

Abstract
We engineered a full-length (8.3-kbp) cDNA coding for fatty acid synthase (FAS; EC 2.3.1.85) from the human brain FAS cDNA clones we characterized previously. In the process of accomplishing this task, we developed a novel PCR procedure, recombinant PCR, which is very useful in joining two overlapping DNA fragments that do not have a common or unique restriction site. The full-length cDNA was cloned in pMAL-c2 for heterologous expression in Escherichia coli as a maltose-binding protein fusion. The recombinant protein was purified by using amylose-resin affinity and hydroxylapatite chromatography. As expected from the coding capacity of the cDNA expressed, the chimeric recombinant protein has a molecular weight of 310,000 and reacts with antibodies against both human FAS and maltose-binding protein. The maltose-binding protein-human FAS (MBP-hFAS) catalyzed palmitate synthesis from acetyl-CoA, malonyl-CoA, and NADPH and exhibited all of the partial activities of FAS at levels comparable with those of the native human enzyme purified from HepG2 cells. Like the native HepG2 FAS, the products of MBP-hFAS are mainly palmitic acid (>90%) and minimal amounts of stearic and arachidic acids. Similarly, a human FAS cDNA encoding domain I (β-ketoacyl synthase, acetyl-CoA and malonyl-CoA transacylases, and β-hydroxyacyl dehydratase) was cloned and expressed in E. coli using pMAL-c2. The expressed fusion protein, MBP-hFAS domain I, was purified to apparent homogeneity (Mr 190,000) and exhibited the activities of the acetyl/malonyl transacylases and the β-hydroxyacyl dehydratase. In addition, a human FAS cDNA encoding domains II and III (enoyl and β-ketoacyl reductases, acyl carrier protein, and thioesterase) was cloned in pET-32b(+) and expressed in E. coli as a fusion protein with thioredoxin and six in-frame histidine residues. The recombinant fusion protein, thioredoxin-human FAS domains II and III, that was purified from E. coli had a molecular weight of 159,000 and exhibited the activities of the enoyl and β-ketoacyl reductases and the thioesterase. Both the MBP and the thioredoxin-His-tags do not appear to interfere with the catalytic activity of human FAS or its partial activities.
The fatty acid synthase (FAS; EC2.3.1.85) is a multienzyme complex that catalyzes the synthesis of the long-chain fatty acid palmitate from acetyl-CoA, malonyl-CoA, and NADPH (1, 2). This biosynthetic process is an important metabolic pathway for the conversion of intermediate metabolites into fatty acids that are essential for the synthesis of cellular lipids and storage fats. In prokaryotes and in higher plants, fatty acid biosynthesis is catalyzed by a type II FAS, a loosely associated multienzyme complex consisting of at least seven protein components with distinct enzyme activities and of the acyl carrier protein [ACP (2–4)]. In animal tissues, the FAS component activities reside on a single multifunctional polypeptide [Mr 272,000 (5–10)]. In 1983, using various proteolytic digestions to isolate the FAS partial activities, we established the following order of the partial activities along the polypeptide subunit: β-ketoacyl synthase; acetyl-CoA and malonyl-CoA transacylases; β-hydroxyacyl dehydratase; enoyl reductase; β-ketoacyl reductase; the site for the prosthetic group, 4′-phosphopantetheine (ACP); and the thioesterase (11–14). Based on the tryptic cleavage of the protein subunit, the component activities were grouped into three subdomains: domain I (ketosynthase and transacylases); domain II (dehydratase, enoyl reductase, ketoreductase, and ACP); and domain III (thioesterase).
Earlier studies showed that native FAS consists of two identical polypeptide subunits that function as a homodimer in palmitate synthesis (15). Dissociation of the two subunits resulted in loss of palmitate synthesis and the β-ketoacyl synthase activity but not the remaining six catalytic activities. Additional investigations led us to propose a head-to-tail model for the organization of the two subunits, generating two palmitate-synthesizing sites that each consists of domain I derived from one subunit and domains II and III derived from the second subunit. In this arrangement, the active cysteine-SH of the ketosynthase of domain I, where the acetyl group is bound, is juxtaposed within bond distance from the cysteamine-SH of domain II, where the malonyl group is bound (14, 15). This arrangement is necessary and a prerequisite for the chain elongation of the acyl group (16).
Although a wealth of biochemical information about FAS has been derived from nonhuman animal studies, very little is known about the human enzyme. Human FAS has been used successfully as a prognostic factor in identifying patients with breast cancer who have a markedly worse prognosis (21). Furthermore, breast tumors marked with high levels of FAS are four times more likely to recur and metastasize than those not so marked. Investigators also found an association between high levels of FAS expression and a worsened prognosis in patients with adenocarcinoma of the prostate (22) or colon (23). Recently, we isolated and purified human FAS from a human cell line (HepG2) and showed that all of its partial activities are comparable to those of other animal FASs (5). We also have cloned and sequenced the human FAS cDNA and identified the active sites of its various partial activities (5).
The predicted amino acid sequences of the human, chicken, rat, and goose synthases show a high degree of homology, especially in the segments assigned for the partial catalytic activities (5–10). However, the similarity in the sequences is significantly less among the sequences of the linking peptides that separate the various catalytic domains, especially in the region between domains I and II. The identification of the active sites of the various catalytic domains along the FAS polypeptide subunits reconfirmed the general order of the partial activities deduced from the proteolytic studies of the FAS subunit protein. The identification by mutagenesis experiments of His878 as an active residue required for the dehydratase activity of rat FAS and the location of His878 in the conserved motif of His-X-X-X-Gly-X-X-X-X-Pro in many dehydratases (17) suggested that this functional unit is located in domain I instead of domain II, as had been proposed earlier (18).
To elucidate the structure and function of the multifunctional FAS and its domains, investigators have tried to express different segments of FAS cDNA in various expression systems. For example, we showed that the ACP and thioesterase components of chicken FAS can be expressed in Escherichia coli (19). Recently, the construction of a full-length cDNA that encodes the rat FAS and the expression of this cDNA in Sf9 insect cells by using baculoviral vectors was reported (17, 20). In this study, we engineered a full-length human FAS cDNA that encodes the entire FAS polypeptide (2504 amino acid residues) and successfully expressed it as a catalytically active recombinant protein in E. coli. We also successfully expressed and purified human FAS domain I (hFAS dI) separately and human FAS domains II and III (hFAS dII-III) jointly and determined their partial activities. The results we obtained reaffirm the distribution of the partial activities on the FAS polypeptide as follows. The ketosynthase, the transacylases, and the dehydratase are grouped within domain I; the enoyl reductase, the ketoreductase, and the ACP are located within domain II; and domains I and II are separated by a sizable interdomain peptide.
MATERIALS AND METHODS
Materials.
The E. coli strains used were TOP10F′ (Invitrogen), BL21(DE3) (Novagen), and DM1 (GIBCO/BRL). The plasmids pMAL-c2 (New England Biolabs) and pET-32 (a, b, and c) (Novagen) were used as expression vectors. The TALON resin was obtained from CLONTECH. The S-tag Western blot kit was from Novagen. The amylose resin and anti-MBP serum were from New England Biolabs. The sources of all other reagents are as described before (5).
Assays of FAS and Its Partial Activities.
The FAS activity was assayed by measuring the rate of oxidation of NADPH or the incorporation of radiolabeled acetyl-CoA or malonyl-CoA into palmitate (24). The partial activities of FAS were assayed as described before (5).
Analysis of Fatty Acids by High-Performance Liquid Chromatography (HPLC).
When FAS activity was assayed with either [1-14C]acetyl-CoA or [2-14C]malonyl-CoA, the fatty acid products were extracted, and the radioactivity was measured as described previously (24). The fatty acids were analyzed as phenacyl esters by HPLC with a slight modification of the method described previously (25). Briefly, the samples of fatty acids dried under partial vacuum were dissolved in 100 μl of 2-butanone, and 150 μl of 2-bromoacetophenone (10 mg/ml in 2-butanone) and 150 μl of triethylamine (10 mg/ml in 2-butanone) were added. The resulting mixture was heated in a heating block at 100°C for 10 min and was cooled to room temperature; 210 μl of acetic acid (2 mg/ml in 2-butanone) were added to the mixture, and it was reheated for 10 min at 100°C. The reaction mixture was then cooled to room temperature, evaporated to dryness under vacuum, and the residue was redissolved in 90% acetonitrile for HPLC analysis. The phenacyl esters of the fatty acids were analyzed by using a Beckman Gradient Liquid Chromatograph (model 332) equipped with a Vydac (201 HS C18) column (pore size, 5 μm; 4.6 × 250 mm) operated at 32°C, a β-ram continuous-flow scintillation counter, and a Spectroflow 773 absorbance detector. The column was washed with 80% acetonitrile for 5 min, and the fatty acyl esters were eluted with a linear gradient of 80 to 95% acetonitrile in water within a period of 45 min at a flow rate of 1.5 ml/min.
Recombinant PCR.
In the process of combining hFAS cDNA clones, we developed a procedure that extends and joins two DNA fragments that have overlapping nucleotide sequences but no convenient common restriction sites that could be used to join them. This procedure, which we call recombinant PCR, was based on the assumption that when two overlapping DNA fragments are included in a PCR reaction, the top strand of the 5′ fragment (5′ → 3′) and the bottom strand of the 3′ fragment (3′ → 5′) hybridize and will be elongated by Taq polymerase to generate a new, longer template that contains the sequences of these two fragments appropriately joined. As shown in Fig. 1, when DNA fragments A and B are denatured and reannealed together, two partially double-stranded DNA hybrids C and D are formed in addition to the double-stranded A and B fragments. Only the hybrid DNA of fragment C, in which the 3′ ends of the two overlapping DNA strands (indicated by an asterisk in Fig. 1) hybridize, can be elongated by Taq DNA polymerase. This process leads to the formation of a new, longer fragment E, which contains the sequences of A and B appropriately joined. Fragment E can then be amplified by appropriate primers. PCR can be initiated by using the primers and the overlapping DNA fragments. In subsequent rounds of amplification, the strands of fragments A and B (indicated by an asterisk in Fig. 1) can also anneal to appropriate strands of fragment E and elongate. We have used this procedure successfully to join two overlapping hFAS cDNA fragments that have no common restriction site (see below). We noticed, however, that if the DNA fragments are too long and have only short overlapping regions, the procedure may not work. To overcome this problem, we initially amplified two short fragments in the overlapping region (Fig. 2) and then elongated them into longer templates.
Figure 1.
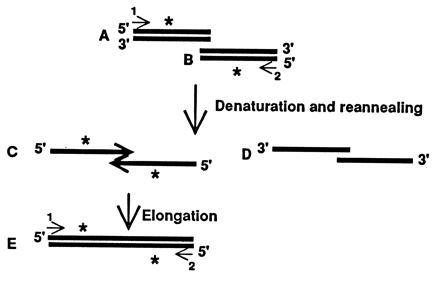
Schematic representation of recombinant PCR (see the text for details).
Figure 2.
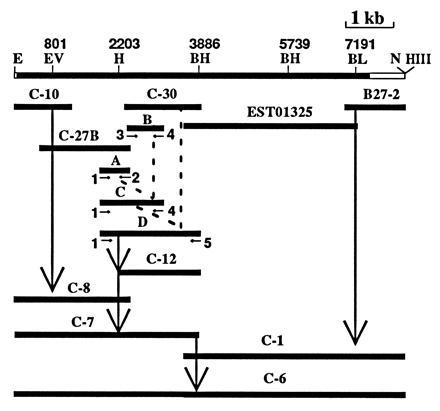
Strategy for engineering a full-length hFAS cDNA clone. The top line shows the location of the restriction sites at the indicated nucleotide number. The cDNA clones C-10 and C-27B were joined together by using EcoRV to generate the N-terminal clone C-8 (nt 1 to 2451). To join the next cDNA clone, C-30, we had to perform recombinant PCR, which generated PCR fragments A, B, C, and D and cDNA clone C-12 as intermediates (see the text for details). The cDNA clone C-7 was obtained by appropriately ligating the HincII fragment from clone C-8 to clone C-12. The cDNA clone C-1, which contained the carboxyl-terminal end of FAS, was generated by joining the EST01325 and B27-2 clones using the common restriction site BclI. To generate the full-length cDNA FAS clone C-6, we had to ligate the BamHI site of C-7 to the BamHI site (nt 5739) of C-1 and then insert the BamHI–BamHI fragment in proper orientation to generate clone C-6. The abbreviations used for the restriction sites are: BH, BamHI; BL, BclI; E, EcoRI; Ev, EcoRV; H, HincII; HIII, HindIII; N, NotI.
Construction of a Full-Length hFAS cDNA.
The strategy for engineering a cDNA construct that contains the 7512-nucleotide open reading frame of hFAS required the assembly of the nucleotides that were distributed in five different clones that had been isolated previously (5) and are shown in Fig. 2. To have a unique restriction site at the beginning of the full-length human FAS cDNA, the cDNA clone C-10 was linearized at the BamHI site that was present in the vector immediately upstream of the ATG codon of human FAS cDNA, was blunt-ended with the Klenow fragment, was ligated with EcoRI linkers, and was cut with EcoRV to release a 0.8-kb fragment. This fragment was cloned into EcoRV-digested clone C-27B to generate clone C-8 (Fig. 2), which contains the hFAS sequence from the start codon ATG (flanked by an EcoRI site on the 5′ side) to base 2451 (5). Although cDNA clones C-8 and C-30 overlapped, they did not have a convenient common restriction site at which they could be joined by using simple cloning procedures. To overcome this problem, we had to extend clone C-30 on the 5′ side to include a HincII site that is unique and is also present in the 3′ end of clone C-8. To achieve this, we used the recombinant PCR procedure described above. To avoid the misincorporation inherent in using Taq polymerase in PCR, and to overcome the relatively short overlapping sequences between C-27B and C-30, we amplified short cDNA fragments with 15 cycles instead of the 30 cycles we routinely use (Fig. 2). Initially, using primer 1 (bases 1806–1823), primer 2 (bases 2434–2451), and 50 ng of clone C-27B cDNA as template DNA, we amplified a 0.6-kb DNA fragment A [Fig. 2 (5)]. Similarly, using primer 3 (bases 2359–2376), primer 4 (bases 3156–3173), and 50 ng of clone C-30 cDNA as template DNA, we amplified a 0.8-kb DNA fragment B (Fig. 2). Using the isolated fragments A and B, which had an overlapping region of 93 bp (bases 2359–2451), and primers 1 and 4, we amplified a 1368-bp PCR fragment C (Fig. 2). This PCR fragment and clone C-30 have an overlapping region of 815 bp (bases 2359–3173). Using PCR fragment C and C-30 as templates and primers 1 and 5 (bases 3872–3896 that overlapped the BamHI site at base 3886), we amplified the 2091-bp fragment D. The PCR fragment D was then cut with HincII and BamHI and cloned into pBluescript KS(+) vector to generate clone C-12. The sequence of clone C-12 was confirmed. The 5′ cDNA fragment was isolated from C-8 as a HincII fragment, thus taking advantage of the SalI/HincII site present in the vector on the 5′ end. This fragment was then cloned into C-12 at the HincII site, resulting in clone C-7. This clone encodes the human FAS sequence from the start codon ATG to base 3890 (5). Clone C-1 was generated by ligating B27-2 and EST01325 using the common BclI site (Fig. 2). Next, a 2.4-kb BamHI–NotI fragment [NotI is located in the multiple cloning site of vector pBluescript SK(−)] was isolated from C-1 and cloned into C-7, generating clone C-15 (data not shown), which lacked the internal BamHI fragment (bases 3886 to 5739). This missing internal 1.8-kb BamHI fragment was then inserted at the unique BamHI site in C-15 in proper orientation, creating the full-length FAS cDNA clone C-6 that has a unique EcoRI site at the 5′ end and a NotI site at the 3′ end. The human FAS cDNA of C-6 was sequenced with T3, T7, and several internal human FAS primers, and the results showed that the sequence exactly matched that of the corresponding overlapping cDNA clones. Moreover, digestion of the C-6 DNA with several restriction enzymes yielded DNA fragments of the sizes that were as predicted from the cDNA sequence (Fig. 2). Digestion with HincII, however, revealed that there is an additional restriction site in C-6 that is inconsequential because it is located in the 3′-untranslated region of hFAS cDNA (data not shown). Altogether, these findings indicated that we had successfully constructed the hFAS cDNA that encodes the 2504 amino acid residues of the FAS subunit polypeptide.
Miscellaneous Procedures.
Measurement of the protein concentration (26), SDS/PAGE analysis (27), DNA sequencing (5), Western blotting (5), and isolation of plasmid DNA (28) were performed as described.
RESULTS AND DISCUSSION
Construction of the Expression Plasmids of Human FAS and Its Domains I, II′-III, and II-III.
We have successfully expressed the chicken FAS-ACP-thioesterase domains in E. coli (19). However, attempts to express the full-length FAS cDNA, or fragments thereof, encoding one or more of the other FAS partial activities were unsuccessful, primarily because the expressed polypeptides were associated with inclusion bodies as insoluble proteins. The recent use of fusion proteins to successfully express functional proteins has encouraged us to express FAS and its domains by using either the pMAL-c2 vector, which was designed for the expression of proteins as MBP-fusion proteins, or pET-32, which produces thioredoxin fusion proteins.
The expression plasmid pMAL-c2-hFAS, which contained the full-length human FAS, was constructed by first isolating the 8.3-kbp EcoRI–NotI fragment from clone C-6 (Fig. 2), cloning it into pFAST-Bac1 vector, and reisolating the 8.3-kbp fragment as an EcoRI–HindIII fragment for the convenience of cloning it into the pMAL-c2 vector. Sequence analyses confirmed that the hFAS cDNA was fused in-frame with that of malE and that the resultant full-length hFAS expression plasmid pMAL-c2-hFAS would express a recombinant protein, MBP-hFAS (∼310 kDa), containing the 42-kDa MBP and the factor Xa cleavage site fused to the 272-kDa human FAS protein.
The pMAL-c2-hFAS-domain I expression plasmid was constructed by cloning the 3.9-kb EcoRI–BamHI fragment from clone C-6 (Fig. 2) into pMAL-c2. Because no translation-termination codons were introduced at the 3′ end, the expressed protein (MBP-hFAS-dI) will have an added sequence of 58 amino acids at the 3′ end that is derived from one of the reading frames of the β-galactosidase component of the plasmid. The MBP-hFAS-dI recombinant protein containing 42 kDa of MBP and the additional 58 amino acids would, therefore, have an approximate mass of 191 kDa.
By cloning the 3′-end, 2.5-kb BamHI–HindIII cDNA fragment from clone pMAL-c2-hFAS (see above) into pET-32c(+), we were able to express parts of domain II and all of domain III (nucleotide 5742 to the 3′ end) as a fusion protein with thioredoxin (TRX-hFAS-dII′-III). This cDNA, which codes for 591 amino acids fused with a TRX-His-tag sequence of 164 amino acids, will produce a protein of 83 kDa.
The TRX-hFAS domains II-III expression plasmid was constructed by first cloning the 3′-end, 2.5-kb BamHI–HindIII cDNA fragment from clone pMAP-c2-hFAS into vector pET-32b(+) and then ligating, in proper orientation, the 1.9-kb BamHI–BamHI fragment (Fig. 2) immediately upstream at the unique BamHI site of this clone. The orientation of the BamHI fragment was verified by sequencing through the ligation sites. This clone is expected to produce a recombinant protein (TRX-hFAS-dII-III) of 159 kDa.
Expression and Purification of Recombinant hFAS and Domains I, II′-III, and II-III.
E. coli TOP10F′ cells containing the expression plasmid MBP-hFAS were grown at 37°C in 10 to 15 liters of LB broth medium containing ampicillin (100 μg/ml) and glucose (2 g/liter) to an optical density of 0.6 to 0.8 at 600 nm. At this point, isopropyl-1-thio-d-galactoside was added to the medium to a final concentration of 1 mM, and the culture temperature was lowered to 30°C. After 3 to 4 hr, the cells were harvested and washed once with a sodium phosphate solution (20 mM, pH 7.4) that contained NaCl (150 mM). The cell pellet was resuspended in lysis buffer (50 mM Tris·HCl, pH 8.0/1 mM EDTA/1 mM DTT/100 μg/ml lysozyme/0.5 μg/ml leupeptin/0.5 μg/ml pepstatin/1 mM benzamidine) at a ratio of 0.33 g (wet weight) per ml. The suspension was kept on ice for 1 hr. To break the DNA and reduce viscosity, the lysed cell suspension was sonicated in 5-ml aliquots by using three pulses (15 sec each) at 50–70 W, and the cell debris was removed by centrifugation at 17,000 × g for 30 min. The cell-free extract was fractionated with solid ammonium sulfate, and the protein fraction precipitating between 25 and 40% saturation was collected by centrifugation, dissolved in a minimal volume of buffer A (50 mM potassium phosphate, pH 7.5/1 mM EDTA/1 mM DTT/10% glycerol), and dialyzed against buffer A for 2 to 4 hr at 4°C. The dialyzed protein (4–6 mg/ml) was loaded onto an amylose-resin column (bed volume, 20 ml), and the column was washed with 60 ml of buffer A. The bound protein was eluted from the resin with 25 ml of buffer A containing 10 mM maltose and 100 mM NaCl. The fractions containing FAS were detected by measuring NADPH oxidation in the presence of acetyl-CoA and malonyl-CoA. The active fractions were pooled, and the protein was precipitated by adding solid ammonium sulfate to a saturation of 55%. The precipitated protein (≈10 mg) was collected by centrifugation, dissolved in 10 mM sodium phosphate (pH 6.8), and dialyzed for 4 hr against the same buffer. The dialyzed proteins from three batches (≈30 mg) were loaded onto a hydroxylapatite column (6.0 × 2.5 cm) that was equilibrated with 10 mM potassium phosphate, pH 6.8. The column was washed successively with 70 ml each of 100-mM and 200-mM sodium phosphate (pH 6.8), followed by a linear gradient of 50 ml each of 200–400-mM sodium phosphate (pH 6.8). Fractions of 3.0 ml each were collected and assayed for FAS activity. Fractions 26 to 28, which contained the active synthase, were pooled, and the protein was precipitated with ammonium sulfate at 50% saturation and redissolved in buffer A. When cell-free extracts were prepared from uninduced E. coli cells that harbored the fusion plasmid and were similarly analyzed, no detectable protein eluted with maltose from the affinity column, suggesting that the FAS activity being measured after affinity chromatography was that of the recombinant protein and not that of the E. coli FAS complex.
MBP-hFAS-dI was expressed in Top10F′ E. coli by essentially the same procedure described for MBP-hFAS. Cell-free extracts of the expressed protein were prepared in the same manner as that of MBP-hFAS, and the active protein was separated by precipitation with ammonium sulfate (0–50% saturation). The protein was collected, dissolved in buffer A, and purified by affinity chromatography using amylose resin in a manner similar to that used for the purification of MBP-hFAS. The eluted protein was precipitated with ammonium sulfate and further purified by chromatography on a Superose 6 column (prepacked HR 10/30 preequilibrated with buffer A). The column was washed with buffer A, and the active proteins were assayed for the activities of the acetyl and malonyl transacylases and the dehydratase. The active fractions were pooled, and the protein was isolated by ammonium sulfate precipitation and dissolved in buffer A.
The TRX-hFAS recombinant proteins, TRX-hFAS-dII′-III and TRX-hFAS-dII-III, were expressed in E. coli BL21(DE3) cells and induced as described above for the MBP-hFAS. Cell-free extracts were also prepared as described above, and the recombinant proteins were partially purified by metal-chelate affinity chromatography using TALON resin as described by the manufacturer. The crude extract (8 ml containing 8–10 mg/ml of protein) was mixed with 2 ml of the settled bed of TALON resin, and the suspension was agitated at 4°C for 20 min. The resin was separated by centrifugation at 700 × g and washed three times with 40 ml of buffer B (20 mM Tris·HCl, pH 8.0/100 mM NaCl) and once with 40 ml of buffer B containing 10 mM imidazole. The resin was then packed in a column, and the proteins were eluted from the resin with 25 ml of buffer B containing 100 mM imidazole. Fractions of 2 ml each were collected and assayed for thioesterase activity. The active fractions were pooled, and the protein was precipitated with ammonium sulfate at 55% saturation. The precipitated protein was collected by centrifugation and dissolved in buffer A.
General Properties and Partial Enzymatic Activities of the Expressed Recombinant Proteins.
The purification procedures used gave variable yields of the expressed proteins, depending upon the size of the expressed proteins. Specifically, there were greater yields of proteins with low molecular weights, as in the case of domain II′-III. Table 1 summarizes the results obtained from a typical purification procedure involving MBP-hFAS-dI. This domain contains the partial activities of the β-ketoacyl synthase, the acetyl and malonyl transacylases, and the β-hydroxyacyl dehydratase; however, the activity of the ketosynthase was not assayed for lack of a specific assay. As shown in Table 1, the activities of the transacylases were relatively high in the cell-free extract due to the presence of the endogenous E. coli transacylases (2, 3). However, the E. coli enzymes were mostly removed by fractionation with ammonium sulfate. Beyond this step, the ratios of the partial activities of the acetyl and malonyl transacylases and the dehydratase remained the same (Table 1), indicating that these activities are associated with the same protein. Similar observations were also made regarding the relative ratio of the partial activities associated with the recombinant proteins MBP-hFAS and TRX-hFAS-dII-III.
Table 1.
Purification of recombinant MBP-hFAS domain I from E. coli
| Step | Total protein (mg) | Specific activities, nmol/min per mg
of protein
|
||
|---|---|---|---|---|
| AT | MT | DH | ||
| Cell-free supernatant | 1200 | 57 | 83 | 0.5 |
| Ammonium sulfate fraction | 400 | 11 | 4 | 0.8 |
| Amylose-resin column | 8 | 516 | 171 | 50.5 |
| FPLC Superose 6 | 4 | 801 | 272 | 65.2 |
AT, acetyl transacylase; MT, malonyl transacylase; DH, β-hydroxyacyl dehydratase; FPLC, fast-protein liquid chromatography.
The purification procedures used yielded partially purified preparations of the recombinant proteins. Based on the nucleotide sequences discussed earlier, the proper expression of MBP-hFAS cDNA should result in the production of a fusion protein that has an approximate molecular weight of 310,000, which indeed is the case as verified by SDS/PAGE analysis of the purified fusion protein (Fig. 3A). Analysis of the protein by Western blotting also confirmed that the 310-kDa protein band reacts with antibodies against human FAS (Fig. 3A, lane 3). Expression of the MBP-hFAS-dI cDNA should yield a protein 191 kDa in size. Analysis of the purified protein by SDS/PAGE verified the size of the protein (Fig. 3B, lane 2) and confirmed its identity as a hFAS dI fusion protein by its reaction with anti-FAS antibodies and anti-MBP serum (Fig. 3B, lanes 3 and 4).
Figure 3.
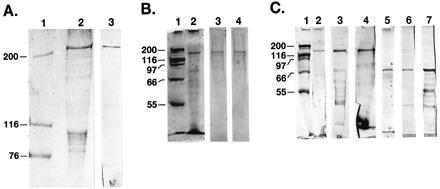
SDS/PAGE and Western blot analyses of partially purified recombinant fusion proteins of hFAS and its subdomains. (A) Protein standards (lane 1) and the MBP-hFAS fusion protein (5 μg) were subjected to SDS/PAGE analysis (4–15% gradient), and the gel was then stained with Coomassie blue (lane 2) or subjected to Western blot analysis using anti-hFAS antibodies (lane 3). (B) and (C) Protein standards (lanes 1) and 5 μg each of fusion proteins MBP-hFAS-dI (B, lane 2), TRX-hFAS-dII-III (C, lanes 2, 3, and 4), and TRX-hFAS-dII′-III (C, lanes 5, 6, and 7) were subjected to 7.5% SDS/PAGE analysis, and the gels were either stained with Coomassie Blue (B, lanes 1 and 2; C, lanes 1, 2, and 5) or subjected to Western blot analysis using either anti-hFAS antibodies (B, lane 3; C, lanes 3 and 6), anti-MBP serum (B, lane 4), or S-protein AP conjugate (C, lanes 4 and 7).
The expressed fusion proteins of the TRX containing the FAS subdomains were readily purified by affinity chromatography with a relatively higher yield than those of the MBP fusion proteins. Based on the nucleotide sequence of the TRX-hFAS domains II-III expression plasmid, the expressed fusion protein is expected to have a molecular mass of 155 kDa. As judged by SDS/PAGE analysis, the recombinant TRX-hFAS-dII-III protein had an estimated molecular mass of 159 kDa (Fig. 3C, lane 2) and reacted positively with anti-hFAS antibodies (Fig. 3C, lane 3) and S-protein alkaline phosphatase conjugate in Western blot analysis (Fig. 3C, lane 4). Similarly, the expression of the cDNA of TRX-hFAS-dII′-III should yield a fusion protein that has a molecular mass of 83 kDa. Analysis of the affinity-purified recombinant fusion protein showed the presence of a polypeptide that had an estimated molecular weight of 83,000 (Fig. 3C, lane 5) and cross-reacted with anti-hFAS antibodies (Fig. 3C, lane 6) and S-protein alkaline phosphatase conjugate in a Western blot (Fig. 3C, lane 7).
The partially purified recombinant proteins were tested for palmitate synthesis and for partial FAS activities. As shown in Table 2, the fusion protein MBP-hFAS catalyzes the synthesis of palmitate as measured by NADPH oxidation and the incorporation of 14C-labeled acetyl-CoA and malonyl-CoA into long-chain fatty acids, which, on analysis by HPLC, showed the presence of [14C]palmitate, [14C]stearate, and [14C]arachidate but very little or no myristate (Fig. 4). By these criteria, the recombinant MBP-hFAS was comparable to the purified FAS that was isolated previously from HepG2 cells grown in culture (5). Nevertheless, the FAS activity of the MBP-hFAS attests well to its expression in E. coli, its posttranslational modification by the attachment of 4′-phosphopantetheine, and its proper folding to form the active FAS dimer. This is a remarkable achievement in E. coli, considering the size of the expressed protein and its multifunctional nature. Moreover, the presence of the MBP fusion protein (≈42 kDa) at the N terminus had little effect on FAS activity.
Table 2.
Comparison of recombinant hFAS and its subdomain activities with native HepG2 FAS
| Functional assays | HepG2 FAS* | MBP-hFAS | MBP-hFAS dI | TRX-hFAS dII-III | TRX-hFAS dII′-III |
|---|---|---|---|---|---|
| FAS activities | |||||
| NADPH oxidation | 462 | 611 | |||
| [1-14C]acetyl-CoA | 29.6 | 43.0 | |||
| [2-14C]malonyl-CoA | 220.6 | 300.0 | |||
| FAS partial activities | |||||
| Acetyl transacylase | 1336.0 | 800.3 | 800.5 | ||
| Malonyl transacylase | 558 | 272 | 272 | ||
| β-Ketoacyl synthase | 0.20 | 0.14 | ND | ||
| β-Hydroxyacyl dehydratase | 57 | 49 | 65 | ||
| β-Ketoacyl reductase | 2514 | 1921 | 2754 | ||
| Enoyl reductase | 89.3 | 34.4 | 95.2 | ||
| Thioesterase | 54.8 | 41.9 | 50.7 | 160.0 |
The assays used to measure the overall and partial activities of hFAS were performed as described in Materials and Methods. The activities were expressed as nmol/min/mg of protein. ND, not determined.
The values given are from our previously published paper (5).
Figure 4.
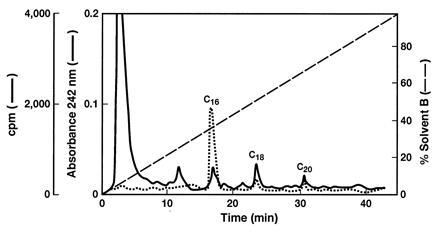
Analysis of phenacyl esters of radioactive fatty acids produced by MBP-hFAS. The fatty acid sample was derivatized with 2-bromoacetophenone and subjected to HPLC analysis. The sample was separated on a Vydac C18 column and eluted with a linear gradient of acetonitrile as described in Materials and Methods.
Testing the recombinant MBP-hFAS for the partial activities associated with FAS showed the presence of all such activities at levels comparable to those of native HepG2 FAS (Table 2). It is not clear why some activities are relatively lower than those in native FAS, but this result may be due to either the use of a model substrate in these assays or the effects of the fusion proteins on these assays. In any case, the activities of these domains are high enough that they may not affect the overall activity of fatty acid synthesis.
As stated above, the order of the partial activities of FAS along the FAS polypeptide were deduced from the proteolytic mapping of subunit protein (11–14, 18). This order was confirmed by the analyses of amino acid sequences derived from the nucleotide sequences of chicken, rat, and human FAS cDNAs (5–10). The location of most of the partial activities of FAS could be readily identified by comparing the already known amino acid sequences of the active sites of the β-ketoacyl synthase and the acetyl-CoA and malonyl-CoA transacylases, the sequence of the binding site of 4′-phosphopantetheine, and the sequence of the thioesterase (1). Moreover, the location of the enoyl and ketoacyl reductases were suggested based on the involvement of a lysine residue in the NADPH binding regions of the protein (29, 30) and the presence of the consensus sequence, Gly-X-Gly-X-X-Gly, which has been found in most dehydrogenase sequences (31). The unavailability of information about the active site sequences and the lack of a discernible similarity in the dehydratase sequences hampered the identification and localization of the dehydratase activity. However, when the His878 of rat FAS was identified in a sequence motif (H878-X-X-X-G-X-X-X-X-P) that is conserved among the cDNA sequences of several bacterial dehydratases and among animal FAS cDNA sequences and was modified to Ala, the dehydratase function was lost, suggesting that the dehydratase active site is located close to the domain of the acetyl-CoA and malonyl-CoA transacylases (17). The presence of the dehydratase active site in the recombinant MBP-hFAS-dI protein, which contains the 1295 N-terminal amino acid residues, and the presence of an equivalent His878 in human FAS (5) clearly indicated that the dehydratase is located in dI and not in dII as suggested before (18). Taking into account our earlier proteolytic mapping of the active functions of chicken FAS (1), the isolation and determination of an N-terminal sequence of a 47-kDa tryptic peptide from rat FAS that contained the transacylase activities, the size of the isolated chicken FAS dehydratase peptide (18), and the results discussed above, we estimated that FAS dI spans about 1000 amino acid residues of the N-terminal region.
The recombinant TRX-hFAS-dII′-III protein, which spans the 590 amino acid residues at the carboxyl-terminal end of human FAS, exhibited only the thioesterase activity. The specific activity of the thioesterase of the expressed purified protein was three times that of the native hFAS prepared from HepG2 cells, a finding that is consistent with the relative sizes of TRX-hFAS-dII′-III (Mr ≈83,000) and human FAS (Mr 272,000). Besides the thioesterase, the TRX-hFAS-dII′-III protein contained the site for the prosthetic group, 4′-phosphopantetheine, and part of the β-ketoacyl reductase domain. Since this expression plasmid contains sequences of human FAS from amino acid residue 1914 to the carboxyl-terminal end, it falls short of residues needed for an active β-ketoacyl reductase domain, which is located between amino acid residues 1870 and 2100 of hFAS (5). Indeed, when the expressed recombinant protein TRX-hFAS-dII-III was extended to cover residue 1296 to the carboxyl-terminal end of FAS as shown in Table 2, the β-ketoacyl reductase and the enoyl reductase are reconstituted. Based on the comparison of the sequences of the various FASs (5–9) and our peptide mapping data (1, 11–14, 18) and the results and the cloning and expression experiments discussed above, recombinant proteins MBP-hFAS-dI and TRX-hFAS-dII-III contain parts of the interconnecting domain region. Since domain II begins near amino acid residue 1620 of the hFAS subunit peptide, the interdomain polypeptide between dII and dIII spans the length of 600 amino acid residues. The considerations for the beginning of domain II and the locations of the ACP and the thioesterase (dIII) are well documented (10, 30, 32–34). Consequently, the cloning and expression of the various components of hFAS further confirmed the domain configuration of the enzyme. The expression, therefore, of the human multifunctional FAS and its subdomains in E. coli represents a leap forward in the study of multifunctional proteins. The folding of the full-length protein subunits into active subdomains and the ease of expressing the full-length protein and its subdomains should facilitate mutation analyses of the structure-function relationship of this complex enzyme.
Acknowledgments
We thank Ms. Pamela Paradis Powell for editing the manuscript. This work was supported by Grant GM19091 from the National Institutes of Health.
Footnotes
Abbreviations: FAS, fatty acid synthase; ACP, acyl carrier protein; hFAS, human FAS; dI, dII, dII′, and dIII, FAS domains I, II, partial II, and III, respectively; MBP, maltose-binding protein; TRX, thioredoxin.
References
- 1.Wakil S J. Biochemistry. 1989;28:4523–4530. doi: 10.1021/bi00437a001. [DOI] [PubMed] [Google Scholar]
- 2.Wakil S J, Stoops J K, Joshi V C. Annu Rev Biochem. 1983;52:537–579. doi: 10.1146/annurev.bi.52.070183.002541. [DOI] [PubMed] [Google Scholar]
- 3.Volpe J J, Vagelos P R. Physiol Rev. 1977;56:339–417. doi: 10.1152/physrev.1976.56.2.339. [DOI] [PubMed] [Google Scholar]
- 4.Shimakata T, Stumpf K. Plant Physiol. 1982;69:1257–1262. doi: 10.1104/pp.69.6.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jayakumar A, Tai M-H, Huang W-Y, Al-Feel W, Hsu M, Abu-Elheiga L, Chirala S S, Wakil S J. Proc Natl Acad Sci USA. 1995;92:8695–8699. doi: 10.1073/pnas.92.19.8695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amy C M, Witkowski A, Naggert J, Williams B, Randhawa Z, Smith S. Proc Natl Acad Sci USA. 1989;86:3114–3118. doi: 10.1073/pnas.86.9.3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kameda K, Goodridge A G. J Biol Chem. 1991;266:419–426. [PubMed] [Google Scholar]
- 8.Holzer K R, Liu W, Hammes G G. Proc Natl Acad Sci USA. 1989;86:4387–4391. doi: 10.1073/pnas.86.12.4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beck K-I, Schreglmann R, Stathopulos I, Klein H, Hoch J, Schweizer M. DNA Seq. 1992;2:359–386. doi: 10.3109/10425179209020817. [DOI] [PubMed] [Google Scholar]
- 10.Huang W-Y, Chirala S S, Wakil S J. Arch Biochem Biophys. 1994;314:45–49. doi: 10.1006/abbi.1994.1410. [DOI] [PubMed] [Google Scholar]
- 11.Mattick J S, Tsukamoto Y, Nickless J, Wakil S J. J Biol Chem. 1983;258:15291–15299. [PubMed] [Google Scholar]
- 12.Mattick J S, Nickless J, Mizugaki M, Yang C-Y, Uchiyama S, Wakil S J. J Biol Chem. 1983;258:15300–15304. [PubMed] [Google Scholar]
- 13.Wong H, Mattick J S, Wakil S J. J Biol Chem. 1983;258:15305–15311. [PubMed] [Google Scholar]
- 14.Tsukamoto Y, Wong H, Mattick J S, Wakil S J. J Biol Chem. 1983;258:15312–15322. [PubMed] [Google Scholar]
- 15.Stoops J K, Ross P R, Arslanian M J, Aune K C, Wakil S J, Oliver R M. J Biol Chem. 1979;254:7418–7426. [PubMed] [Google Scholar]
- 16.Stoops J K, Wakil S J. J Biol Chem. 1981;256:5128–5133. [PubMed] [Google Scholar]
- 17.Joshi A K, Smith S. J Biol Chem. 1993;268:22508–22513. [PubMed] [Google Scholar]
- 18.Tsukamoto Y, Wakil S J. J Biol Chem. 1988;263:16225–16229. [PubMed] [Google Scholar]
- 19.Pazirandeh M, Chirala S S, Huang W-Y, Wakil S J. J Biol Chem. 1989;264:18195–18201. [PubMed] [Google Scholar]
- 20.Joshi A K, Smith S. Biochem J. 1993;296:143–149. doi: 10.1042/bj2960143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuhajda F P, Jenner K, Wood F D, Hennigar R A, Jacobs L B, Dick J D, Pasternack G R. Proc Natl Acad Sci USA. 1994;91:6379–6383. doi: 10.1073/pnas.91.14.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shurbaji M S, Kuhajda F P, Pasternack G R, Thurmond T S. Am J Clin Pathol. 1992;97:686–691. doi: 10.1093/ajcp/97.5.686. [DOI] [PubMed] [Google Scholar]
- 23.Redston M S, Kern S E, Vogelstein B, Hamilton S R. Lab Invest. 1992;66:47A. (abstr.). [Google Scholar]
- 24.Arslanian M J, Wakil S J. Methods Enzymol. 1975;35:59–65. doi: 10.1016/0076-6879(75)35138-0. [DOI] [PubMed] [Google Scholar]
- 25.Wood R, Lee T. J Chromatogr. 1983;254:237–246. [Google Scholar]
- 26.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 27.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 28.Maniatis T, Fritsch E F, Sambrook J. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1982. [Google Scholar]
- 29.Poulose A J, Kolattukudy P E. Arch Biochem Biophys. 1983;220:652–656. doi: 10.1016/0003-9861(83)90459-9. [DOI] [PubMed] [Google Scholar]
- 30.Chirala S S, Kasturi R, Pazirandeh M, Stolow D T, Huang W-Y, Wakil S J. J Biol Chem. 1989;264:3750–3757. [PubMed] [Google Scholar]
- 31.Eventoff W, Rossman M G. Crit Rev Biochem. 1975;3:111–140. doi: 10.3109/10409237509102554. [DOI] [PubMed] [Google Scholar]
- 32.Rangan V S, Witkowski A, Smith S. J Biol Chem. 1991;266:19180–19185. [PubMed] [Google Scholar]
- 33.Yang C-Y, Huang W-Y, Chirala S S, Wakil S J. Biochemistry. 1988;27:7773–7778. doi: 10.1021/bi00420a028. [DOI] [PubMed] [Google Scholar]
- 34.Huang W-Y, Stoops J K, Wakil S J. Arch Biochem Biophys. 1988;270:92–98. doi: 10.1016/0003-9861(89)90011-8. [DOI] [PubMed] [Google Scholar]