

Abstract
The role of the abundant stress protein Hsp90 in protecting cells against stress-induced damage is not well understood. The recent discovery that a class of ansamycin antibiotics bind specifically to Hsp90 allowed us to address this problem from a new angle. We find that mammalian Hsp90, in cooperation with Hsp70, p60, and other factors, mediates the ATP-dependent refolding of heat-denatured proteins, such as firefly luciferase. Failure to refold results in proteolysis. The ansamycins inhibit refolding, both in vivo and in a cell extract, by preventing normal dissociation of Hsp90 from luciferase, causing its enhanced degradation. This mechanism also explains the ansamycin-induced proteolysis of several protooncogenic protein kinases, such as Raf-1, which interact with Hsp90. We propose that Hsp90 is part of a quality control system that facilitates protein refolding or degradation during recovery from stress. This function is used by a limited set of signal transduction molecules for their folding and regulation under nonstress conditions. The ansamycins shift the mode of Hsp90 from refolding to degradation, and this effect is probably amplified for specific Hsp90 substrates.
Exposure of prokaryotic and eukaryotic cells to heat and other stresses induces several classes of highly conserved stress proteins, including the members of the Hsp70, Hsp60, and Hsp90 families (1–3). These proteins are generally thought to act as molecular chaperones in preventing the aggregation of nonnative polypeptides and in aiding their correct folding. Although significant progress has been made in understanding the chaperone mechanisms in de novo protein folding, surprisingly little is known about the role of chaperones under stress conditions. This lack of knowledge is particularly apparent for the Hsp90s, the most abundant constitutively expressed stress proteins in the eukaryotic cytosol. Although Hsp90 can prevent protein aggregation in vitro (4–6) and is required for the survival of yeast at elevated temperature (7), its actual role in protein refolding and repair under stress has remained elusive (2, 8). Instead, current thinking views Hsp90 as part of a specific chaperone system for the conformational maturation and regulation of signal transduction molecules, such as several potentially oncogenic protein kinases and the nuclear receptors of steroid hormones (8–11). In the mammalian cytosol, these proteins are found in heterocomplexes containing Hsp90, Hsp70/Hsc70, the Hsp70 regulator Hip (p48), p60, various immunophilins, and the small acidic protein p23.
A recent study proposed that the benzoquinone ansamycins geldanamycin (GA) and herbimycin A (HA), originally classified as tyrosine kinase inhibitors (12), do not execute their biological effects directly by inhibiting kinase activities, but rather indirectly by acting on Hsp90 (13). Thus, these agents provide a powerful tool to explore the physiological role of Hsp90. We show that Hsp90, in cooperation with Hsc70, p60, and other factors, mediates the refolding of thermally denatured proteins, such as firefly luciferase, in vivo and in vitro. Unfolded luciferase bound by this multichaperone complex is either released in an ATP-dependent manner for refolding or is presented for degradation. The ansamycins, by binding to Hsp90, shift this chaperone system from protein refolding to degradation mode. Our results suggest that Hsp90 and its cofactors function as a quality control system in the refolding or degradation of thermally labile proteins.
MATERIALS AND METHODS
Experiments with Cells in Culture.
SW620 colon carcinoma cells and MCF-7 human breast carcinoma cells were maintained at 37°C (14). SW620 cells constitutively expressing firefly luciferase under the control of the lck proximal promoter (SW620-Luc) were kindly provided by R. Muise-Helmericks (Memorial Sloan–Kettering Cancer Center). Cells were incubated with either 1.7 μM HA (GIBCO) in 0.1% dimethyl sulfoxide (DMSO) or with 0.1% DMSO alone in the presence or absence of 40 μg/ml cycloheximide. SW620-Luc cells were radiolabeled overnight at 37°C with [35S]methionine/cysteine ProMix (50 μCi/ml, 1000 Ci/mmol; Amersham). Before the experiment, cells were transferred for 1 h into [35S]methionine/cysteine-free media containing 150 μg/ml each of methionine and cysteine. Cell monolayers were washed twice in PBS, solubilized at 4°C in lysis buffer containing 1% Nonidet P-40 (14). Luciferase activity was assayed (15) in aliquots of cell extracts, and luciferase was immunoprecipitated after a clarifying spin (10 min, 14,000 × g) using a polyclonal antiserum (Promega) and protein A-Sepharose (Pharmacia). Pellets were solubilized in 2% SDS buffer (10 min at 95°C) diluted 20-fold with lysis buffer and were also immunoprecipitated with luciferase antibody. Immunoprecipitates were subjected to SDS/PAGE phosphorimager analysis. Raf-1 was immunoprecipitated from MCF7 lysates (2 mg of protein) by 2 μg of anti-Raf-1 rabbit polyclonal antibody (Santa Cruz Biotechnology, C-12) for 90 min at 4°C. Immunocomplexes were analyzed by immunoblotting with anti-Raf-1 and anti-Hsp90 using purified bovine Hsp90 and human Raf-1 as standards (kindly provided by J. Young and U. Hämmerling, Memorial Sloan–Kettering Cancer Center).
Luciferase Refolding in Reticulocyte Lysate (RL).
Luciferase-myc-His was generated from the firefly luciferase gene and purified upon overexpression in Escherichia coli. Luciferase-myc-His (23 μM) was denatured in buffer A (6 M guanidinium-HCl/50 mM potassium acetate/5 mM DTT) for 20 min at 25°C. RL (Green Hectares, Oregon, WI) was desalted on a 9.1-ml G25-Sephadex column (Pharmacia) into buffer B (20 mM Hepes·KOH, pH 7.4/100 mM potassium acetate/5% glycerol) and incubated for 30 min at 25°C either with 0.2% DMSO (control), 36 μM HA, or 36 μM GA (GIBCO) (added from a 17-mM solution in DMSO), or with the ansamycin concentrations indicated (0.2% DMSO had no effect on refolding). Unfolded luciferase was diluted to 0.23 μM into desalted RL containing 1 mM ATP and 5 mM magnesium acetate when indicated, and reactivation was followed at 25°C. To measure the renaturation of thermally denatured luciferase, native luciferase-myc-His (0.23 μM) was added to RL, incubated for 5 min at 42°C, and refolding followed at 25°C. Ansamycin effects are reduced in the presence of DTT, which was therefore partially removed from RL by desalting (see above). This did not affect the refolding capacity of RL in the absence of drug.
Luciferase Translation.
Nuclease-treated RL (Promega) was desalted into buffer C (25 mM potassium acetate/10 mM NaCl/1 mM magnesium acetate/0.1 mM EGTA/10 mM Hepes·KOH, pH 7.4), concentrated, and resubstituted with 0.4 mM spermidine, 20 μM hemin, 1 ng/10 μl tRNA, 120 mM potassium acetate, 0.6 mM magnesium acetate, 0.5 mM ATP, and 0.1 mM GTP. Lysates were then incubated for 20 min at 25°C either with 0.2% DMSO or with increasing concentrations of GA added in DMSO (see above). Translation was for 60 min at 30°C (16) and analyzed by enzyme assay and SDS/PAGE/fluorography.
Isolation of Chaperone Complexes.
Luciferase:chaperone complexes were generated either by dilution of denatured luciferase-myc-His into RL (see above) followed by incubation for 2 min at 30°C and treatment with apyrase (20 units/ml apyrase; Sigma, grade VIII) for 10 min at 30°C, or by incubation of native luciferase in RL for 5 min at 42°C, followed by apyrase treatment at 30°C. Reactions were centrifuged for 5 min at 16,000 × g and cooled on ice. Supernatants were diluted 2-fold with buffer B, and c-myc antibodies crosslinked to protein G-Sepharose were added (≈80% of the total luciferase was precipitated). After 1 h at 4°C, beads were washed with 1.8 ml of buffer B/150 mM potassium acetate, followed by 1.8 ml of buffer B. Beads were incubated with 1 mM ATP/5 mM Mg2+ in buffer B for 5 min at 25°C, and supernatants were prepared by a short spin. Beads were then resuspended in 2% SDS buffer and incubated for 40 min at 25°C. ATP eluates were precipitated with trichloroacetic acid and analyzed by SDS/PAGE, together with the SDS eluates. To release luciferase:chaperone complexes from beads, washed beads were incubated with 0.05 mg/ml factor Xa protease (New England Biolabs) and 2 mM CaCl2 for 10 min at 25°C.
Luciferase Degradation.
When indicated, ATP was removed from hemin-free RL by incubation with 12 mM glucose, 5 mM MgCl2, and 50 units/ml hexokinase (Boehringer Mannheim) for 6 min at 25°C. Control RL received glucose and MgCl2. Unfolded luciferase-myc-His (23 nM, final concentration) was diluted as described above into 250-μl reactions containing 100 μl of lysate, 5 mM MgCl2, 40 mM Tris·HCl (pH 7.6), 10 mM creatine phosphate, and 200 μg/ml creatine kinase (Boehringer Mannheim) at 4°C. After centrifugation for 10 min at 16,000 × g, equal aliquots were removed from the supernatants. When indicated, reactions received 0.5 mM ATP, and degradation was initiated at 37°C. Degradation was stopped by adding an equal volume of 2% SDS buffer and heating for 5 min at 95°C. Reactions were analyzed by SDS/PAGE and immunoblotting with anti-luciferase antibody. When indicated, 50 μM hemin, 50 μM methylated ubiquitin (17), or 50 μM ubiquitin was added to the reaction prior to luciferase.
RESULTS AND DISCUSSION
When mammalian cells expressing firefly luciferase in the cytosol are heat stressed, preexistent luciferase denatures but refolds upon temperature downshift in an ATP-dependent manner (18). To test whether Hsp90 is involved in this process, we analyzed the effects of ansamycins on luciferase renaturation. Human colon carcinoma cells expressing luciferase were incubated at 37°C with cycloheximide to inhibit de novo protein synthesis and then shifted for 10 min to 42°C to deactivate luciferase (Fig. 1A). Upon downshift to 37°C, luciferase renatured, reaching 70% of control activity within 60 min. It is striking that this renaturation was inhibited by HA (Fig. 1A). HA had no effect on the activity of luciferase at 37°C. It is interesting that in the presence of HA, most of the luciferase protein was rapidly degraded during and after heat stress, as shown by immunoprecipitation of luciferase from 35S-labeled cells (Fig. 1B). The bulk pattern of 35S-labeled polypeptides was not significantly changed (not shown), suggesting that under moderate heat stress the majority of cellular proteins remained stable. Luciferase protein in control cells decreased by ≈20%, equivalent to the fraction that did not renature. Incubation of lysates from radiolabeled cells with ansamycin attached to agarose beads served to establish the high specificity of these drugs for Hsp90. As reported earlier for the RL (13), Hsp90 was the only detectable protein that bound to immobilized GA stably and with high affinity (Fig. 2). Thus, our observations suggest that Hsp90 plays a critical role in the refolding or proteolytic disposal of denatured proteins under heat stress.
Figure 1.
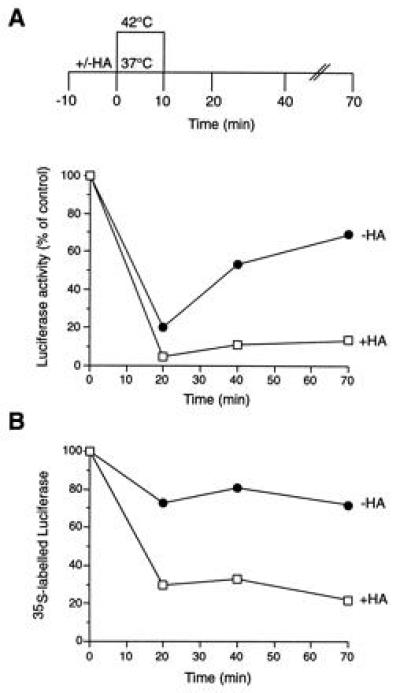
Effect of heat shock on firefly luciferase expressed in vivo in the presence of HA. Luciferase activities (A) and levels of 35S-labeled luciferase protein (B) in SW620 colon carcinoma cells subjected to heat shock at 42°C and recovery at 37°C in the presence (□) and absence (•) of HA. Ten minutes before heat shock, cultures received 40 μg/ml cycloheximide. Values measured in DMSO-treated control cells maintained at 37°C throughout are set to 100%. Note that, although normally localized in peroxisomes, more than 90% of luciferase expressed in SW620 cells was localized in the soluble supernatant fraction of a high-speed centrifugation (18), as uptake into peroxisomes is inefficient. Luciferase did not form insoluble aggregates during incubation at 42°C.
Figure 2.
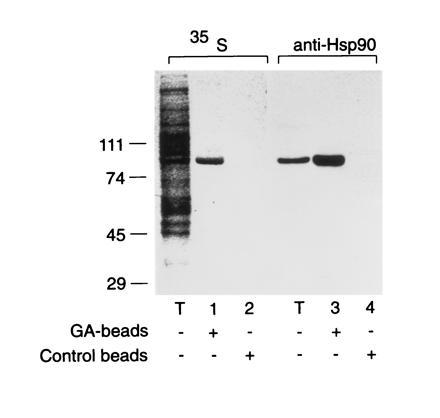
Specific binding of Hsp90 in cell extracts to GA affinity beads. Extracts of 35S-labeled SW620 cells (see Fig. 1) were incubated with GA coupled to Affi-Gel 10 (13) or Affi-Gel 10 alone, as indicated, followed by 10 washes in lysis buffer (14) and by elution with SDS buffer. Eluates were analyzed by SDS/PAGE and autoradiography (35S) or immunoblotting with anti-Hsp90 antibody (anti-Hsp90). Total protein (25 μg) was analyzed in lane T. In lanes 1–4, 875 μg of total protein was incubated with 10-μl beads for 60 min at 25°C.
Luciferase refolding was analyzed further in rabbit RLs (15), following the suggestion that refolding in this system involves Hsp90 (19). A luciferase fusion protein, luciferase-myc-His (Fig. 3A), was added to the lysate, either in native form followed by heat treatment or by dilution from denaturant. Efficient ATP-dependent refolding was observed under both conditions (Fig. 3B); refolding was faster upon thermal denaturation (shift from 42°C to 25°C). Micromolar concentrations of HA and GA inhibited renaturation by 50–70%, whereas the remaining refolding capacity was ansamycin-insensitive (Fig. 3 B and C). Order-of-addition experiments demonstrated that the drugs did not act directly on luciferase (see legend to Fig. 3). This conclusion was supported by the finding that, in contrast to denatured protein, folding of newly translated luciferase in RL was completely unimpaired by ansamycin (shown for GA in Fig. 3C). De novo folding of luciferase in RL involves Hsp70 and the chaperonin TRiC (16). These chaperones may also be responsible for the residual, ansamycin-insensitive refolding.
Figure 3.
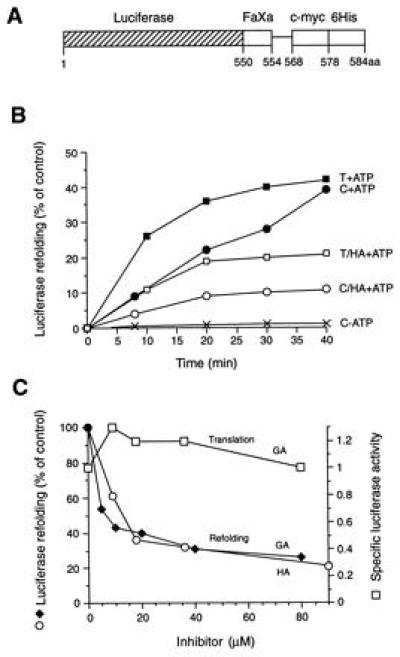
Refolding of firefly luciferase in RL. (A) Schematic representation of the luciferase-myc-His fusion construct used as a substrate (FaXa, factor Xa cleavage site; c-myc, c-myc epitope; 6His, 6Histidine-tag). The factor Xa cleavage site is separated from the c-myc epitope by a 14-amino acid spacer. (B) Refolding of thermally denatured (T) and chemically denatured (C) luciferase in control lysate (RL) in the presence and absence of ATP and in HA-treated lysate as indicated. The activity of an equivalent amount of native luciferase is set to 100%. (C) Effect of increasing concentrations of HA and GA on the refolding of chemically denatured luciferase (○, ♦) and on the de novo folding of newly translated luciferase (□). Enzyme activities were measured after 40 min of refolding or after 60 min of translation. Specific luciferase activities were calculated as the ratio of enzyme activity:full-length luciferase with the specific activity in untreated RL set to 1.0. Note that refolding was not inhibited when purified luciferase was incubated with HA or GA before or during chemical denaturation, and free drug was removed by gel filtration before refolding; inhibition persisted, however, when drug-treated RL was gel-filtered before the addition of unfolded luciferase (not shown). In contrast to Hsp90, luciferase itself did not bind to GA-agarose beads.
Proteins that interact with luciferase during refolding were coimmunoprecipitated. Denatured luciferase was diluted into RL, and refolding was interrupted by ATP depletion with apyrase (Fig. 4Ai). Alternatively, native protein was incubated in lysate at 42°C, followed by apyrase treatment (Fig. 4B). Luciferase and associated proteins were precipitated with anti c-myc antibody linked to protein G-beads and successively eluted with Mg2+-ATP and SDS. Three prominent polypeptides of approximately 90, 70, and 60 kDa were specifically associated with unfolded luciferase and eluted with ATP (Fig. 4 A and B). Immunoblotting identified the 90- and 70-kDa bands as Hsp90 and Hsc70/Hsp70, respectively (Fig. 4Aii), and microsequencing identified the 60-kDa band as p60 (see legend to Fig. 4A), a close homologue of the yeast stress protein Sti1 (20) that is thought to mediate an interaction between Hsc70 and Hsp90 (21). The isolated luciferase:chaperone complex fractionated at ≈500 kDa upon gel filtration (not shown), as determined after releasing luciferase-myc-His plus associated components from the beads by cleavage with factor Xa (Fig. 3A). Although the exact stoichiometry of the chaperone components relative to luciferase remains to be established, immunoprecipitation with anti-Hsp70 demonstrated that more than 50% of luciferase was associated simultaneously with Hsp70, Hsp90, and p60 (not shown). Notably, efficient formation of the multichaperone complex with luciferase was only observed when ATP was present (Fig. 4B). Thus, both formation and dissociation of the complex is ATP-dependent.
Figure 4.
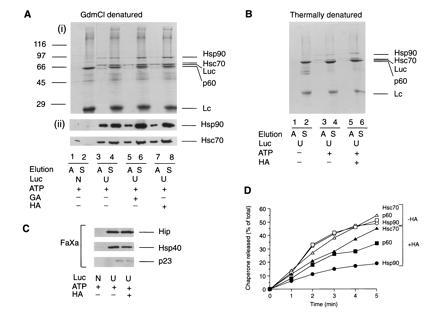
Isolation and characterization of chaperone-bound luciferase from RL. Immunoisolation of chaperone complexes containing unfolded luciferase-myc-His (U) from control lysate and from lysate treated with HA or GA using chemically denatured (A) and thermally denatured (B) luciferase. In A, lanes 1 and 2, native luciferase (N) was added. In B, lanes 1 and 2, thermal denaturation of luciferase was carried out in ATP-depleted lysate. Complexes bound to protein G-Sepharose were eluted with ATP (A) or SDS (S). Eluted fractions were analyzed by SDS/PAGE followed by Coomassie blue staining (Ai and B) or immunoblotting with anti-Hsp90 and anti-Hsc70 (Aii). (C) Luciferase complexes were released from Sepharose beads with factor Xa and eluates immunoblotted with antibodies against Hip, Hsp40, and p23. (D) Time course of ATP-dependent elution of chaperones from complexes with luciferase isolated as in A from control lysate (−HA) and HA-treated lysate (+HA). Sepharose beads were incubated for 1 min at 25°C in 200 μl of buffer B with 1 mM ATP/5 mM Mg2+, and the supernatant was removed. This procedure was repeated five times, followed by a final elution with SDS. ATP eluates were analyzed as in A. Proteins were quantified by densitometry and plotted as the amount of total chaperone protein released up to a given time.
Three additional proteins, Hip (p48), Hsp40, and p23, were present in the luciferase:Hsp90 heterocomplex in substoichiometric amounts (Fig. 4C). These proteins tended to adsorb nonspecifically to protein G-beads, but their specific association with unfolded luciferase was revealed after release of the complex by factor Xa cleavage. Hip is a cochaperone of Hsp70/Hsc70 (22, 23) and is present in the complexes of immature progesterone receptor with Hsp90, Hsc70, p60, and p23 (23). Hsp40, a mammalian DnaJ homolog, is required for complex formation between Hsc70 and Hip (22). A DnaJ homologue is also required for Hsp90 function in yeast (24).
Similar amounts of chaperones bound to unfolded luciferase in ansamycin-treated lysates (Fig. 4 A–C). However, only a fraction of Hsp90 dissociated from the complex with ATP, thus explaining the inhibition of luciferase refolding by GA and HA. In the absence of ansamycin, Hsp90, Hsc70, and p60 dissociated from luciferase with indistinguishable kinetics (Fig. 4D) at a rate severalfold faster than refolding. This suggested that folding involves multiple cycles of chaperone release and rebinding. In contrast, Hsp90 dissociation was markedly slowed from the complex formed in the presence of HA, only 15% of the bound Hsp90 being released from luciferase within 5 min (Fig. 4D). No more than 20–30% of the total Hsp90 was released within 30 min (not shown). The dissociation rate of Hsc70 and p60 was reduced to a lesser extent than that of Hsp90. We conclude that the ansamycins prevent the coordinated dissociation of the luciferase:chaperone complex by inhibiting the release of Hsp90 from unfolded protein. Normal dissociation of Hsp90 from luciferase probably depends on the ATPase activity of Hsp70 and may be mediated by p60.
Cycling of substrate protein on and off the Hsp90 complex, when refolding is inefficient, may target proteins for degradation. The ansamycins would induce degradation by trapping the unfolded protein in a chaperone-bound state. To test this hypothesis, we analyzed the fate of unfolded luciferase in RL at 37°C, which reduced the efficiency of refolding compared with incubation at 25°C (not shown). Accelerated degradation of luciferase by the ansamycins was indeed observed (shown for HA in Fig. 5A). Proteolysis occurred without the accumulation of degradation products, suggesting the involvement of the highly processive ubiquitin/proteasome system (25). This was supported by the following findings: (i) Degradation was ATP-dependent (Fig. 5 A and B) and was inhibited by hemin, an inhibitor of the proteasome pathway (26), as well as by methylated ubiquitin, a competitive inhibitor of protein polyubiquitination (27) (Fig. 5B). (ii) Inhibition by methylated ubiquitin was partially reversed by excess unmodified ubiquitin. (iii) Polyubiquitinated luciferase could indeed be detected by probing luciferase immunoprecipitates with antiubiquitin antibody (not shown). However, some luciferase may be degraded in a ubiquitin-independent pathway.
Figure 5.
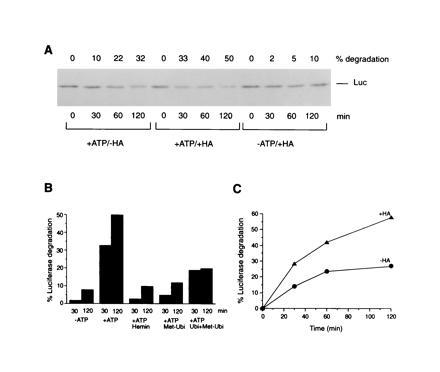
Protein degradation from HA-trapped Hsp90 complexes. (A) Time course of luciferase degradation in control RL (−HA) and in HA-treated (+HA) RL with and without ATP. Luciferase immunoblots are shown. Amounts of luciferase protein degraded were determined by densitometry. (B) Inhibition of luciferase degradation by hemin and methylated ubiquitin (Met-Ubi) and partial reversal of inhibition by ubiquitin (Ubi) in HA-treated RL. (C) Degradation of luciferase immunoisolated as a chaperone complex from control (−HA) and HA-treated (+HA) RL upon transfer into untreated, degradation-competent RL. Time-dependent degradation is shown in percent of the total luciferase protein in the reaction.
We established further that increasing the time a protein spends in association with the Hsp90 chaperone complex is sufficient to cause its degradation: Luciferase:chaperone complex was isolated from HA- treated or control RLs, released from the protein G-beads by factor Xa cleavage, and transferred to untreated, degradation-competent lysate (Fig. 5C). Degradation of luciferase occurred under both conditions, but was significantly accelerated with the HA-modified complex. Apparently, the HA-modified Hsp90 that was bound to luciferase did not readily exchange with the large excess of unmodified Hsp90 in the added lysate, consistent with the observation that the inhibition of luciferase refolding persisted (not shown). This data, therefore, suggested that the substrate protein can be presented to the degradation machinery directly from its chaperone-bound state.
The ansamycins reverse certain transformed-growth phenotypes of cells (28). Because several protooncogenic protein kinases are destabilized upon ansamycin treatment (13, 14, 29), these proteins may also be trapped in an Hsp90-bound state when cycling on and off their respective Hsp90 heterocomplexes. We tested this hypothesis for the serine/threonine kinase Raf-1, which associates in the cytosol with Hsp90 and other factors in a manner similar to luciferase and progesterone receptor (30, 31). Raf-1 and associated components were immunoprecipitated from human breast cancer cells at different times during incubation with or without HA at 37°C (Fig. 6 A and B). Before HA treatment, only ≈10% of the precipitated Raf-1 was associated with Hsp90. [Although “stable” Hsp90 complexes are assumed in the case of hormone receptors, it has been shown that these complexes are highly dynamic (21, 23).] In any case, between 6 and 9 h the amount of Hsp90-bound Raf-1 increased markedly, reaching essentially 100% at 12 h (Fig. 6A and B). It is striking that binding to Hsp90 coincided with the disappearance of nearly all cellular Raf-1 (Fig. 6A and legend). Similar observations were made for two transmembrane tyrosine kinase receptors, the proteasome-dependent degradation of which is also induced by HA (14) (data not shown). Unlike luciferase (Fig. 1), however, degradation of Raf-1 and the receptor kinases set in several hours after the beginning of HA treatment. Under steady-state conditions, most preexistent Raf-1 apparently is not bound to Hsp90 (Fig. 6A) and may interact with Hsp90 at a slow rate determined by its conformational stability. In the presence of HA, normal dissociation of the Raf-1:Hsp90 chaperone complex is inhibited, resulting in the degradation of Raf-1.
Figure 6.
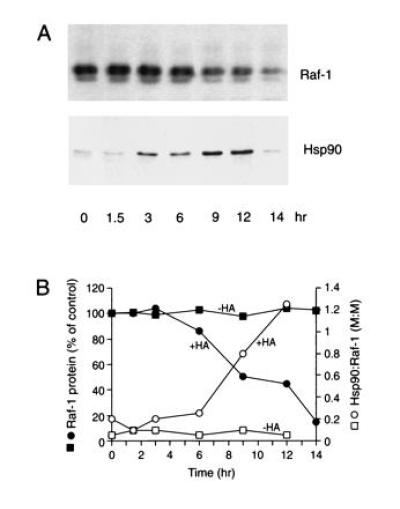
HA-induced degradation of Raf-1 in human breast carcinoma cells. (A) Raf-1 immunoprecipitates from MCF-7 cells, cultured at 37°C in the presence of HA, were analyzed by SDS/PAGE and immunoblotting with anti-Raf-1 and anti-Hsp90 antibodies. Raf-1 (74 kDa) migrates as a double-band due to phosphorylation. Direct immunoblotting of cells confirmed the degradation of essentially all Raf-1. (B) Time-dependent formation of Hsp90:Raf-1 complex and Raf-1 degradation. Depending on the efficiency of Hsp90 coimmunoprecipitation with anti-Raf-1 antibody (probably less than 100%), the results are consistent with a 1:1 or 1:2 stoichiometry of Raf-1:Hsp90 in the complex. Note that HA caused an approximately 2-fold increase in total Hsp90 (32) (not shown).
Our results suggest that Hsp90, in cooperation with Hsp70, p60, and other factors, functions as a quality control system in the refolding or degradation of thermally labile proteins. The components of this protein repair/degradation pathway have been described in the regulation of signal transduction molecules (8, 10, 11). These specific Hsp90 substrates may have an intrinsic structural instability under normal cellular conditions, and this may render them especially sensitive to ansamycin effects on Hsp90. Future studies will have to define how the Hsp90 complexes acting in refolding and in signal transduction are related.
While the extent of protein degradation mediated by the Hsp90 multichaperone machine normally may depend on the relative rates of refolding and rebinding to chaperones, Hsp90 substrates become inevitably directed toward degradation by the ansamycins. These drugs represent the first specific chaperone inhibitors. Binding of HA or GA shifts Hsp90 from refolding to degradation mode by preventing release of Hsp90 (and indirectly of other chaperones) from the polypeptide substrate. This mechanism of interference with the dynamic nature of Hsp90-substrate interactions (21, 23) provides a plausible explanation for the drug-induced degradation of several protooncogenic protein kinases (13, 14, 29), as well as steroid receptors (L.S.L. and N.R., unpublished observations). It is distinct from the previous view that the ansamycins induce the dissociation of Hsp90 from its target proteins (13, 29). Our observations suggest that the chaperone-bound polypeptide is the substrate for proteolysis. Notably, this presentation mechanism would differ from others, as in mitochondria, in which stable chaperone:polypeptide complexes have been shown to inhibit degradation (33).
Acknowledgments
We thank M. Timaul-Holmes and Z. Ma for expert technical assistance, R. Muise-Helmericks, J. Young, and U. Hämmerling for providing cell lines and purified proteins, respectively, and B. Netzer for critically reading this manuscript. This work was supported by grants from National Institutes of Health and National Cancer Institute, and by the Howard Hughes Medical Institute. C.S. is a fellow of the German Center of Cancer Research.
Footnotes
Abbreviations: GA, geldanamycin; HA, herbimycin A; DMSO, dimethyl sulfoxide; RL, reticulocyte lysate.
References
- 1.Craig E A, Gross C A. Trends Biochem Sci. 1991;16:135–140. doi: 10.1016/0968-0004(91)90055-z. [DOI] [PubMed] [Google Scholar]
- 2.Parsell D A, Lindquist S. Annu Rev Genet. 1993;27:437–496. doi: 10.1146/annurev.ge.27.120193.002253. [DOI] [PubMed] [Google Scholar]
- 3.Georgopoulos C, Liberek K, Zylicz M, Ang D. Properties of the Heatshock Proteins of Escherichia coli and the Autoregulation of the Heat Shock Response. Plainview, NY: Cold Spring Harbor Lab. Press; 1994. pp. 209–249. [Google Scholar]
- 4.Wiech H, Buchner J, Zimmermann R, Jacob U. Nature (London) 1992;358:169–170. doi: 10.1038/358169a0. [DOI] [PubMed] [Google Scholar]
- 5.Yonehara M, Minami Y, Kawata Y, Nagai J, Yahara I. J Biol Chem. 1996;271:2641–2645. doi: 10.1074/jbc.271.5.2641. [DOI] [PubMed] [Google Scholar]
- 6.Freeman B C, Morimoto R I. EMBO J. 1996;15:2969–2979. [PMC free article] [PubMed] [Google Scholar]
- 7.Borkovich K A, Farelly F W, Finkelstein D B, Taulien J, Lindquist S. Mol Cell Biol. 1989;9:3919–3930. doi: 10.1128/mcb.9.9.3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jakob U, Buchner J. Trends Biochem Sci. 1994;19:205–211. doi: 10.1016/0968-0004(94)90023-x. [DOI] [PubMed] [Google Scholar]
- 9.Bohen S P, Yamamoto K R. In: The Biology of Heat Shock Proteins and Molecular Chaperones. Morimoto R, Tissieres A, Georgopoulos C, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1994. pp. 313–334. [Google Scholar]
- 10.Smith D F. Mol Endocrinol. 1993;7:1418–1429. doi: 10.1210/mend.7.11.7906860. [DOI] [PubMed] [Google Scholar]
- 11.Pratt W B, Welsh M J. Semin Cell Biol. 1994;5:83–93. doi: 10.1006/scel.1994.1012. [DOI] [PubMed] [Google Scholar]
- 12.Uehara Y, Hori M, Takeuchi M, Umezawa H. Jpn J Cancer Res (Gann) 1985;76:672–675. [PubMed] [Google Scholar]
- 13.Whitesell L, Mimnaugh E G, De Costa B, Myers C E, Neckers L M. Proc Natl Acad Sci USA. 1994;91:8324–8328. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sepp-Lorenzino L, Ma Z, Lebwohl D E, Vinitsky A, Rosen N. J Biol Chem. 1996;270:16580–16587. doi: 10.1074/jbc.270.28.16580. [DOI] [PubMed] [Google Scholar]
- 15.Nimmesgern E, Hartl F U. FEBS Lett. 1993;331:25–30. doi: 10.1016/0014-5793(93)80290-b. [DOI] [PubMed] [Google Scholar]
- 16.Frydman J, Nimmesgern E, Ohtsuka K, Hartl F U. Nature (London) 1994;370:111–117. doi: 10.1038/370111a0. [DOI] [PubMed] [Google Scholar]
- 17.Hershko A, Heller H. Biochem Biophys Res Commun. 1985;128:1079–1086. doi: 10.1016/0006-291x(85)91050-2. [DOI] [PubMed] [Google Scholar]
- 18.Nguyen V T, Bensaude O. Eur J Biochem. 1994;220:239–246. doi: 10.1111/j.1432-1033.1994.tb18619.x. [DOI] [PubMed] [Google Scholar]
- 19.Schumacher R J, Hurst R, Sullivan W P, McMahon N J, Toft D O, Matts R L. J Biol Chem. 1994;269:9493–9499. [PubMed] [Google Scholar]
- 20.Nicolet C M, Craig E A. Mol Cell Biol. 1989;9:3638–3646. doi: 10.1128/mcb.9.9.3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen S, Prapapanich V, Rimerman R A, Honore B, Smith D F. Mol Endocrinol. 1996;10:682–693. doi: 10.1210/mend.10.6.8776728. [DOI] [PubMed] [Google Scholar]
- 22.Höhfeld J, Minami Y, Hartl F U. Cell. 1995;83:589–598. doi: 10.1016/0092-8674(95)90099-3. [DOI] [PubMed] [Google Scholar]
- 23.Prapapanich V, Chen S, Nair S, Rimerman R A, Smith D F. Mol Endocrinol. 1996;11:420–428. doi: 10.1210/mend.10.4.8721986. [DOI] [PubMed] [Google Scholar]
- 24.Kimura Y, Yahara I, Lindquist S. Science. 1995;268:1362–1365. doi: 10.1126/science.7761857. [DOI] [PubMed] [Google Scholar]
- 25.Jentsch S. Annu Rev Genet. 1992;26:179–207. doi: 10.1146/annurev.ge.26.120192.001143. [DOI] [PubMed] [Google Scholar]
- 26.Haas A L, Rose I A. Proc Natl Acad Sci USA. 1981;78:6845–6850. doi: 10.1073/pnas.78.11.6845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hershko A, Ganoth D, Pehrson J, Palazzo R E, Cohen L H. J Biol Chem. 1991;266:16376–16381. [PubMed] [Google Scholar]
- 28.Uehara Y, Murakami Y, Mizuno S, Kawai S. Virology. 1988;164:294–298. doi: 10.1016/0042-6822(88)90649-6. [DOI] [PubMed] [Google Scholar]
- 29.Schulte T W, Blagosklonny M V, Ingui C, Neckers L. J Biol Chem. 1995;270:24585–24588. doi: 10.1074/jbc.270.41.24585. [DOI] [PubMed] [Google Scholar]
- 30.Davis R J. J Biol Chem. 1993;268:14553–14556. [PubMed] [Google Scholar]
- 31.Stancato L F, Chow Y-H, Hutchinson K A, Perdew G H, Jove R, Pratt W B. J Biol Chem. 1993;268:21711–21716. [PubMed] [Google Scholar]
- 32.Hegde R S, Zuo J R, Voellmy R, Welch W J. J Cell Physiol. 1995;165:186–200. doi: 10.1002/jcp.1041650122. [DOI] [PubMed] [Google Scholar]
- 33.Wagner I, Arlt H, van Dyck L, Langer T, Neupert W. EMBO J. 1994;13:5135–5145. doi: 10.1002/j.1460-2075.1994.tb06843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]