

Abstract
Understanding the mechanisms of action of membrane proteins requires the elucidation of their structures to high resolution. The critical step in accomplishing this by x-ray crystallography is the routine availability of well-ordered three-dimensional crystals. We have devised a novel, rational approach to meet this goal using quasisolid lipidic cubic phases. This membrane system, consisting of lipid, water, and protein in appropriate proportions, forms a structured, transparent, and complex three-dimensional lipidic array, which is pervaded by an intercommunicating aqueous channel system. Such matrices provide nucleation sites (“seeding”) and support growth by lateral diffusion of protein molecules in the membrane (“feeding”). Bacteriorhodopsin crystals were obtained from bicontinuous cubic phases, but not from micellar systems, implying a critical role of the continuity of the diffusion space (the bilayer) on crystal growth. Hexagonal bacteriorhodopsin crystals diffracted to 3.7 Å resolution, with a space group P63, and unit cell dimensions of a = b = 62 Å, c = 108 Å; α = β = 90° and γ = 120°.
Keywords: bacteriorhodopsin structure, bicontinuous phases, lipidic matrices, x-ray crystallography
Membrane proteins, residing in highly insulating lipid bilayers, catalyze vital reactions such as solute transport, charge separation and conversion of energy, as well as signal transduction. Understanding such fundamental vectorial processes at a molecular level requires knowledge of the structures of these hydrophobic proteins at high resolution, a challenge that has met with limited success thus far. Three general methods are currently in use: electron microscopy, NMR, and x-ray crystallography. Due to the regular arrays of the proton-pumping bacteriorhodopsin (BR) in two-dimensional crystals (purple patches), electron microscopy and image reconstruction have yielded the first insight into the structural organization of a transmembrane protein at a resolution of 7 Å (1). Since then, electron crystallography of two-dimensional crystals has developed significantly (2–5), but attaining resolutions beyond 3 Å by this method remains a goal for the future (6). NMR has been applied successfully to BR (7), yet most membrane proteins exist in complexes exceeding in size the current limitations of this method. As to x-ray crystallography, a mere handful of structures of membrane proteins, belonging to only four families (8–16), have been solved to 1.8–2.7 Å, and all but one originates from bacterial sources (the exception being a mitochondrial protein; ref. 15). These proteins exhibit unusual stability (17, 18) and often have a propensity to form highly ordered two-dimensional lattices spontaneously in vivo or in vitro (19–23).
The hydrophobic surfaces and anisotropic orientation of membrane proteins are serious obstacles in producing well-ordered three-dimensional crystals suitable for x-ray analysis. The most critical element, which is unique to the crystallization of membrane proteins, is the necessity to remove them from their native lipid bilayers by detergent solubilization, thus exposing them to solution conditions that have properties drastically different from those existing in native membranes: indeed, most membrane proteins are vulnerable once they are solubilized, and even slight perturbations of their structures may lead to denaturation, aggregation, and sometimes degradation by proteases. Since upon reconstitution, they often regain stability (24), we argue that it is the release of lateral pressure of the membrane (25) that causes lability upon solubilization. We therefore sought to devise an approach that would keep these proteins in a quasisolid membrane environment throughout crystallization, setting the criteria for such a novel system as follows. (i) Its viscoelastic properties should be comparable to those existing in biological membranes. (ii) It should be capable of incorporating large amounts of proteins, detergents and precipitants without perturbation of the matrix. (iii) Membrane proteins incorporated into the system should retain their activity and structural integrity, and these properties should be amenable to noninvasive spectroscopic tests. (iv) The system should provide a structured yet flexible matrix facilitating crystal nucleation and growth.
Lipid polymorphism gives rise to a complex phase behavior (26). Among the multitude of phases, the two highly viscous cubic phases, one micellar and one bicontinuous, appear most promising to fulfill the requirements set out above. Both are macroscopically stable, solid-like, and fully transparent materials, facilitating diffusion of both polar and lipidic components (27, 28). In the former array, the lipids are packed in micelles arranged in a cubic lattice, whereas in the bicontinuous cubic phase, a curved bilayer, extending in three dimensions, forms the diffusion space (Fig. 1). In both types, the lipidic compartments are interpenetrated by a freely communicating system of aqueous channels. Soluble and membrane proteins, incorporated into these matrices, have been shown to retain enzymatic activity and native conformation (29–31), and protein insertion did not impair the rigidity and transparency of the host materials. Neither did high concentrations of salts, detergents and precipitating agents affect the stability of such cubic phases (E.M.L., G. Rummel, S. W. Cowan-Jacob, and J.P.R., unpublished data). In the present report, we describe the batch-crystallization of the membrane protein BR from two homologous bicontinuous cubic phases, with the pigment of BR providing a convenient intrinsic spectral probe. Preliminary x-ray diffraction data of two crystal habits are presented.
Figure 1.
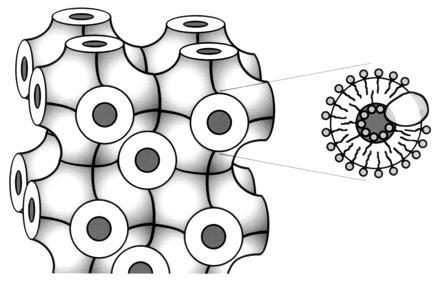
Schematic model of a bicontinuous cubic phase composed of monoolein, water, and a membrane protein. The matrix consists of two compartments, a membrane system with an infinite three-dimensional periodic minimal surface (Left), interpenetrated by a system of continuous aqueous channels (shown in black). The enlarged section (Right) shows the curved lipid bilayer (with an inserted membrane protein molecule) enveloping a water conduit. In a cubic phase consisting of 60–70% (wt/wt) monoolein or monopalmitolein and water, hydrophobic proteins diffuse laterally in the bilayer, while water-soluble components diffuse freely through the intercommunicating aqueous channel system (see text). Adapted from plate 9, ref. 27, with kind permission of Elsevier Science–NL, Sara Burgerhartstraat 25, 1055 KV Amsterdam, The Netherlands.
MATERIALS AND METHODS
Lipidic Components.
Bicontinuous cubic phases consisted of monoolein (1-monooleoyl-rac-glycerol, C18:1c9, or MO, Sigma) or monopalmitolein (1-monopalmitoleyl-rac-glycerol, C16:1c9, or MP, Sigma) and water (buffers). Micellar type cubic phases contained palmitoyl-lysophosphatidylcholine (Avanti Polar Lipids) and water (buffer). Intrinsic bacterial lipids were not assayed.
BR.
The protein was purified, dissociated, and delipidated essentially as described (32). It was isolated from purple patches of Halobacterium halobium (strain S9), kindly provided by G. Büldt (Strukturforschungszentrum, Jülich, Germany). Purple membranes (20 mg) were suspended in water and adjusted to final concentrations of 1.2% (wt/vol) β-octylglycopyranoside (OG) and 0.025 M Na/K-Pi, pH 6.9. The detergent-to-protein ratio used for solubilization was 20:1. Following sonication (1 min, 22 kHz), the clear solution was incubated for 24 hr in the dark. The pH was then adjusted with 0.1 N HCl to 5.5, the solution centrifuged (45 min at 200,000 × g), and the supernatant collected. BR concentrations were determined spectrophotometrically at 550 nm (ɛ = 5.8 × 104 M−1·cm−1). Concentration to ≈2 ml in an Amicon cell (PM 10) was followed by gel filtration chromatography (BioGel A-0.5 m; diameter 1 cm, column volume 90 ml, flow rate 10 ml/hr) in 1.2% OG and 0.025 M Na/K-Pi at pH 5.5 (column buffer). Bleached BR, eluting with the excluded volume, was discarded. The fractions in the second peak contained native BR and were pooled. All operations were carried out under red light at minimal intensity. Storage was at −70°C in the dark.
Preparation of Cubic Phases and Crystallization Experiments.
Cubic phases were prepared by mixing lipids with aqueous buffer containing freshly thawed monomeric BR, salts, detergents, and precipitants (methylpentanediol), to yield a final volume of 10–20 μl. This was followed by centrifugation (10,000 × g) in an Eppendorf desk top centrifuge for 150 min. Typically, bicontinuous cubic phase preparations yielding crystals contained 60–70% (wt/wt) lipids, either MO or MP. The components used were in the following ranges: 2.5–4.5 mg BR/ml; 0.7–4.0 M Na/K-Pi; 1.5–3.75% methylpentanediol; 0.36–0.48% OG; final pH 5.6. Components were added in the following sequence: (i) MO, (ii) Na/K-Pi (weighed as dry powders) to yield 3.3 M and pH 5.6, (iii) BR (stock solution 18 mg/ml in column buffer with 1.5% OG) to give a final protein concentration of 3.5 mg/ml, and (iv) buffer containing methylpentanediol (0.05%) and OG (1.2%). MP was used analogously: MP, BR (3.3 mg/ml), Na/K-Pi (1 M; pH 5.6), and methylpentanediol (2.5%). The preparations were thermostated at 20°C in the dark throughout crystallization.
X-Ray Diffraction Experiments.
Crystals were mounted with the surrounding host cubic phase in thin glass capillaries (typically 0.3 mm inner diameter) that were sealed in larger capillaries (0.7 mm). Diffraction experiments were performed in collaboration with E. Pebay-Peyroula at the European Synchrotron Radiation Facility (Grenoble, France) on the D2AM beamline. The wave length used was λ = 1.012 Å; beam collimation was 100 μm, and the crystal-to-detector distance was 400 mm.
RESULTS
BR-containing bicontinuous cubic phases, formed with either monoolein or monopalmitolein, yielded preparations of initially uniformly purple color, indicating a homogeneous protein distribution. After several days, plates of hexagonal morphology appeared inside the MO matrix (Fig. 2a), while the MP system yielded rhombic crystals (Fig. 2b). In both cases, the intensely purple colored crystals reached their mature sizes (≤0.1 × 0.1 × 0.03 mm) within 14 days. Using light microscopy, the morphology and size of individual immobilized crystals could be easily monitored. Growth was uniform, with ratios between the long and short axes remaining constant throughout. The transparency and viscoelasticity, characteristic of cubic phases, were not impaired by the growing crystals, implying a remarkable plasticity of the matrix. Fig. 3 shows the spectra of the initial and the final stages of this process. During growth, the absorption intensity of BR at 550 nm decreased gradually in the host phase, eventually approaching baseline, while a concomitant gradual increase of the absorbance of the crystals was observed. The absence of a peak at λ = 380 nm, corresponding to bleached BR, demonstrated that the color change was due to protein incorporation into growing crystals rather than to bleaching of the protein in the “mother” cubic phase. X-ray crystallographic analysis of the hexagonal BR crystals, grown in the MO phase at room temperature, revealed diffraction to 3.7 Å resolution in all dimensions (Fig. 4a). The space group is P63, with unit cell dimensions of a = b = 62 Å, c = 108 Å; α = β = 90°, γ = 120°, and an Rsym-factor of 15–20% at ≈4Å, as determined from a 20° scan. Rhombic BR crystals, grown in the MP cubic phase, were less ordered, with a resolution limit currently at ≈9 Å (Fig. 4b). Extensive crystallization attempts in micellar type cubic phases, composed of palmitoyl-lysophosphatidylcholine, failed to yield crystals under analogous conditions.
Figure 2.
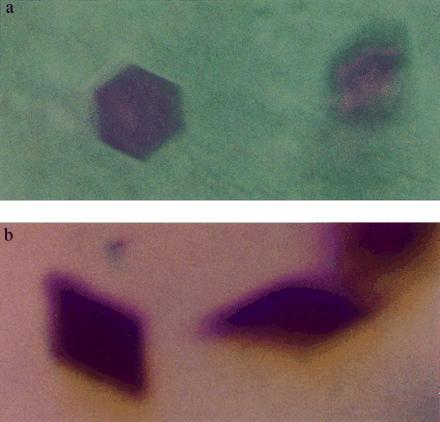
Morphology of BR crystals. (a) Hexagonal crystals grown in a monoolein cubic phase. (b) Rhombic crystals grown in a monopalmitolein cubic phase. In both photographs, crystals can be seen that, due to the depth in which they are embedded in the lipidic materials, are out of focus.
Figure 3.
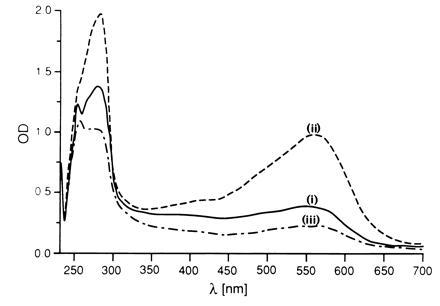
Optical spectra of BR in a cubic phase. (i) Initial absorption spectrum of the uniformly colored purple cubic phase (60% MO). (ii) Spectrum obtained by focusing the beam on a BR crystal at the final stages of crystal growth in the cubic phase after 14 days. (iii) Spectrum focused on the cubic phase surrounding the crystals. Spectra were recorded in both the visible and ultraviolet range using a Zeiss microspectrophotometer, kindly made available by J. N. Jansonius (Basel, Switzerland).
Figure 4.
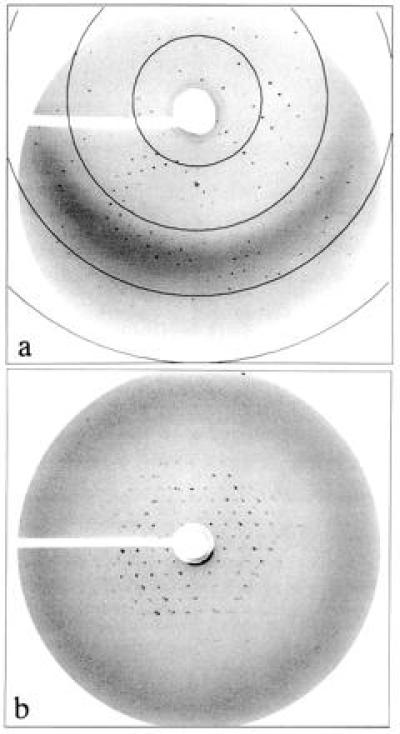
X-ray diffraction patterns of BR crystals. (a) Diffraction of a hexagonal crystal grown in MO cubic phase. The circles drawn indicate resolutions of 3.1, 4.1, 6.2, and 12.4 Å, respectively, at 2ω = 6.1°. The diffraction limit of the crystal is at a resolution of 3.7 Å. (b) Diffraction of a rhombic crystal grown in MP cubic phase. The diffraction limit of this crystal is at 9 Å. For details, see Materials and Methods.
DISCUSSION
Our basic tenet of the crystallization of membrane proteins in quasisolid lipidic cubic phases implies that such proteins partition into the hydrophobic bilayer of a bicontinuous system, which also serves as a sink for detergent monomers. Nucleation, which may be favored by the extremely large area formed by the interface between lipidic and aqueous compartments, is followed by growth to mature crystals, with the supply of proteins afforded by lateral diffusion in the bilayer. The fact that BR reproducibly yields well-ordered crystals, as is described here, demonstrates the validity and efficiency of the approach. Our hypothesis is supported by the observation that the crystal habits formed are critically determined by the packing arrangement of the constituent lipids. Both cubic systems, consisting of the homologous monoglycerides, MO and MP, allow free lateral diffusion of their lipid components (27), but exhibit different radii of curvature (33). BR crystallizing in either of the two bicontinuous phases thus assumed distinctly different crystal habits, hexagonal and rhombic. In contrast to these bilayer matrices, the micellar cubic system, in which lateral diffusion is markedly hindered (27), did not promote BR crystal formation. Unlike membrane proteins, the water-soluble lysozyme, expected to reside in the aqueous channel system, crystallized independently of the cubic phase type and packing of the lipids (E.M.L., G. Rummel, S. W. Cowan-Jacob, and J.P.R., unpublished data). These independent and complementary lines of evidence strongly support the conclusion that the process of nucleation and growth of three-dimensional BR crystals in bicontinuous cubic phases is governed by the composition and structure of the lipidic compartments, and that the bilayers are necessary elements for crystallization to occur.
The hexagonal space group (P63), and the unit cell dimensions (a = b = 62Å; c = 108Å) of BR crystals grown from bicontinuous MO cubic phases (E. Pebay-Peyroula, G. Rummel, J.P.R., and E.M.L., unpublished results) tally with the results obtained earlier by electron crystallography (1, 34, 35) and x-ray diffraction studies (34, 36) using two-dimensional crystals with a space group P3, thus validating our results. The value of the c-axis is likely to correspond to the height of two BR molecules (34). The resolution obtained (3.7 Å in all dimensions) is comparable to that observed in the a and b directions of a three-dimensional, detergent-grown orthorhombic crystal form of BR (37). The resolutions observed with both BR and lysozyme (2.0 Å) (E.M.L., G. Rummel, S. W. Cowan-Jacob, and J.P.R., unpublished data) suggest, moreover, that no fragments of lipid bilayers are incorporated into the crystals, and that the bilayer system, due to its plasticity, recedes during crystal growth.
In conclusion, the results reported here demonstrate the potentials of lipidic cubic phases as novel matrices for obtaining three-dimensional crystals of membrane proteins amenable to x-ray analysis. Detailed investigations of crystallization conditions and an in-depth understanding of the structural and dynamic properties of these complex systems are now called for to allow further explorations of the applicability of the method. There is also a need to investigate the structure of cubic phases as a function of lipid composition and protein content, using freeze-fracture electron microscopy, small angle x-ray diffraction, and the assessment of detailed phase diagrams. We are using fluorescent probes to monitor bilayer fluidity and lateral diffusion of the lipid and protein components (unpublished results). Attempts to induce specific crystal habits as a function of designed alterations of the microenvironment will be carried out with the two crystal forms of porin (OmpF), the structures of which have been determined to high resolution with crystals grown from detergent solutions (10, 38). The crucial test for the applicability of this approach will be the extension from a conceptual to a general method. To this end, very labile membrane proteins, as they exist in cytoplasmic membranes of prokaryotic and eukaryotic cells, are currently being studied. The lactose permease from Escherichia coli (39), which thus far has proven notoriously reluctant to yield crystals (24, 40), may be considered as a paradigm. And while limited exposure to solution conditions during purification is difficult to avoid altogether, modern, rapid purification methods, followed by swift reconstitution into cubic phase bilayers, in which the lateral pressure is comparable to that in native membranes, may allow the crystallization of such labile proteins. In conjunction with the current capacity of acquisition and processing of x-ray data, this approach thus promises high resolution structural studies of membrane proteins to become more rational and more routine.
Note Added in Proof.
Recently, a hexagonal BR crystal diffracted to a resolution of 2.0 Å at the European Synchrotron Radiation Facility in Grenoble (Pebay–Peyroula, E., Rummel, G., J.P.R., and E.M.L., unpublished data).
Acknowledgments
It is a pleasure to thank Dr. E. Pebay-Peyroula (Grenoble, France) for her guidance with the x-ray experiments and her interest and Drs. G. Büldt (Jülich, Germany) and B. Witholt (Zürich, Switzerland) for their generosity. To them and to Drs. T. Bickle (Basel, Switzerland), D. L. Dorset (Buffalo, NY), R. S. Eisenberg (Chicago), and J. Kyte (San Diego), we are indebted for stimulating discussions and critical comments on the manuscript. The highly dedicated and expert help of G. Rummel and A. Hardmeyer is most gratefully acknowledged. We thank Drs. M. Roth and E. Fanchon, European Synchrotron Radiation Facility, for their help at the synchrotron. This project was supported by the Swiss National Foundation, Priority Program in Biotechnology 5002–037911. Beam time at the European Synchrotron Radiation Facility, Grenoble, France, was granted for experiment LS 435.
Footnotes
Abbreviations: BR, bacteriorhodopsin; MO, 1-monooleoyl-rac-glycerol, C18:1c9; MP, 1-monopalmitoleyl-rac-glycerol, C16:1c9; OG, β-octylglycopyranoside.
References
- 1.Henderson R, Unwin P N T. Nature (London) 1975;257:28–32. doi: 10.1038/257028a0. [DOI] [PubMed] [Google Scholar]
- 2.Henderson R, Baldwin J M, Ceska T A, Zemlin F, Beckmann E, Downing K H. J Mol Biol. 1990;213:899–929. doi: 10.1016/S0022-2836(05)80271-2. [DOI] [PubMed] [Google Scholar]
- 3.Kühlbrandt W. Q Rev Biophys. 1992;25:1–49. doi: 10.1017/s0033583500004716. [DOI] [PubMed] [Google Scholar]
- 4.Unwin N. J Mol Biol. 1993;229:1101–1124. doi: 10.1006/jmbi.1993.1107. [DOI] [PubMed] [Google Scholar]
- 5.Karrasch S, Bullough P A, Ghosh R. EMBO J. 1995;14:631–638. doi: 10.1002/j.1460-2075.1995.tb07041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henderson R. Q Rev Biophys. 1995;28:171–193. doi: 10.1017/s003358350000305x. [DOI] [PubMed] [Google Scholar]
- 7.Thompson L K, McDermott A E, Raap J, van der Wielen C M, Lugtenberg J, Herzfeld J, Griffin R G. Biochemistry. 1992;31:7931–7938. doi: 10.1021/bi00149a026. [DOI] [PubMed] [Google Scholar]
- 8.Deisenhofer J, Michel H. Science. 1989;245:1463–1473. doi: 10.1126/science.245.4925.1463. [DOI] [PubMed] [Google Scholar]
- 9.Weiss M S, Abele U, Weckesser J, Welte W, Schiltz E, Schulz G E. Science. 1991;254:1627–1630. doi: 10.1126/science.1721242. [DOI] [PubMed] [Google Scholar]
- 10.Cowan S W, Schirmer T, Rummel G, Steiert M, Ghosh R, Pauptit R A, Jansonius J N, Rosenbusch J P. Nature (London) 1992;358:727–733. doi: 10.1038/358727a0. [DOI] [PubMed] [Google Scholar]
- 11.Kreusch A, Neubuser A, Schiltz E, Weckesser J, Schulz G E. Protein Sci. 1994;3:58–63. doi: 10.1002/pro.5560030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schirmer T, Keller T A, Wang Y-F, Rosenbusch J P. Science. 1995;264:914–916. [Google Scholar]
- 13.McDermott G, Prince S M, Freer A A, Hawthornethwaite-Lawless A M, Papiz M Z, Cogdell R J, Isaacs N W. Nature (London) 1995;374:517–521. doi: 10.1016/s0969-2126(96)00050-0. [DOI] [PubMed] [Google Scholar]
- 14.Iwata S, Ostermeier C, Ludwig B, Michel H. Nature (London) 1995;376:660–668. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 15.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinazawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. Science. 1995;269:1069–1074. doi: 10.1126/science.7652554. [DOI] [PubMed] [Google Scholar]
- 16.Dutzler R, Wang Y-F, Rizkallah P J, Rosenbusch J P, Schirmer T. Structure (London) 1996;4:127–134. doi: 10.1016/s0969-2126(96)00016-0. [DOI] [PubMed] [Google Scholar]
- 17.Rosenbusch J P. J Biol Chem. 1974;249:8019–8029. [PubMed] [Google Scholar]
- 18.Jackson M B, Sturtevant J M. Biochemistry. 1978;17:911–915. doi: 10.1021/bi00598a026. [DOI] [PubMed] [Google Scholar]
- 19.Henderson R, Capaldi R A, Leigh J S. J Mol Biol. 1977;112:631–648. doi: 10.1016/s0022-2836(77)80167-8. [DOI] [PubMed] [Google Scholar]
- 20.Steven A C, ten Heggeler B, Müller R, Kistler J, Rosenbusch J P. J Cell Biol. 1977;72:292–301. doi: 10.1083/jcb.72.2.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller K R. Proc Natl Acad Sci USA. 1979;76:6415–6419. doi: 10.1073/pnas.76.12.6415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engel A, Massalski A, Schindler H, Dorset D L, Rosenbusch J P. Nature (London) 1985;317:643–645. doi: 10.1038/317643a0. [DOI] [PubMed] [Google Scholar]
- 23.Kühlbrandt W, Wang D N, Fujiyoshi Y. Nature (London) 1994;367:614–621. doi: 10.1038/367614a0. [DOI] [PubMed] [Google Scholar]
- 24.Page M G P, Rosenbusch J P, Yamato I. J Biol Chem. 1988;263:15897–15905. [PubMed] [Google Scholar]
- 25.Chapman D, Benga G. In: Biological Membranes. Chapman D, editor. Vol. 5. London: Academic; 1984. pp. 1–56. [Google Scholar]
- 26.Luzzati V, Gulik-Krzywicki T, Tardieu A. Nature (London) 1968;218:1031–1034. doi: 10.1038/2181031a0. [DOI] [PubMed] [Google Scholar]
- 27.Lindblom G, Rilfors L. Biochim Biophys Acta. 1989;988:221–256. [Google Scholar]
- 28.Cribier S, Gulik A, Fellmann P, Vargas R, Deveaux P F, Luzzati V. J Mol Biol. 1993;229:517–525. doi: 10.1006/jmbi.1993.1051. [DOI] [PubMed] [Google Scholar]
- 29.Portmann M, Landau E M, Luisi P L. J Phys Chem. 1991;95:8437–8440. [Google Scholar]
- 30.Landau E M, Luisi P L. J Am Chem Soc. 1993;115:2102–2106. [Google Scholar]
- 31.Hochkoeppler A, Landau E M, Venturoli G, Zannoni D, Feick R, Luisi P L. Biotechnol Bioeng. 1995;46:93–98. doi: 10.1002/bit.260460202. [DOI] [PubMed] [Google Scholar]
- 32.Dencher N, Heyn M. Methods Enzymol. 1982;88:5–10. [Google Scholar]
- 33.Briggs J, Chung H, Caffrey M. J Phys II. 1996;6:723–752. [Google Scholar]
- 34.Henderson R. J Mol Biol. 1975;93:123–138. doi: 10.1016/0022-2836(75)90123-0. [DOI] [PubMed] [Google Scholar]
- 35.Grigorieff T, Ceska T A, Downing K H, Baldwin J M, Henderson R. J Mol Biol. 1996;259:393–421. doi: 10.1006/jmbi.1996.0328. [DOI] [PubMed] [Google Scholar]
- 36.Blaurock A E. J Mol Biol. 1975;93:139–158. doi: 10.1016/0022-2836(75)90124-2. [DOI] [PubMed] [Google Scholar]
- 37.Schertler G F X, Bartunik H D, Michel H, Oesterhelt D. J Mol Biol. 1993;234:156–164. doi: 10.1006/jmbi.1993.1570. [DOI] [PubMed] [Google Scholar]
- 38.Cowan S W, Garavito R M, Jansonius J N, Jenkins J, Karlsson R, Koenig N, Pai E F, Pauptit R A, Rizkallah P J, Rosenbusch J P, Rummel G, Schirmer T. Structure. 1995;3:1041–1050. doi: 10.1016/s0969-2126(01)00240-4. [DOI] [PubMed] [Google Scholar]
- 39.Wu J, Sun J, Kaback H R. Biochemistry. 1996;35:5213–5219. doi: 10.1021/bi960064f. [DOI] [PubMed] [Google Scholar]
- 40.Li J, Tooth P. Biochemistry. 1987;26:4816–4823. doi: 10.1021/bi00389a032. [DOI] [PubMed] [Google Scholar]