

Abstract
It has been shown with lipid layers and more recently with purple membranes that protons have slow surface-to-bulk transfer. This results in long-range proton lateral conduction along membranes. We report here that such lateral transfer can take place along a pure protein film. It is strongly controlled by the packing. Subtle reorganizations of the protein–protein contact can be biological switches between interfacial and delocalized proton pathways between sources and sinks.
The occurrence of barriers preventing free exchange of protons from membranes to the bulk phase has been the subject of debate in membrane biology for more than 30 years (1–4). The barriers are of primary relevance to the occurrence of an interfacial pathway in chemioosmotic coupling and to other reactions at the membrane/solution interface. In the mid-1980s, experiments using fluorescence spectroscopy with lipid monolayers proved that facilitated interfacial proton conduction was present with this simple model (5). This observation was confirmed on the same system by surface potential, pressure, and electrical conductance measurements (6–9). A thermodynamic implication from such conductions was that barriers prevented the free leakage of protons from the membrane interface to the bulk phase (10). Last year, two groups reported that delayed transfer of proton from surface to bulk was taking place in purple membrane systems after they were energized by a light flash (11, 12). These results indicated that proton transfer was taking place between the proton channel and the membrane/solution interface. It was suggested that this took place along the lipid headgroups as described with monolayers.
The results on lipid monolayers were tentatively explained either by coupled protonation/deprotonation reactions of the lipid headgroups (13) or by the presence of a hydrogen bond network along the film. This was created by the polar headgroup region of phospholipids and by the interfacial water molecules (9, 14). This was borne out by electrical measurements on lipid multilayers (15) in which it was shown that the degree of hydration was a key parameter (16). This was proved by recent results using scanning tunneling microscopy that showed lateral conductivity of ultrathin water films (17). Such conductivity was observed both on hydrated mica samples and on dipalmitoylphosphatidylcholine Langmuir Blodgett films that were formed on mica sheets (18). This was suggested to be due to proton movement (19).
An unsolved problem for this surface conduction is knowing whether it is restricted to lipid domains or whether proteins play a similar role or prevent it. To mimic membrane interfaces, molecular assemblies such as monolayers are a useful tool, as shown with lipids. They can be built by the self-assembly of proteins on the air/water interface. Bovine serum albumin (BSA) was chosen because it is known to form monolayers when spread on the air/water interface while conserving residual native structures (20). The film can be compressed with no abrupt conformational change. It is rigid as shown by dilatometric studies (21).
Investigation of proton conduction by BSA films was carried out by two complementary methods, using either a direct approach by using the pH dependence of fluorescein isothiocyanate (FITC)-BSA fluorescence (5) or an indirect one by monitoring the surface electrical conductance (8).
MATERIALS AND METHODS
Chemicals.
Mops, BSA, and FITC-BSA were purchased from Sigma. Salts were analytical grade. Ultrapure water was obtained from a Milli-Q column (Millipore).
Monolayers Preparation.
The troughs were milled in Plexiglas to minimize light scattering. Proteins were dissolved in Milli-Q water (0.3 g/ml). The protein film was obtained by pouring a small volume of protein solution along a glass rod implanted across the water/air interface (Fig. 1A) (22, 23). The subphase was 1 M NaCl and 1 mM Mops (pH 7.4) for fluorescent measurements, and pure Milli-Q water for conductance assays. The film compression was obtained by moving a Teflon barrier to change the total surface area of the monolayer. The film surface pressure was monitored by means of a platinum plate connected to a force transducer that was designed in our laboratory. For lateral proton conduction assays, the film surface pressure was changed by increasing the number of protein molecules spread on the air/subphase interface for a constant total surface area of the film (5).
Figure 1.
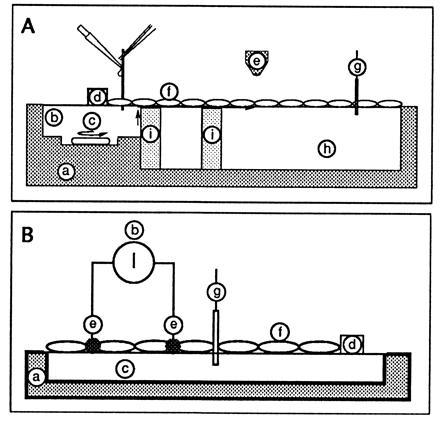
Schematic drawing of the monolayer trough. (A) Fluorescence detection. This was the setup described in ref. 5. Sites: a, Plexiglas trough (to reduce light scattering); b, injection of acid; c, stirrer to homogenize the injection compartment; d, Teflon barrier (to limit the film and to define the part in contact with the acidic subphase); e, fluorescence observation area (detection was either with a PM tube or with an intensified video camera); f, monolayer (the protein film was obtained by pouring a small volume of a protein solution along a glass rod implanted across the water/air interface); g, surface pressure transducer; h, aqueous subphase; i, glass barrier to prevent proton diffusion in the bulk. The fluorescence baseline was obtained without spreading film. The 100% fluorescence was given after spreading the protein film on a 1 M NaCl/1 mM Mops buffer (pH 7.4). Acid was then injected in b (bringing the local bulk pH to 2). The transfer of protons to site h was detected by the associated decrease in fluorescence emission. (B) Conductance detection. The procedure and setup are described in the text.
Fluorescence Studies.
Upon protonation, the fluorescence of fluorescein is known to decrease. The apparent pK of the FITC adduct of BSA taken as the subphase pH giving 50% in the total emission was around 7 whatever the surface pressure (Table 1). Fluorescence emission changes upon compression of the monolayer of FITC-BSA or of BSA containing the pH indicator FITC-BSA (20 mol percent) were observed using two approaches.
Table 1.
Apparent pK values of the FITC adduct of BSA obtained for different surface pressure of the labeled protein film
| Surface pressure, mN/m | Apparent pKa |
|---|---|
| 0.5 | 7 |
| 1 | 6.9 |
| 2 | 6.95 |
| 3 | 6.9 |
| 4 | 6.95 |
(i) An interface fluorimeter constructed in the laboratory was used to monitor the front face fluorescence (5). The fluorescence from a small illuminated area (about 2 mm in radius) was measured by means of a photomultiplier tube (Thorn EMI Electron Tubes model 9558) connected to a data acquisition unit. The light source was an Osram (Berlin) model XBO 75W/4 xenon lamp. Wavelengths were selected by means of optical filters (λex = 462 ± 11 nm, λem = 519 ± 8 nm) (Métallisations et Traitements Optiques, France).
(ii) The monolayer interface was observed under an inverted fluorescence microscope (Leitz Fluovert). The light source was an Osram model HBO 100W/2 mercury arc lamp. Wavelengths were selected by an H3 filter block (390 nm ≤ λex ≤ 490 nm, 515 nm ≤ λem) (Leitz). A video monitoring device was connected to the microscope: a light-intensifying camera (Lhesa) associated with a black-and-white monitor (RCA). Using a 32× objective, the system provided depth of focus of about 1–2 μm. The video device was connected to a digitizer (Info’Rop, France) driven by a computer (Motorola). The video signal was then digitized at 8 bits (256 light levels) with a pixel definition of 3 μm2. Image analysis was performed by means of a software library (Trimago, Ifremer) containing the major routines for digital image processing.
Proton Conduction.
The fluorescence baseline was obtained without spreading the film. The 100% fluorescence was given after spreading the protein film on a 1 M NaCl/1 mM Mops buffer (pH 7.4). The experiment was performed in the steepest part of the pH response of the dye. Acid was then injected (bringing the local bulk pH to 2) (Fig. 1A, site b). The transfer of protons to the observation area (Fig. 1A, site e), i.e., on a distance of 4 cm, was detected by the associated decrease in fluorescence emission. Two key data set the parameters of the proton conduction: TH+, the delay between H+ injection and the beginning of the decrease in fluorescence, and ΔF, the change in fluorescence emission.
Surface Electrical Measurements.
As shown in Fig. 1B, two very thin platinum electrodes (site e: diameter, 0.2 mm; length, 2 cm; width, 5 mm) were brought into contact with the air/water interface formed in a plexiglas trough (site a) (24). Ultrapure water subphase (site c) was used to obtain a very low background signal. A small voltage was applied and the resulting current intensity was recorded (site b). This background was reduced by the use of very thin interfacial electrodes. As other resistances in the network were negligible when compared with the monolayer, the current was directly related to the film conductance. The protein film was spread (site f) and then compressed by moving the Teflon barrier (site d). The associated change in the current was recorded as a function of the protein packing [monitored by the film pressure (site g)]. The conductance between the two electrodes was due to a contribution from the film and from a constant background signal associated with the bulk phase (8).
RESULTS
Compression Isotherms.
Compression of the film induced sequential changes in surface pressure and fluorescence as shown in Fig. 2A. A dramatic increase in emission was observed around 550 nm2 per spread FITC-BSA molecule, followed by a plateau. A decrease in emission was present below 350 nm2 where the surface pressure started to increase. Direct observation of the film organization by interfacial digitized video fluorescence microscopy showed that no macroscopic domain formation was present during film compression (Fig. 2B). Quantification of the average light levels of the digitized images was shown in Fig. 2C. When working with a mixture of BSA and FITC-BSA (3/1 molar ratio), the same behavior was observed except that the fluorescence intensity was reduced to about 25% of what was observed with a pure FITC-BSA monolayer.
Figure 2.
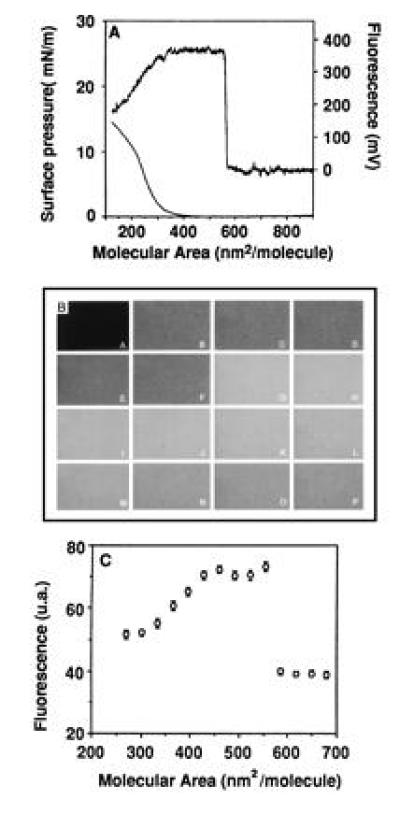
Fluorescence emission change of FITC-BSA films on the air/water interface upon compression. The film was compressed after spreading the protein solution by a slow lateral movement of a Teflon barrier. The subphase was 1 M NaCl/1 mM Mops, pH 7.3. The protein molecular area was obtained from the spread volume. By comparing with previous data (20), it was concluded that only 33% of the proteins remained at the interface by this spreading procedure. (A) The detection was obtained by a photomultiplier tube on an interface fluorimeter specially designed for monolayer investigation (5). (B) The detection was obtained on an inverted microscope (Leitz, H3 block filter, 32 × objective) by epifluorescence using video detection (Lhesa camera, Optimas software). (A) Free air/water interface. (B–P) Compression of the film (B, 730 nm2/mol; C, 680 nm2/mol; D, 650 nm2/mol; E, 617 nm2/mol; F, 586 nm2/mol; G, 554 nm2/mol; H, 523 nm2/mol; I, 491 nm2/mol; J, 460 nm2/mol; K, 428 nm2/mol; L, 397 nm2/mol; M, 365 nm2/mol; N, 334 nm2/mol; O, 302 nm2/mol; P, 271 nm2/mol). Micrograph was scanned with a SprintScan 35 (Polaroid) and printed using a XLS 8600 PS printer (Kodak). (C) The emission detected in B was quantified by pixel averaging (4 × 4, sampling 30).
As previously described with lipid films (5, 25), by changing the pH of the subphase, a rough estimation of the probe apparent pKs was obtained for different packings of the film where the fluorescence is detectable (Table 1). A value close to 7 was detected in all cases, showing that no surface potential effect was present.
Proton Surface Conduction.
Proton lateral movement was monitored by the dissipation of a local pH jump (5) (Fig. 3A) and by comparing the movement in the bulk and in the film by the associated fluorescein emission drop. The fluorescence approach shows that a fast lateral proton diffusion (a low TH+ value, about 200 s) was present when the film packing density was between 400 and 300 nm2 per molecule, but disappeared as soon as the film was compressed below 300 nm2 per molecule (Fig. 3B). The associated relative fluorescence change ΔF was fairly constant for the different film packings. The movement was much slower in the bulk phase. When no film was spread on the air/water interface and the water soluble form of FITC was present in the bulk, no fluorescence change was detected before 40 min after acid injection.
Figure 3.
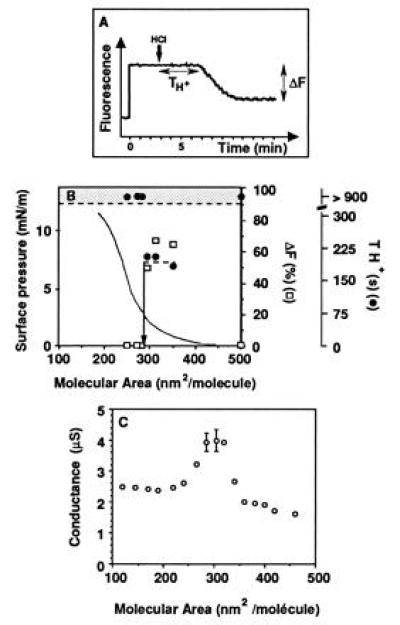
Detection of the change of proton lateral conduction along the BSA film with the monolayer compression. (A) In the fluorescence approach, the dissipation of the pH jump is described by the two parameters TH+ (delay between the acid injection and the decrease in fluorescence) and ΔF (the relative amplitude in fluorescence decrease) (see text). In this experiment, the film packing density was 350 nm2/mol. (B) Dependence of the proton lateral conduction on the molecular area. Conduction was observed by the fluorescence approach. TH+ in the bulk phase without spreading film was larger than 2000 s. (C) In the conductance method, the change is recorded with a background signal associated with the free air/water interface equivalent to 1.7 ± 0.1 μS. (When not shown, error bars are in the symbols.)
No dramatic change in surface pressure of the BSA film took place when the proton conductivity was switched off. When present, the conduction was not modulated by film compression. The same kinetic of fluorescence change was obtained when working on a film that was formed with a mixture of free and labeled BSA (3:1 mol/mol). The only difference was that the intensity of the emission was only 25% of what was observed with a pure FITC-BSA film, reflecting the dilution effect. The FITC labeling was therefore not involved in proton surface conduction.
Surface Electrical Conductance.
No drift in surface electrical conductance was observed on a free solution during 2000 s as would has been the case if contamination had been created either by detergent present in the subphase or by aerosol deposition (data not shown). When the BSA film was spread with low packing, no change in conductance was detected when compared with the subphase. Upon film compression, a slow increase in conductance was observed up to 350 nm2/mol, where a dramatic increase (more than twofold) was detected (Fig. 3C). A plateau region was then present down to 290 nm2/mol, where the conductance was observed to decrease again and to reach a value close to that of the free solution, at a packing of 220–200 nm2/mol.
DISCUSSION
The conclusion of the present study is that interfacial electrical conductance is present along a BSA film spread on the air/water interface. It reflects a motion of protons along the film as shown by fluorescence investigation. The film organization controls this conduction (Fig. 4). The elementary conducting unit is the surface of the protein as shown with BSA (26) and lysozyme (27). As no conduction is observed for a loosely packed film, contact between proteins must take place so that the long-distance transfer occurs as we showed previously in the case of phospholipids (5, 13). A drop in proton conduction and in the associated interfacial electrical conductance was observed at high packing as in some cases with lipid films (8, 28). This was associated with the organizational change due to phase or structural transitions (28). In the present investigation, we made the same observation but another explanation must be given which take into account the rigidity of a BSA molecule (21). The disappearance of proton conduction must be explained by the reorganization of the molecular assembly. This is borne out by the observation that in the BSA experiments the fluorescence isotherm shows that a change in emission takes place for packing densities lower than 300 nm2/mol. This reorganization should result in a mismatch in the neighbor to neighbor transfer. A decrease in the transfer efficiency (i.e., in electrical conductivity) is observed. It decreases as the packing increases and film conductivity collapses.
Figure 4.
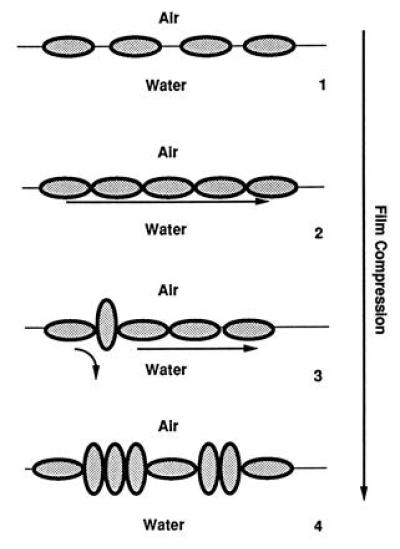
Molecular structural events of the protein film during its compression and their consequences on the proton lateral conduction. BSA molecules are drawn as grey ellipsoids. (1) At a low packing density, the molecules lie flat at the air/water interface. There is no molecular assembly, i.e., no contact between molecules. No proton conduction can take place. (2) When the film is compressed, a critical phenomenon takes place, giving the formation of a molecular assembly. A continuous layer of interfacial water molecules is created which facilitates the lateral proton transfer over a long distance (long arrow). (3) With a further film compression, some protein molecules are tilted to an upright position giving local breaks in the molecular assembly. These defects prevent the lateral conduction locally (curved arrow). (4) When the film is highly packed, more and more protein molecules are brought to the upright orientation. The associated increase in defects prevents the long-range proton conduction.
The convergence of the observations obtained by two completely different methodologies rules out any criticisms in the case of lipid films (29). It was suggested that convection induced by stirring played some role in proton movement when detected by fluorescence, but no stirring is present in the electrical experiments. Local heating due to the dilution of the 3 M acid solution may take place in the fluorescence experiments but is present whatever the film packing. Furthermore it does not affect the electrical measurements. Very thin electrodes were used to avoid any change in their contact with the film as suggested previously (24).
Some differences are present in the results obtained with the two methods, mostly in the high packing region. We suggest that this difference between the fluorescence and the electrical approaches is due to the methodologies for film compression or/and to a difference in sensitivity in the two assays. In a previous work, we showed that the proton transfer depended on the length of the pathway between the acid reservoir and the detection area (10). This could explain the dramatic character of the changes in the fluorescence experiments. A more progressive change is observed by the electrical method as suggested by the simulation previously described (10). There is another experimental difference between the two methods. Electrical conductance measurements are performed on pure water, whereas a buffered subphase is used with the fluorescence method.
This study shows that when proteins are spread as monolayers, surface proton conduction occurs over a long distance through a cooperative organization of the molecular assembly as described in the case of phospholipid monolayers (13).
A previous model suggested that protonation of fluorescein bound on BSA by bulk protons proceed along a pathway that is built by the multitude of carboxylate groups covering the protein surface (26). In this case, a deprotonation of the surface would occur due to the presence of proton acceptors in the bulk, preventing the long-range movement that we observed.
Proton transfer along protein channels was described as being supported by a hydrogen bond network built between amino acids (30, 31). But as such a model was rigid, a more realistic description was proposed in which some flexibility was present by involving hydration water molecules in the network (32). Proton conductivity along single protein surface was indeed observed in the case of lysozyme (27) and purple membrane (33). Conductivity was detected only when a critical level of protein hydration was present. The conclusion was that it was assisted by water molecules bound to the protein surface. The conductivity reflects motion of protons along threads of hydrogen-bonded water molecules supporting a long-range proton transfer. From percolation theory, it was concluded that conductivity results from the assembly of conducting elements. All these descriptions bear out the occurrence of proton conduction on the surface of proteins. Proton transfer will take place along a protein film if and only if there is a continuity of the hydrogen bond network between the proteins. We suggested that this was due to a bidimensional network of hydrogen bonds involving interfacial water molecules and film molecules.
From these observations, the mechanism of proton conduction along protein films therefore appears to present similarities to what was observed with lipid monolayers. This is borne out by the analogy of the observed TH+. Mismatches between the individual conducting units due to either orientational or conformational changes modulate the proton transfer as previously shown in the case of phospholipids (13). A key condition for lateral conduction to occur along interfaces is that an energy barrier prevents the free diffusion of protons from the interfacial pathways to the bulk phase (10). The same conclusion was suggested for other systems involving phospholipids (11, 12, 34). The present study definitively demonstrates that this energy barrier is present with protein layers but is controlled by changes in packing. This control can play a regulatory role in coupling in the case of biological membranes by controlling the proton surface exchange between sources and sinks. Two pathways can be present: (i) the classical delocalized one through the bulk phase and (ii) an interfacial one, when the membrane packing permits it. A final conclusion is that the interfacial proton conduction is supported not only by lipids as already shown but that proteins can play an active role in its support.
Acknowledgments
We thank Prof. A. Milon, Drs. M. P. Rols, N. Eynard, and P. Demange for their comments, Mr. D. Villa for his skillful assistance for the micrographs, and Mr. J. Robb for re-reading the manuscript.
Footnotes
Abbreviation: FITC, fluorescein isothiocyanate.
References
- 1.Boyer P D, Chance B, Ernster L, Mitchell P, Racker E, Slater E. Annu Rev Biochem. 1977;46:955–1026. doi: 10.1146/annurev.bi.46.070177.004515. [DOI] [PubMed] [Google Scholar]
- 2.Williams R J P. Proc R Soc London Ser B. 1978;200:353–380. doi: 10.1098/rspb.1978.0023. [DOI] [PubMed] [Google Scholar]
- 3.Williams R J P. Annu Rev Biophys Biophys Chem. 1988;17:71–97. doi: 10.1146/annurev.bb.17.060188.000443. [DOI] [PubMed] [Google Scholar]
- 4.Kell B B. Biochim Biophys Acta. 1979;549:55–99. doi: 10.1016/0304-4173(79)90018-1. [DOI] [PubMed] [Google Scholar]
- 5.Teissié J, Prats M, Soucaille P, Tocanne J F. Proc Natl Acad Sci USA. 1985;82:3217–3221. doi: 10.1073/pnas.82.10.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prats M, Teissié J, Tocanne J F. Nature (London) 1986;322:756–758. [Google Scholar]
- 7.Prats M, Tocanne J F, Teissié J. J Membr Biol. 1987;99:225–227. doi: 10.1007/BF01995703. [DOI] [PubMed] [Google Scholar]
- 8.Morgan H, Taylor D M, Oliveira O N. Biochim Biophys Acta. 1991;1062:149–156. doi: 10.1016/0005-2736(91)90386-m. [DOI] [PubMed] [Google Scholar]
- 9.Teissié J, Gabriel B, Prats M. Trends Biochem Sci. 1993;18:243–246. doi: 10.1016/0968-0004(93)90171-i. [DOI] [PubMed] [Google Scholar]
- 10.Prats M, Tocanne J F, Teissié J. Eur J Biochem. 1985;149:663–668. doi: 10.1111/j.1432-1033.1985.tb08975.x. [DOI] [PubMed] [Google Scholar]
- 11.Heberle J, Riesle J, Thiedemann G, Oesterhelt D, Dencher N A. Nature (London) 1994;370:379–382. doi: 10.1038/370379a0. [DOI] [PubMed] [Google Scholar]
- 12.Scherrer P, Alexiev U, Marti T, Khorana H G, Heyn M P. Biochemistry. 1994;33:13684–13692. doi: 10.1021/bi00250a019. [DOI] [PubMed] [Google Scholar]
- 13.Haines T M. Proc Natl Acad Sci USA. 1983;80:160–164. doi: 10.1073/pnas.80.1.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prats M, Tocanne J F, Teissié J. Eur J Biochem. 1987;162:379–385. doi: 10.1111/j.1432-1033.1987.tb10612.x. [DOI] [PubMed] [Google Scholar]
- 15.Leslie R B, Chapman D, Hart C J. Biochim Biophys Acta. 1967;135:797–811. doi: 10.1016/0005-2736(67)90050-8. [DOI] [PubMed] [Google Scholar]
- 16.Jendrasiak G L, Mendible J C. Biochim Biophys Acta. 1976;484:133–158. doi: 10.1016/0005-2760(76)90183-1. [DOI] [PubMed] [Google Scholar]
- 17.Guckenberger R, Heim M, Cecv G, Knapp H F, Wiegräbe W, Hillebrand A. Science. 1994;266:1538–1540. doi: 10.1126/science.7985024. [DOI] [PubMed] [Google Scholar]
- 18.Heim M, Cevc G, Guckenberger R, Knapp H F, Wiegräbe W. Biophys J. 1995;69:489–497. doi: 10.1016/S0006-3495(95)79922-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kell D B. Bioelectrochem Bioenerg. 1992;27:235–237. [Google Scholar]
- 20.Graham D E, Phillips M C. J Colloid Interface Sci. 1979;70:427–439. [Google Scholar]
- 21.Graham D E, Phillips M C. J Colloid Interface Sci. 1980;76:240–239. [Google Scholar]
- 22.Verger R, Pattus F. Chem Phys Lipids. 1976;16:285–291. doi: 10.1016/0009-3084(76)90023-2. [DOI] [PubMed] [Google Scholar]
- 23.Douillard R, Teissié J. J Colloid Interface Sci. 1991;143:111–119. [Google Scholar]
- 24.Widrig C A, Miler C J, Majda M. J Am Chem Soc. 1988;110:2009–2011. [Google Scholar]
- 25.Soucaille, Prats M, Tocanne J F, Teissié J. Biochim Biophys Acta. 1988;939:289–294. doi: 10.1016/0005-2736(88)90073-9. [DOI] [PubMed] [Google Scholar]
- 26.Yam R, Nachliel E, Gutman M. J Am Chem Soc. 1988;110:2636–2640. [Google Scholar]
- 27.Careri G, Geraci M, Giansanti A, Rupley J A. Proc Natl Acad Sci USA. 1985;82:5342–5346. doi: 10.1073/pnas.82.16.5342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gabriel B, Teissié J. J Am Chem Soc. 1991;113:8818–8821. [Google Scholar]
- 29.Gutman M, Nachliel E. Biochim Biophys Acta. 1995;1231:123–138. [Google Scholar]
- 30.Nagle J F, Morowitz H J. Proc Natl Acad Sci USA. 1978;75:298–302. doi: 10.1073/pnas.75.1.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagle J F, Tristam-Nagle S. J Membr Biol. 1983;74:1–14. doi: 10.1007/BF01870590. [DOI] [PubMed] [Google Scholar]
- 32.Schulten Z, Schulten K. Eur Biophys J. 1985;11:149–155. doi: 10.1007/BF00257393. [DOI] [PubMed] [Google Scholar]
- 33.Rupley J A, Siemankowski L, Careri G, Bruni F. Proc Natl Acad Sci USA. 1988;85:9022–9025. doi: 10.1073/pnas.85.23.9022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Antonenko Y N, Kovbasnjuk O N, Yaguzhinsky L S. Biochim Biophys Acta. 1993;1150:45–50. doi: 10.1016/0005-2736(93)90119-k. [DOI] [PubMed] [Google Scholar]