Dürnten, Switzerland) was connected to a microreaction chamber containing 50 mM TEA buffer (pH 7.0) equilibrated with NOx-free air, which was also passed over the stirred sample (total volume 1.0 ml). Reactions were started by addition of NOS or Angeli’s salt. Calibration and measurements were performed at 37°C as described (36). The detection limit was 1 pmol of ·NO min−1.
Stoichiometric cooxidation of ·NO and HbO2 to methemoglobin (Hb+) and NO3− was monitored spectrophotometrically (27) at 37°C (5 μM HbO2, 0.67 μg of NOS). Reactions were started by addition of Arg (in some experiments, by addition of NOS) or solvent (blanks) and monitored for 5–15 min.
Other NOS Products.
NO2− and NO3− (NOx−) were determined by ion-exchange HPLC with UV detection (220 nm). Aliquots (20 μl) of enzyme incubation mixtures (8 μg of NOS ml−1) were applied directly to a LCA-A08 column (Sykam, Gilching, Germany) with acetonitrile/methanol/20 mM aqueous NaCl (70:10:20; vol/vol) as mobile phase at 1.4 ml·min−1 and under argon. For calibration, enzyme incubation mixtures were spiked with known amounts of NOx−. The detection limits were 20 and 2 pmol for NO2− and NO3−, respectively.
GSNO formation was quantified by reversed-phase HPLC as described (37). To increase GSNO recovery (47.0%), NOS was incubated in the presence of 2 instead of 7 mM GSH.
Nitrous oxide (N2O) was determined by gas chromatography using a Porapak Q column [2.4 mm × 12 feet (3.6 m), 80/100 mesh] operated at 50°C with nitrogen as carrier gas and electron capture detection (38). Purified and rechromatographed NOS (6.22 μg) was incubated in sealed glass vials at 37°C in a total volume of 1 ml. Recovery of Angeli’s salt-derived NO− as N2O under NOS assay conditions was 57.8% ± 12.6% of control incubations in the absence of NOS and cofactors (n = 3).
Hydroxylamine (NH2OH) was accumulated as cyclohexanone oxime with 10–100 μM cyclohexanone, released with 0.01 M HCl (15 min) as stable hydroxylammonium chloride, and determined spectrophotometrically (refs. 39 and 40; recovery 42.7% ± 7.7%).
Conversion of 3H-labeled Arg to Cit was measured by liquid scintillation counting (4, 35). NOS (0.04 μg) was incubated at 37°C in a total volume of 0.1 ml containing 5.55 kBq of l-[2,3,4,5-3H]arginine. In NADPH:Cit stoichiometry experiments, the incubation protocol was matched to the NADP+ assay (see below) except for nicotinamide.
Formation of NADP+ during NOS (0.4 μg)-catalyzed Arg turnover was measured fluorimetrically (22). Reaction mixtures (100 μl) containing also 3 mM nicotinamide were incubated for 15 min at 37°C. The difference in NADP+ fluorescence observed with and without 1 mM Nω-methyl-l-arginine (MeArg) was defined as specific for NOS-catalyzed Arg turnover. Oxidation of NADPH in the absence or presence of ONOO− was followed spectrophotometrically at 340 nm.
H2O2 formation by 0.5 μg of NOS was determined spectrophotometrically as Fe(III) thiocyanate complex (25). Contamination of ONOO− stock solutions with H2O2 was determined by horseradish peroxidase-catalyzed 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) cation radical formation (24).
Protein.
Protein concentrations were determined spectrophotometrically according to Bradford (41).
RESULTS
Guanylyl Cyclase Activation.
Purified NOS (428 nmol of Cit mg−1·min−1) catalyzed the conversion of Arg into a soluble guanylyl cyclase-activating-factor (Fig. 2). The NOS-induced cGMP response was similar to the response to 10 μM SNP, a ·NO donor.
Figure 2.
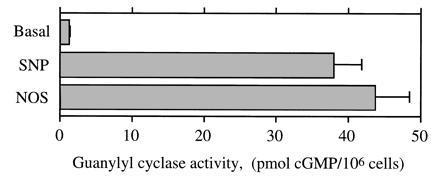
Soluble guanylyl cyclase activation by NOS. Intracellular cGMP levels in 106 RFL-6 detector cells were measured under basal conditions and in the presence of either 10 μM sodium nitroprusside (SNP), a ·NO donor compound, or 3 μg of purified rat NOS-I (incubated for 10 min at 37°C in the presence of 1 mM isobutylmethylxanthine. Mean ± SEM (n = 3).
SOD Is Required to Detect NOS-Derived ·NO.
When the electrochemical ·NO sensor was used, no ·NO signal was observed during NOS-catalyzed Arg turnover (Fig. 3A, •), unless SOD was added. The combined NOS- and SOD-derived signal was fully dependent on Arg and NADPH (not shown) and partially dependent on H4Bip (Fig. 3A). Furthermore, it was protein-dependent and was abolished by HbO2 (not shown). These observations were confirmed when the chemiluminescence method was used (Fig. 3 B and C). Here, addition of SOD to NOS produced an immediate ·NO signal which remained stable for 30 min and was abolished by addition of the NOS inhibitors MeArg (not shown) or NO2Arg (Fig. 3B). With less NOS, the signal was not only smaller but also less persistent (Fig. 3C). ·NO production was maximal when SOD, CaM, and NOS were added simultaneously to the reaction mix. When SOD was added 7, 12, or 17 min after NOS, total ·NO formation was reduced to 44%, 18%, and 14% of control, respectively. However, the concentration of SOD required to allow ·NO detection from NOS differed profoundly with and without H4Bip. Whereas with H4Bip nanomolar concentrations of SOD were sufficient (○ in Fig. 4A), micromolar concentrations were required in its absence (□ in Fig. 4). In contrast, the rate of HbO2 oxidation by NOS was not affected by SOD (control, 385 nmol·mg−1·min−1; 5000 units of SOD ml−1, 375 nmol·mg−1·min−1; n = 2, not significant).
Figure 3.
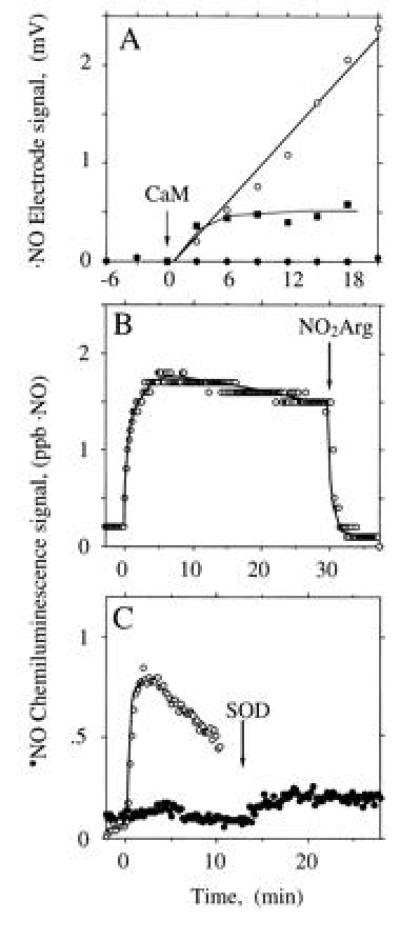
Dependence of the ·NO signal derived from NOS on H4Bip, Arg, and SOD. CaM was added at 0 min. (A) ·NO formation from CaM-activated NOS (30 μg) expressed in mV of the electrode signal either in the presence of 35.7 μM SOD, with (○) or without (▪) 10 μM H4Bip, or in the absence of SOD (•), when no signal was observed, irrespective of whether H4Bip was present. Tracings are representative of six experiments with similar results. (B) ·NO formation from NOS (1.7 μg) in the presence of 35.7 μM SOD and abolition upon addition of 1 mM Nω-nitro-l-arginine (NO2Arg). (C) Dependence of ·NO formation from NOS (0.7 μg) on time of SOD addition, which was at 0 (○) or 12 min (•).
Figure 4.
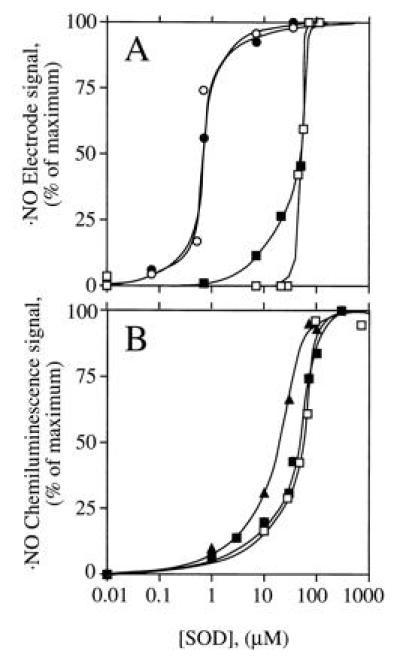
SOD dependence of ·NO detection. NOS (○, □, up to 24 μg), the NO donor SperNO (•, 40 μM), and the NO− donor Angeli’s salt (▴, 1 μM; ▪, 10 μM) were incubated with different SOD concentrations in the absence (□, ▪, ▴) or presence (○, •) of 10 μM H4Bip and otherwise standard assay conditions (i.e., with Arg). ·NO was detected electrochemically (A) or by chemiluminescence (B); n = 3.
SOD Is Not Absolutely Required to Recover Internal ·NO Standards.
Recovery of internal ·NO standards (SperNO) depended on H4Bip in the incubation buffer. In its absence, NOS generated about 918 nmol of H2O2 mg−1·min−1 upon addition of CaM but ·NO detection (Fig. 5A) was unaffected by coincubation with CaM-activated NOS (Fig. 5 B and C). However, when H4Bip was present, the ·NO signal was quenched and required nanomolar SOD to be recovered (Fig. 4A; ref. 19).
Figure 5.
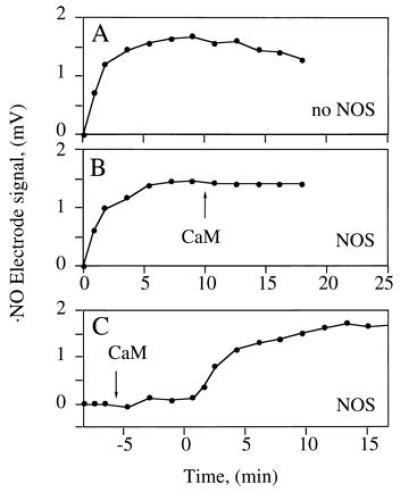
·NO recovery in the presence of NOS. The SperNO-derived NO signal in the absence of H4Bip was recorded in the absence (A) and presence (B and C) of NOS (24 μg). SperNO was added at 0 min; NOS was activated by addition of CaM at the indicated times (arrow). Representative data of three experiments are shown.
SOD Is Required to Detect NO−-Derived ·NO.
To examine whether NO− could be converted by SOD to ·NO, Angeli’s salt was used to generate NO−. SOD was required at micromolar concentrations to generate ·NO; the EC50 for SOD was similar to that observed for detection of NOS-derived ·NO in the absence of H4Bip. The different dependencies on SOD to detect ·NO could thus be grouped into two categories (Fig. 4): nanomolar SOD was sufficient to recover internal ·NO standards and to generate ·NO during NOS-catalyzed Arg turnover in the presence of H4Bip, whereas micromolar SOD was required to generate ·NO from NOS in the absence of H4Bip and to convert NO− to ·NO.
Detection of Other N-Oxides.
These concentration differences for SOD to detect ·NO suggested that SOD may act upon NOS also by at least two mechanisms, one of which may include conversion of NO− to ·NO. We therefore examined whether NO−-related products could be detected upon NOS-catalyzed Arg turnover. Indeed, substoichiometric amounts of N2O and NH2OH were detected in NOS incubation mixtures (see Table 1). NH2OH formation was absolutely dependent on Ca2+ and was abolished by MeArg or NO2Arg but only marginally affected by GSH. Similarly, the recovery of both N2O and NH2OH from Angeli’s salt was independent of GSH (n = 2; not shown). Consistent with the formation of N oxides other than ·NO, formation of NOx− was substoichiometric with respect to Cit (see Table 1). When coincubated with 7 mM GSH, total NOx− formation from NOS decreased by about 13%, possibly by scavenging ONOO− to yield GSNO. Consistent with this interpretation, significant GSNO formation was detected in the presence of 2 mM GSH, provided SOD was present to prevent O2⨪-mediated nitrosothiol decomposition. Concentrations >2 mM GSH were found to accelerate the breakdown of internal GSNO standards.
Table 1.
Arg-derived products from NOS
| Product | GSH, mM | n | Amount, % of Cit |
|---|---|---|---|
| Cit | 7 | 3 | 100 ± 2.3 |
| HbO2/Hb+ assay | 7 | 3 | 87.5 ± 0.9 |
| NOx− | 0 | 3 | 68.3 ± 2.1 |
| 7 | 3 | 61.2 ± 2.2 | |
| GSNO* | 2 | 8 | 5.0 ± 1.1 |
| NH2OH* | 7 | 3 | 13.3 ± 3.5 |
| N2O* | 0 | 3 | 5.3 ± 0.9 |
| 7 | 3 | 6.9 ± 1.4 |
Purified porcine cerebellum NOS (up to 6.22 μg; specific activity 428 nmol of Cit min−1 per mg of protein) prepared in the absence of thiols was incubated under Vmax conditions in the presence of 7 mM GSH in a final volume of 0.1–1 ml at 37°C. Arg-derived products are expressed in % of Cit formation.
Corrected for incomplete recovery as described in the text.
Interaction of ONOO− with NADPH and NOS Stoichiometry.
Because ·NO recovery in the presence of H4Bip appeared to be diminished by O2⨪ and subsequent ONOO− formation, we also examined the presence of ONOO− in NOS incubation mixtures and its possible interactions with other assay components. SOD or bovine serum albumin readily became NO2Tyr positive upon exposure to 4.6 μM ONOO−. However, when coincubated with NOS to generate a final concentration of 109 μM Cit, none of the proteins became tyrosine-nitrated. NOS itself remained free of NO2Tyr both upon Arg turnover and after exposure to exogenous ONOO− (unpublished results). However, NADPH was found to react with ONOO− in a stoichiometric manner (Fig. 6) and independent of the buffer system used (TEA, Mops, and phosphate, all 50 mM and pH 7.0). NADPH consumption by ONOO− was rapid, linear up to 20 μM ONOO− (Fig. 6A), and prevented by excess thiol (e.g., 2-mercaptoethanol; Fig. 6B). When exogenous ONOO− was allowed to decompose, no NADPH loss was observed. To examine whether this chemical interaction between NADPH and ONOO− also interferes with the determination of NADPH stoichiometry of NOS, NADP+ and Cit formation were measured in the presence of H4Bip, with and without addition of SOD. Although SOD dose-dependently lowered NOS activity (Cit formation) due to the autoinhibitory effect of ·NO (H.H., M.F., and H.H.H.W.S., unpublished results), the SOD-induced drop in NADP+ formation was always much more pronounced so that SOD (35.7 μM) lowered the overall NADP+:Cit ratio by almost a factor of 1 (Table 2).
Figure 6.
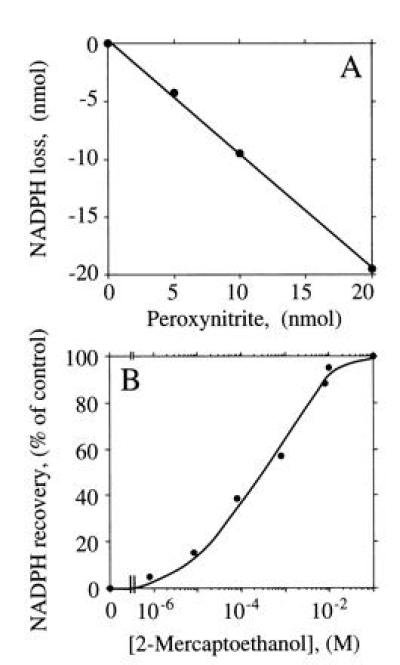
NADPH consumption by ONOO−. Recovery of NADPH was determined spectrophotometrically under NOS assay conditions in the presence of increasing concentrations of ONOO−. (A) Stoichiometric loss of NADPH (100 μM) by ONOO− corrected for autoxidation of NADPH. (B) Protection of NADPH (25 μM final concentration) from ONOO− (12.5 μM)-induced loss in the presence of increasing concentrations of 2-mercaptoethanol.
Table 2.
Effect of SOD on the NADPH:Cit stoichiometry of NOS-I
| NOS-I | SOD, μM | Product formation,
nmol·mg−1·h−1
|
Ratio NADPH:Cit | Difference +SOD | |||
|---|---|---|---|---|---|---|---|
| n | Cit | n | NADP+ | ||||
| Porcine | 0 | 6 | 983.8 ± 45.1 | 6 | 1951 ± 145 | 1.98 | |
| 35.7 | 6 | 528.7 ± 9.9 | 6 | 603 ± 107 | 1.14 | −0.84 | |
| Human | 0 | 17 | 176.8 ± 7.4 | 4 | 290.7 ± 24.1 | 1.64 | |
| 35.7 | 3 | 67.9 ± 7.9 | 3 | 51.5 ± 13.7 | 0.76 | −0.88 | |
Native NOS-I purified from porcine cerebellum (0.4 μg) or recombinant NOS-I extracted as crude cytosolic fraction of Sf9 cells overexpressing the human NOS1 gene (30 μl) was incubated in the absence of GSH for 15 min as described in the text (NADP+ formation); for measurement of Arg-to-Cit conversion, nicotinamide was unnecessary and therefore omitted.
DISCUSSION
The key findings of the present study are as follows: (i) no ·NO can be detected as a product of the action of NOS on Arg unless high, though physiological, amounts of SOD are present; (ii) lack of ·NO detection in the absence of SOD cannot be attributed to lack of ·NO recovery; (iii) oxidation of NADPH by ONOO−, a reaction product of NOS-related chemistry, appears to have caused overestimation in previous attempts to establish the NADPH stoichiometry of the NOS reaction; and (iv) detection of lower oxidation state N oxides (N2O, NH2OH) possibly derived from NO−. Thus, alternative stoichiometries and proximal products of the NOS reaction have to be considered.
Lack of ·NO Formation or Recovery?
The question of whether NOS-derived ·NO could not be detected because it was not formed, or simply because of technical limitations, is a crucial one. Consistent with published literature, purified NOS converted Arg into Cit (4) and a bioactive N oxide which activated guanylyl cyclase and decomposed to NOx− (35, 42). For a further analysis of the present data it is essential to differentiate between two conditions—i.e., presence (standard assay condition) and absence of H4Bip in the incubation mixture. Both conditions have the potential to interfere with the detection of ·NO by generation of O2⨪. However, recovery of internal ·NO standards was incomplete only when H4Bip was present. Even under this condition, which does not allow one to differentiate between lack of ·NO formation and lack of ·NO recovery, NOS would not form ·NO but ONOO−.
Evidence for ONOO− Formation from NOS.
One reason for the lack of ·NO recovery may be that in vitro NOS functions as an ONOO− synthase. This is consistent with the finding that rates of Hb+ formation in the absence and presence of SOD were similar when the HbO2 technique was used for ·NO detection, as this assay cannot discriminate between ·NO and ONOO−. In addition, during NOS turnover both NO2− and NO3− were detected. Decomposition of ONOO− is known to yield predominantly nitrate (29). However, when ·NO reacts with enzymatically generated O2⨪ (from xanthine oxidase), a condition which may resemble NOS catalysis more closely, the major decomposition product of ONOO− is NO2− (43).
Thus, detection of both NOx− species is also in agreement with ONOO− formation. The fact that during NOS-catalyzed Arg turnover no Tyr nitration was detected in proteins that were otherwise readily Tyr nitrated by exogenous ONOO− suggests an alternative scavenging mechanism for ONOO−. ONOO− may have been scavenged by protein thiols, resulting in S-nitrosylation (44) or S-oxidation (29) and explaining GSNO detection. Alternatively, chemical interaction with NADPH may have contributed to ONOO− scavenging even more effectively (Fig. 7). The mechanism of this tertiary reaction of a secondary reaction product of NOS is unknown; however, a similar reaction between ONOO− and NADH has recently been described to follow second-order rate laws (4.0 × 103 M−1·s−1), yielding NO2− and NAD+ (45).
Figure 7.
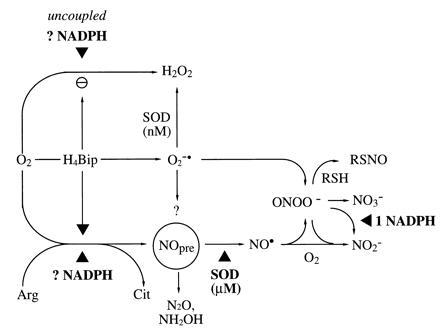
Modified hypothesis on the NOS reaction mechanism and interaction of NOS reaction products. In the absence of H4Bip, uncoupled NADPH consumption and O2 activation appears to yield primarily H2O2, not O2⨪. Thus, analytical limitations cannot explain the absence of a ·NO signal from NOS-catalyzed Arg turnover under these conditions. SOD converts a proximal NOS product, the ·NO precursor (NOpre), to ·NO. Candidate molecules for NOpre include NH2O· and NO−, yielding N2O and NH2OH as by-products. Generated ONOO− (e.g., from H4Bip-derived O2⨪ + ·NO or O2 + ·NO−) will, in a secondary reaction, consume additional NADPH equivalents. ONOO− can be scavenged by high thiol concentrations, providing an alternative source for RSNO, and yield not only NO3− but also NO2−. ▴ depicts requirement for redox equivalents (NADPH, SOD, and possibly also H4Bip).
Implications for Cofactor Stoichiometry of the NOS Reaction.
The ONOO− and NADPH interaction during Arg-dependent NADPH consumption by NOS is important for the NOS reaction mechanism. Thus, the true NADPH:Cit stoichiometry in each reported case may be up to 1 mol NADPH lower, depending on the amount of ONOO− generated and the presence of other nucleophiles, such as thiols that compete with NADPH for ONOO−. In two cases, thiols were deliberately omitted from the incubation mixture (13, 21) and a stoichiometry of 1.5 consistent with ·NO formation was found. Our present results warrant careful reinvestigation of this important issue of NOS biochemistry. With rat and human NOS-I, SOD lowered the NADPH:Cit ratio by a factor of 0.84–0.88, which is within the range of the theoretical value for the reaction between ONOO− and NADPH. Correcting published stoichiometry values (1.5–2) accordingly, ≤1 mol of NADPH is likely to be consumed per mol of Cit, which fits with the values of 0.76–1.14 we have found in the present study. Also, overestimating NADPH consumption on the one hand means underestimating the requirement for additional reducing equivalents on the other hand. Despite the notion that NOS activity fully depends on H4Bip, the current stoichiometry (see Eq.1) has no requirement for a redox-active cofactor besides NADPH. It has not been ruled out that NOS uses redox equivalents of H4Bip in a manner different from aromatic amino acid hydroxylases, which oxidize H4Bip to the quinoid form of dihydrobiopterin (H2Bip). One possibility would be hydrogen abstraction to yield H3Bip+·. This hypothesis is in agreement with the known ability of NOS to catalyze single-electron reductions (16, 46, 47) and the observation that neither H2Bip nor quinoid H2Bip can be detected as intermediates during NOS catalysis (46).
In the absence of added H4Bip, NOS utilizes the protein-associated pterin cofactor, amounts of which are, however, subsaturating (0.2–0.3 mol per monomer) and thus insufficient to prevent uncoupling of NADPH oxidation from Arg turnover (Figs. 1 and 7). Internal ·NO standards producing a ·NO signal of duration and height similar to those of the NOS and SOD-derived signal were readily recovered even in the absence of SOD. However, NOS did not produce an ·NO signal unless micromolar concentrations of SOD were added. These data cannot be explained by an ·NO-consuming reaction, such as formation of O2⨪ and ONOO−, as this should have affected the detection limit for ·NO in the incubation mixtures, which was not the case. Complete recovery of ·NO is expected if the primary product of uncoupled oxygen reduction in the absence of added H4Bip were not O2⨪ but H2O2, which does not interfere with ·NO detection. Low levels of O2⨪ in the absence of H4Bip would also be consistent with the finding that CaM-dependent cytochrome c reduction by NOS is not SOD-inhibitable (48). Thus, in the absence of added H4Bip, one can differentiate between lack of ·NO formation and lack of ·NO recovery, and NOS clearly does not form ·NO as proximal product.
Which Reaction Then Does NOS Catalyze?
The detection of NH2OH and N2O cannot be explained by nonenzymatic degradation of ·NO. On the basis of the consumption of 1 mol of NADPH, NO− is a conceivable product. We here show that SOD is capable of converting NO− from Angeli’s salt into ·NO, as suggested by others (49). In the reaction
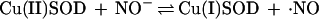 |
2 |
SOD is not a catalyst but a reactant. In the absence of H4Bip, the NOS-derived and the Angeli’s salt-derived signals have similar concentration dependencies for SOD. Formation of HNO/NO− as an alternative NOS product or ·NO precursor could explain the detection of both NH2OH and N2O according to Eqs. 3a–c.
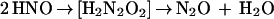 |
3a |
 |
3b |
 |
3c |
NO− shares many functional effects with ·NO, including vascular relaxation (31, 50). Moreover, triplet NO− can rapidly combine with O2 and would thus provide an alternative source for ONOO− and NOx− (51). However, selective methods for the detection of NO− under aerobic conditions are not available. At present, formation of NO− can therefore neither be proven nor be rejected. The fact that detection of N2O from NOS was independent of thiols (Table 1) and no N2O was detected upon incubation of ·NO with GSH (not shown) suggests that thiol-mediated reduction of ·NO as an alternative nonenzymatic source for NO− did not occur.
Matters are further complicated by the fact that NOS, like other cytochrome P450 enzymes (52), may function as an ·NO reductase, forming N2O from exogenous ·NO (S. Shoun and H.H.H.W.S., unpublished results), possibly via intermediate NO−. Such a reaction could in principle also take place at the enzyme during Arg turnover. Therefore, the possibility that NO− and N2O arise as secondary rather than primary products from NOS-derived ·NO cannot be entirely ruled out. However, the fact that iron in resting NOS is in the ferric state going to a ferrous nitrosyl state within seconds of Arg turnover (53) would be consistent with NO− being released to leave NOS in the ferric state (Eq. 4).
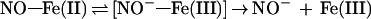 |
4 |
Another important question is why SOD facilitates the detection of ·NO from NOS in nanomolar concentrations in the presence of H4Bip while it requires micromolar concentrations of SOD in the absence of H4Bip. Interestingly, compounds such as N-hydroxyguanidines are converted nonenzymatically by O2⨪ to NO (54). Thus, in the presence of H4Bip as effective radical generator (O2⨪ and H3Bip+·), the NO precursor may be directly converted to ·NO (Fig. 7) by a similar mechanism, which then would elude detection due to immediate conversion to ONOO− unless nanomolar SOD is present. In the absence of H4Bip, the NO precursor requires direct conversion to ·NO by micromolar concentrations of SOD as for NO−.
In conclusion, the primary product of NOS-catalyzed Arg turnover is not ·NO but probably NO−. It will now be of key importance to define the redox role of H4Bip in the conversion of Arg before the NOS reaction and its intermediates can be established.
Acknowledgments
The present work is part of a Ph.D. thesis to be submitted (by H.H.) to the Faculty for Chemistry and Pharmacy, Julius Maximilian University. We thank G. Alt, A. Grave, I. Jasch, K. Knüttel, H. Sennefelder, and R. Spahr for expert technical assistance; C. Kristmann and K. Rehse for help with the N2O measurements; and J. S. Beckman, H. G. Korth, and M. E. Murphy for stimulating discussions. This work was supported by the Thyssen foundation (1994/1/45). H.H. and Z.S.S. received fellowships from the Hilmer Foundation (T146/02.008/93) and the Alexander v. Humboldt/Soros Foundation, respectively.
Footnotes
Abbreviations: Arg, l-arginine; CaM, calmodulin; Cit, l-citrulline; GSH, glutathione; GSNO, S-nitrosoglutathione; H2Bip, (6R)-dihydro-l-biopterin; H3Bip+·, (6R)-trihydro-l-biopterin cation radical; H4Bip, (6R)-tetrahydro-l-biopterin; Hb+, methemoglobin [Fe(III)]; HbO2, oxyhemoglobin [Fe(II)]; MeArg, Nω-methyl-l-arginine; Mops, 3-(N-morpholino)propanesulfonic acid; NH2O·, dihydronitroxide; NH2OH, hydroxylamine; N2O, nitrous oxide (synonym dinitrogen monoxide); ·NO, nitric oxide (synonym nitrogen monoxide); NO−, nitroxyl; NO+, nitrosonium; NO2, nitrogen dioxide; NO2−, nitrite; NO3−, nitrate; NOx−, oxides of nitrogen anions, here sum of NO2− and NO3−; NO2Arg, Nω-nitro-l-arginine; NOS, NO synthase; NO2Tyr, nitrotyrosine; OHArg, Nω-hydroxy-l-arginine; O2⨪, superoxide anion; SNP, sodium nitroprusside; SOD, superoxide dismutase; SperNO, spermine NONOate {N-[4-(1-aminopropyl-2-hydroxy-2-nitrosohydrazino)butyl]-1,3-propanediamine}; TEA, triethanolamine.
References
- 1.Schmidt H H H W, Walter U. Cell. 1994;78:919–925. doi: 10.1016/0092-8674(94)90267-4. [DOI] [PubMed] [Google Scholar]
- 2.Palmer R M J, Ashton D S, Moncada S. Nature (London) 1988;333:664–666. doi: 10.1038/333664a0. [DOI] [PubMed] [Google Scholar]
- 3.Schmidt H H H W, Nau H, Wittfoht W, Gerlach J, Prescher K-E, Klein M M, Niroomand F, Böhme E. Eur J Pharmacol. 1988;154:213–216. doi: 10.1016/0014-2999(88)90101-x. [DOI] [PubMed] [Google Scholar]
- 4.Bredt D S, Snyder S S. Proc Natl Acad Sci USA. 1990;87:682–685. doi: 10.1073/pnas.87.2.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nathan C, Xie Q-w. Cell. 1994;78:915–918. doi: 10.1016/0092-8674(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 6.Abu-Soud H M, Yoho L L, Stuehr D J. J Biol Chem. 1994;269:32047–32050. [PubMed] [Google Scholar]
- 7.Marletta M A. J Biol Chem. 1993;268:12231–12234. [PubMed] [Google Scholar]
- 8.Hofmann H, Schmidt H H H W. Biochemistry. 1995;34:13443–13452. doi: 10.1021/bi00041a023. [DOI] [PubMed] [Google Scholar]
- 9.Tayeh M A, Marletta M A. J Biol Chem. 1989;264:19654–19658. [PubMed] [Google Scholar]
- 10.Kwon N S, Nathan C F, Stuehr D J. J Biol Chem. 1989;264:20496–20501. [PubMed] [Google Scholar]
- 11.Mayer B, Werner E R. Naunyn–Schmiedebergs Arch Pharmacol. 1995;351:453–463. doi: 10.1007/BF00171035. [DOI] [PubMed] [Google Scholar]
- 12.Stuehr D J, Kwon N S, Nathan C F, Griffith O W, Feldman P L, Wiseman J. J Biol Chem. 1991;266:6259–6263. [PubMed] [Google Scholar]
- 13.Mayer B, John M, Heinzel B, Werner E R, Wachter H, Schultz G, Böhme E. FEBS Lett. 1991;288:187–191. doi: 10.1016/0014-5793(91)81031-3. [DOI] [PubMed] [Google Scholar]
- 14.Leone A M, Palmer R M J, Knowles R G, Francis P L, Ashton D S, Moncada S. J Biol Chem. 1991;266:23790–23795. [PubMed] [Google Scholar]
- 15.Pou S, Pou W S, Bredt D S, Snyder S S, Rosen G M. J Biol Chem. 1992;267:24173–24176. [PubMed] [Google Scholar]
- 16.Klatt P, Schmidt K, Uray G, Mayer B. J Biol Chem. 1993;268:14781–14787. [PubMed] [Google Scholar]
- 17.Blough N V, Zafiriou O C. Inorg Chem. 1985;24:3502–3504. [Google Scholar]
- 18.Beckman J S, Beckman T W, Chen J, Marshall P A, Freeman B A. Proc Natl Acad Sci USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mayer B, Klatt P, Werner E R, Schmidt K. J Biol Chem. 1995;270:655–659. doi: 10.1074/jbc.270.2.655. [DOI] [PubMed] [Google Scholar]
- 20.Pfleiderer W. In: Folates and Pterins. Blakely R L, Benkovic S J, editors. New York: Wiley; 1984. pp. 43–114. [Google Scholar]
- 21.Hobbs A J, Fukuto J M, Ignarro L J. Proc Natl Acad Sci USA. 1994;91:10992–10996. doi: 10.1073/pnas.91.23.10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y, Rosazza J P N. Biochem Biophys Res Commun. 1994;203:1251–1258. doi: 10.1006/bbrc.1994.2317. [DOI] [PubMed] [Google Scholar]
- 23.Wang W, Inoue N, Nakayama T, Ishii M, Kato T. Anal Biochem. 1995;227:274–280. doi: 10.1006/abio.1995.1280. [DOI] [PubMed] [Google Scholar]
- 24.Huie R E, Padjama S. Free Radical Res Commun. 1993;18:195–199. doi: 10.3109/10715769309145868. [DOI] [PubMed] [Google Scholar]
- 25.Pryor W A, Cueto R, Jin X, Koppenol W H, Ngu-Schwemlein M, Squadrito G L, Uppu P L, Uppu R M. Free Radical Biol Med. 1995;18:75–83. doi: 10.1016/0891-5849(94)00105-s. [DOI] [PubMed] [Google Scholar]
- 26.Heinzel B, John M, Klatt P, Böhme E, Mayer B. Biochem J. 1992;281:627–630. doi: 10.1042/bj2810627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feelisch M, Kubitzek D, Werringloer J. In: Methods in Nitric Oxide Research. Feelisch M, Stamler J S, editors. Chichester, U.K.: Wiley; 1996. pp. 455–478. [Google Scholar]
- 28.Feelisch, M. (1991) J. Cardiovas. Pharmacol. 17, Suppl. 3, S25–S33.
- 29.Beckman J S, Chen J, Ischiropoulos H, Crow J P. Methods Enzymol. 1994;233:229–240. doi: 10.1016/s0076-6879(94)33026-3. [DOI] [PubMed] [Google Scholar]
- 30.Beckman J S, Ye Y Z, Anderson P G, Chen J, Accavitti M A, Tarpey M M, White C R. Biol Chem Hoppe-Seyler. 1994;375:81–88. doi: 10.1515/bchm3.1994.375.2.81. [DOI] [PubMed] [Google Scholar]
- 31.Zamora R, Feelisch M. Biochem Biophys Res Commun. 1994;201:54–62. doi: 10.1006/bbrc.1994.1668. [DOI] [PubMed] [Google Scholar]
- 32.Hart T W. Tetrahedron Lett. 1985;26:2013–2016. [Google Scholar]
- 33.Nakane M, Schmidt H H H W, Pollock J S, Förstermann U, Murad F. FEBS Lett. 1993;316:175–180. doi: 10.1016/0014-5793(93)81210-q. [DOI] [PubMed] [Google Scholar]
- 34.Ishii K, Sheng H, Warner T D, Förstermann U, Murad F. Am J Physiol. 1991;261:H598–H603. doi: 10.1152/ajpheart.1991.261.2.H598. [DOI] [PubMed] [Google Scholar]
- 35.Schmidt H H H W, Pollock J S, Nakane M, Gorsky L D, Förstermann U, Murad F. Proc Natl Acad Sci USA. 1991;88:365–369. doi: 10.1073/pnas.88.2.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knüttel K, Feelisch M. In: Biology of Nitric Oxide. Moncada S, Feelisch M, Busse R, Higgs E A, editors. Vol. 4. London: Portland; 1994. pp. 209–212. [Google Scholar]
- 37.Moro M A, Darley-Usmar V M, Goodwin D A, Read N G, Zamora-Pino R, Feelisch M, Radomski M W, Moncada S. Proc Natl Acad Sci USA. 1994;91:6702–6706. doi: 10.1073/pnas.91.14.6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elkins J W. Anal Chem. 1980;52:263–267. [Google Scholar]
- 39.Frear D S, Burrel R C. Anal Chem. 1955;27:1644–1665. [Google Scholar]
- 40.Schmidt H H H W, Zernikow B, Baeblich S, Böhme E. J Pharmacol Exp Ther. 1990;254:591–597. [PubMed] [Google Scholar]
- 41.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 42.Mayer B, Böhme E. FEBS Lett. 1989;256:211–214. doi: 10.1016/0014-5793(89)81750-8. [DOI] [PubMed] [Google Scholar]
- 43.Furchgott, R. F., Jothianandan, D. & Freay, A. D. (1990) in Nitric Oxide froml-Arginine: A Bioregulatory System, eds. Moncada, S. & Higgs, A. E. (Excerpta Medica Elsevier, Amsterdam), pp. 5–14.
- 44.Stamler J S, Singel D J, Loscalzo J. Science. 1992;258:1898–1902. doi: 10.1126/science.1281928. [DOI] [PubMed] [Google Scholar]
- 45.Kobayashi, K., Miki, M. & Tagawa, S. (1995) Endothelium 3, Suppl., S65.
- 46.Giovanelli J, Campos K L, Kaufman S. Proc Natl Acad Sci USA. 1991;88:7091–7095. doi: 10.1073/pnas.88.16.7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmidt H H H W, Smith R M, Nakane M, Murad F. Biochemistry. 1992;31:3243–3249. doi: 10.1021/bi00127a028. [DOI] [PubMed] [Google Scholar]
- 48.Klatt P, Heinzel B, John M, Kastner M, Böhme E, Mayer B. J Biol Chem. 1992;267:11374–11378. [PubMed] [Google Scholar]
- 49.Murphy M E, Sies H. Proc Natl Acad Sci USA. 1991;88:10860–10864. doi: 10.1073/pnas.88.23.10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fukuto J M, Chiang K, Hszieh R, Wong P, Chaudhuri G. J Pharmacol Exper Ther. 1992;263:546–551. [PubMed] [Google Scholar]
- 51.Yagil G, Anbar M. J Inorg Nucl Chem. 1964;26:453–460. [Google Scholar]
- 52.Shiro Y, Fujii M, Iizuka T, Adachi S, Tsukamoto K, Nakahara K, Shoun H. J Biol Chem. 1995;270:1617–1628. doi: 10.1074/jbc.270.4.1617. [DOI] [PubMed] [Google Scholar]
- 53.Wang J, Abu-Soud H M, Rousseau D L, Fukuto J M, Ignarro L J, Stuehr D J. J Biol Chem. 1995;270:22997–23006. doi: 10.1074/jbc.270.39.22997. [DOI] [PubMed] [Google Scholar]
- 54.Sennequier N, Boucher J-L, Battioni P, Mansuy D. Tetrahedron Lett. 1995;36:6059–6062. [Google Scholar]