

Abstract
One of the best-described transmembrane signal transduction mechanisms is based on receptor activation of the α subunit of the heterotrimeric G protein Gs, leading to stimulation of adenylyl cyclase and the production of cAMP. Intracellular cAMP is then thought to mediate its effects largely, if not entirely, by activation of protein kinase A and the subsequent phosphorylation of substrates which in turn control diverse cellular phenomena. In this report we demonstrate, by two different methods, that reduction or elimination of protein kinase A activity had no effect on phenotypes generated by activation of Gsα pathways in Drosophila wing epithelial cells. These genetic studies show that the Gsα pathway mediates its primary effects by a novel pathway in differentiating wing epithelial cells. This novel pathway may in part be responsible for some of the complex, cell-specific responses observed following activation of this pathway in different cell types.
Keywords: epithelial cells, integrins, signal transduction, cell adhesion, G-protein
One of the simplest and best-understood transmembrane signal transduction pathways in eukaryotic cells involves a family of heptahelical receptors that are coupled by the heterotrimeric GTP binding protein complex Gs, to the activation of adenylyl cyclase (AC). In this pathway, the initial binding of extracellular ligands to these receptors results in the activation of the Gs complex by promoting the exchange of GTP for GDP on the α subunit and the dissociation of Gsα from βγ. The GTP-bound, activated Gsα then mediates the activation of a family of ACs, resulting in elevation of the intracellular levels of the second messenger, cAMP (1–5). Termination of the signal occurs when GTP bound by the α subunit is hydrolyzed to GDP by an enzymatic activity intrinsic to the α subunit (6).
The role of intracellular cAMP is also thought to be well understood. A large number of genetic and biochemical studies in cultured mammalian cells have led to the view that cAMP evokes its intracellular effects primarily, if not exclusively, through activation of protein kinase A (PKA) (7–12). In all metazoan systems, PKA is a tetrameric complex composed of two regulatory subunits and two catalytic subunits. Upon elevation of intracellular cAMP, cAMP binds to regulatory subunits, releasing them from catalytic subunits which are then free to phosphorylate substrate proteins. Thus, the traditional transduction pathway is based in receptor activation of Gsα leading to stimulation of AC, the production of cAMP, activation of PKA, and the phosphorylation of substrates that in turn control diverse cellular phenomena such as metabolism, cell proliferation, gene transcription, and learning and memory. Studies in cultured cell systems have also demonstrated a wide variety of complex cell-specific responses to activation of Gsα pathways (13–16). Given the traditional view of the Gsα transduction pathway, specificity has been thought to be based on a number of factors such as selective expression of receptors and AC isoforms in the responding cell and/or selective activation of specific intracellular pools of PKA (17). More recently, a widely distributed family of Ca2+-permeable ion channels that are directly activated by cyclic nucleotides has also been described (18, 19). Signals arising from activation of these channels by cAMP are almost certainly integrated with PKA-generated signals to produce specific cellular responses to Gsα activation, although the exact contribution of these channels to any particular cellular response has yet to be clearly defined.
Aside from genetic studies in Dictyostelium, virtually all evidence underlying this traditional pathway of Gsα-activated signal transduction derives from the study of cultured cells. Recently, we have begun to use the genetic tools available in Drosophila to ask how activation of Gsα impacts on the development and function of specific cell types in vivo and to define the pathway by which these effects are mediated. Each of the components of the traditional Gsα signaling pathway has been identified in Drosophila (20–23). For example, a single Drosophila gene encoding both a structural and functional homolog of mammalian Gsα has been identified (DGsα; refs. 24–26). Drosophila genes encoding isoforms of PKA catalytic and regulatory subunits have also been identified and subjected to mutational analysis (27–29). Biochemical and genetic analysis of PKA activity in flies carrying mutations in one catalytic subunit gene, dco, indicate that this gene encodes the sole cAMP-activated protein kinase activity in postembryonic flies (28, 29). Consistent with this data, these studies have demonstrated that only the dco gene is required for the viability of the whole organism and is involved in mediating vital intercellular communication during oogenesis, embryogenesis and larval development (28, 30–33).
In this report we demonstrate by two different methods that elimination of PKA activity had no effect on the development of phenotypes generated by activation of Gsα pathways in Drosophila wing epithelial cells. These genetic studies show that the Gsα pathway generates this phenotype in differentiating wing epithelial cells by a mechanism that does not involve the traditional transduction pathway developed on the basis of studies in cultured cells. This novel pathway may in part be responsible for some of the complex, cell-specific responses observed following activation of this pathway in different cell types.
MATERIALS AND METHODS
Fly Stocks.
Vectors containing cDNAs or genes encoding the short form (25) of wild-type Drosophila Gsα (DGsαWT) and site-directed mutant (Q215L) Drosophila Gsα (DGsα*; ref. 26), wild-type hemagglutinin (HA)-tagged rat GsαWT, and site-directed mutant HA-tagged (Q227L) rat Gsα (Gsα*; ref. 34), dominant-negative regulatory subunits of PKA (Rdn; gift of Dan Kalderon, Columbia University; ref. 33) and β-galactosidase (β-gal) were subcloned into plasmid pUAST (35) by standard procedures. In these constructs, coding sequences are located downstream of five consensus binding sites for the yeast GAL4 protein. Resulting plasmids were used to generate multiple Drosophila transgenic lines for each construct by standard methods (35).
The GAL4-30A and 71B enhancer trap lines were a gift from Andrea Brand (35). GAL4-10 was recovered in a screen carried out in one of our laboratories (K. Moffat, J. Connoly, J. Keane, S. Sweeney, and C.O., unpublished data). HEM/TM3 flies were produced by recombining GAL4-10 onto the same chromosome as a UAS-DGsα* transgene. yw;FRT40@ DCOH2/CyO, and y122, p[hsp-flp];FRT@40A, p[y+]25F;plz-lacz flies were gifts from Dan Kalderon (Columbia University) and vn1, rhoVE flies were the gift of Seth Blair (University of Wisconsin).
β-Gal and Antibody Staining.
Expression of β-gal was detected using 5-bromo-4-chloro-3-indolyl β-d-galactoside (X-Gal) as a chromogenic substrate. Staining of imaginal discs was done as described by Brand and Perrimon (35). X-Gal staining of pharate adult wings was similar except that before fixation, freshly removed wings were placed in Drosophila Ringers, diluted 4-fold with water until the wing had unfolded (about 2–3 min) and then immediately transferred to 1% glutaraldehyde in PBS and processed as described for discs (35).
For antibody staining, pharate adults of the desired genotype were removed from the pupal case and an incision was made along the dorsal midline of the abdomen and thorax. In addition, the tips of the wings were cut off to facilitate penetration of fixative. The tissue was rocked in 4% paraformaldehyde in PBS for 2–3 h at room temperature. Five micron cryostsat sections were cut through the thorax and wings, collected, and returned to fixative for at least another 20 min. After washing three times in PBT (PBS/0.1% Triton X-100), sections were blocked for 1 h in 10% horse serum in PBS. To detect the HA epitope, the 12CA5 mAb (Boehringer Mannheim) was diluted to 1 μg/ml in 10% horse serum. Endogenous Gsα was detected using the RM antibody (36) diluted to 2 μg/ml. Integrin was detected with a specific PS-β-integrin mAb (gift of D. Brower, University of Arizona) and F-actin detected using rhodamine-phalloidin (Molecular Probes). Antibody binding was localized with biotinylated secondary antibodies (Vector Laboratories) followed by either fluoresceinated avidin or peroxidase ABC reagent (both from Vector Laboratories). Slides were then processed essentially as describe in Wolfgang et al. (36). Confocal images were recorded on a Bio-Rad 6000 microscope.
Generation of dco Mutant Clones.
First instar larvae, generated by crossing y122, p[hsp-flp]; FRT@40A, p[y+]25F;plz-lacz females to y, w;FRT@40A, dcoH2/CyO; HEM/TM2 males, were heat shocked for 1 h at 38°C to induce mitotic recombination, and adult flies were examined for clones of y− wing tissue. Clones extending over both dorsal and ventral surfaces were examined for blistering. Wings were removed in Ringers solution, dehydrated in ethanol and mounted in cedar wood oil.
RESULTS
GAL4 Mediated Expression of DGsα*.
Given the potential for cell-specific responses to activation of Gsα pathways, we have taken advantage of recently developed expression systems based in the yeast GAL4 protein and GAL4 upstream activating control elements (UAS) (35) to examine the effect of expression of an activated form of Drosophila Gsα subunit (Q215L; DGsα*) in specific cell types at a variety of times in development. Biochemical studies have shown that mutation of this position in DGsα and in the equivalent position in mammalian Gsα (i.e., Q227L; Gsα*), results in a greatly reduced GTP hydrolytic rate and thus, in receptor-independent activation of the α subunit and downstream components of this pathway (26, 37). Expression of Gα proteins containing such activating mutations has been demonstrated in many situations to provide a valid system for activating appropriate intracellular signaling pathways in the absence of any knowledge of upstream receptors. Wild-type DGsα cDNAs and those containing the Q215L activating mutation were subcloned into a Drosophila transformation vector downstream from 5 UAS sequences recognized by the yeast GAL4 transcriptional activator. Expression of each of these molecules during development was then mediated by crossing resulting transformed lines to 22 different “enhancer trap” lines in which GAL4 protein expression is controlled by the insertion of transposable elements carrying the GAL4 gene adjacent to promoters and enhancers that now direct its expression. Expression of DGsα* results in different phenotypes (eight lethal, four wing, three smaller adult flies, and seven no phenotype; data not shown) depending on the temporal and spatial pattern of expression dictated by individual Gal4 lines and appear to have their basis in the alteration of a number of cellular properties (e.g., cell adhesion, proliferative potential).
One consistent effect of DGsα* was the formation of wing blisters by GAL4 lines that mediate expression in wing epithelium during late pupal periods. For example, when GAL4-30A and GAL4-10 lines are used to drive expression of DGsα*, the epithelia that form the dorsal and ventral surfaces of the wing no longer adhere to one another and separate following emergence from the pupal case, causing the wing to appear ballon-like rather than flat (Fig. 1; for a review of wing morphogenesis, see ref. 38). Nonneural wing tissue is organized into the patterned distribution of two cell types, intervein (90%) and vein (10%). During wing development, intervein cells are responsible for connecting and holding the two surfaces of the wing together and, to that end, differentiate a highly specialized system of cytoskeletal supports (the transalar array) anchored in integrin-mediated basal adhesions. Wing blisters (e.g., Fig. 1) can arise from (i) failure of the dorsal and ventral wing surfaces to adhere properly during pupal development, (ii) defects in the formation or organization of the transalar array, or (iii) defects in the specification of vein and intervein cells in the developing wing disc. For example, flies carrying mutations in the blistered gene have enlarged veins, and thus blistered wings, due to the misspecification of intervein cells into vein cells (39). However, blisters still form on expression of DGsα* in the wings of flies carrying mutations in the vein and rhomboid genes (vn1, rhoVE). This combination of mutations essentially eliminates wing veins (not shown) (40). These results demonstrate then that the blisters resulting from expression of DGsα* is specifically due to alterations in the structure or function of intervein cells.
Figure 1.

Phenotypes produced when Gal4-30A is used to drive expression of (A) wild-type DGsα or (B) DGsα*. Expression of wild-type DGsα produces no visible phenotype while DGsα* results in wing blisters.
The pattern of GAL4 expression directed by lines which generate wing blisters on expression of DGsα* was determined by using each to direct expression of β-gal placed under UAS control. In late third instar larvae, GAL4-30A drives strong expression in a group of cells largely restricted to a ring of wing disc tissues that will form the most basal/proximal portions of the wing blade (Table 1) while GAL4-10 directs a weak cruciform expression pattern in the blade region (Table 1). Later, during the second half of the pupal stage and in pharate adult wings, β-gal expression in both GAL4 lines is detected throughout the entire wing blade in both epithelial layers which secrete the cuticular surface of the wing (Table 1).
Table 1.
Pattern of GAL4 expression in wing discs and late pupal wings
| GAL4 line | Late third instar wing discs | Late pupal wing |
|---|---|---|
| 10 | Weak expression in prospective wing blade | Strong expression throughout wing blade |
| 30 | Strong expression in prospective wing base | Strong expression throughout wing blade |
| 71b | Strong expression in prospective wing blade | Strong expression restricted to wing base |
A number of observations indicate that the blistering phenotype is a specific response of wing epithelial cells to activation of DGsα pathways. First, wing blistering depends on expression of DGsα* because wings appear normal when these GAL4 lines are used to drive expression of wild-type DGsα (DGsαWT) (Fig. 1A) or in flies containing DGsα* transgenes in the absence of a source of Gal4 protein (data not shown). Consistent with this observation, wing blisters are observed when these GAL4 lines are used to drive expression of activated forms of mammalian Gsα (Q227L; Gsα*), and no phenotype is observed following expression of wild-type mammalian Gsα (GsαWT) (data not shown). Second, no phenotype or a different phenotype is observed following expression by these GAL4 lines of activated or wild-type forms of Drosophila Giα and Goα or mammalian Giα proteins (data not shown), indicating that this phenotype does not depend on nonspecific components of this pathway like βγ subunits. Finally, the blistering phenotype depends on expression of DGsα* at specific times during development. For example, one specific Gal4 line, Gal4-71b, mediates expression in wing discs (Table 1) but only in a small region in the most proximal portion of the wing in late stage pupae. No wing blisters are observed when GAL4-71b is used to drive expression of DGsα* (data not shown). Thus, expression primarily in third instar wing discs leads to wild-type wings while expression during the later half of pupal wing development generates blisters. These observations indicate that blistering is a specific response of wing epithelial cells to activation of DGsα pathways and cannot be due to nonspecific responses produced by overexpression of activated Gα proteins. In addition, it appears that the sensitivity of wing epithelial cells to activation of this pathway changes as they undergo their developmental program.
Cellular Distribution of Gsα Produced from UAS Transgenes Is Similar to That of the Endogenous DGsα Protein.
To address the possibility that wing blistering is a consequence of aberrant DGsα* localization in pharate wing epithelia or to activation of this pathway in a cell type that does not normally express the DGsα protein, we used confocal microscopy to localize both endogenous DGsα and exogenous forms of activated and wild-type mammalian Gsα expressed from GAL4-sensitive transgenes. To distinguish mammalian Gsα from endogenous DGsα, the mammalian forms contained a peptide epitope taken from the influenza HA protein. Previous studies have shown that these “tagged forms” of the mammalian Gsα proteins (HA-GsαWT and HA-Gsα*) function normally (34, 41). In Fig. 2A, a conventional differential interference contrast image of a section through a pharate adult wing reveals the two epithelial surfaces that are connected at their basal surfaces by transalar elements (38). Blistering must result from failure of these specializations to form or the rupture of these connections during wing expansion. In Fig. 2B, a confocal image of the same section localizes endogenous DGsα to the basal membranes of these cells as well as membranes surrounding individual transalar elements connecting the two epithelial surfaces. Using antibodies specific for the peptide epitope, exogenously expressed HA-GsαWT appears to be present in identical subcellular locations in wings that will not contain blisters (Fig. 2C). The distribution of HA-Gsα* in these cells in wings that will contain blisters is similar to DGsαWT and HA-GsαWT in that the staining is associated with the basal and transalar membranes of the epithelium (Fig. 2D). However, whereas both endogenously expressed DGsα and exogenously expressed HA-GsαWT appear tightly associated with these membranes (note the bright and sharp line of staining), HA-Gsα* staining is somewhat more diffuse suggesting that this activated form of Gsα may not be as tightly associated with these cell membranes, as would be predicted from a number of biochemical studies (34, 41). Since the samples in Fig. 2 C and D were processed together (i.e., fixed, sectioned, stained, and photographed) under identical conditions, the similar levels of staining observed reflect similar levels of expression of each HA-tagged protein. Thus, exogenously expressed Gsα proteins, whether wild type or activated, are localized to the same subcellular domains and expressed at roughly the same level as the endogenous protein. In addition, at least at this level, the overall morphology of wing epithelial cells is not overtly altered by expression of activated forms of Gsα. Consistent with this conclusion, the distribution of F-actin, as revealed by phalloidin staining, is similar in cells expressing DGs* when compared with cells expressing DGs (not shown). Since F-actin distribution serves to accurately reflect the overall architecture of the transalar apparatus (38), these results demonstrate that the blistering observed on expression of DGsα* in pharate wings is not due to incorrect formation of transalar connections.
Figure 2.
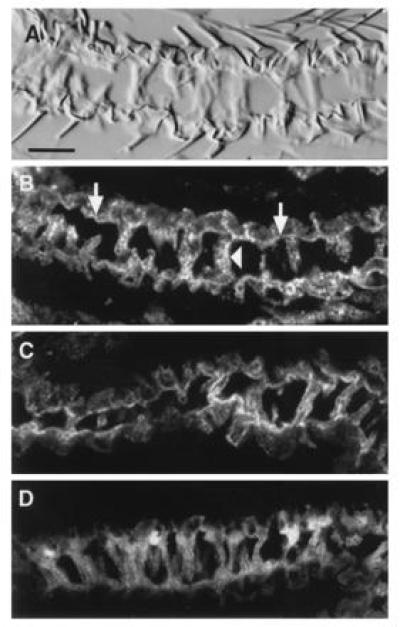
Localization of endogenous DGsα and HA-tagged mammalian Gsαs, expressed from transgenes in pharate adult wings, by immunofluorescence confocal microscopy. (A and B) Transverse sections through pharate adult wings of a Gal4-30A fly in the absence of an inducible transgene. (A) Bright field image. (B) The same section viewed in a confocal microscope and immunostained for endogenous DGsα protein. Note that immunostaining is most intense on the basal surface of the wing epithelia (arrows) and along the transalar elements (arrowheads). (C) Localization of wild-type rat HA-Gsα expressed under Gal4-30A control from transgenes and detected using an antibody to the HA epitope. The wild-type rat Gsα mimics the distribution of endogenous DGsα. (D) Localization of rat HA-Gsα* expressed under Gal4-30A control. (Bar = 10 μm.)
Blistering Does Not Result from Inappropriate Localization of Integrin Complexes.
High levels of β-integrin are detected in the transalar elements which connect the basal surfaces of the dorsal and ventral epithelia of the wings (38). In addition, wing blisters indistinguishable from those generated by expression of DGs* in late pupal wings can arise from the loss of integrin function through mutations in genes encoding either the α or β subunit of the PS-integrins (42–45). These observations suggest that integrins in these regions serve as a critical component of the mechanism which holds the opposing epithelia together and prevent their separation during the process of wing inflation.
To test whether expression of DGsα* produces blisters by loss or inappropriate localization of integrin complexes, we examined the distribution of β-integrin in sections of pharate adult wings from flies expressing DGsαWT and DGsα*. No difference could be detected in the levels or distribution of β-integrin in flies expressing DGsα*, which will have wing blisters (Fig. 3B), compared with the DGsαWT expressing flies, which will have wild-type wings (Fig. 3A). Thus, blistering must result from the rupture of these connections during wing expansion.
Figure 3.
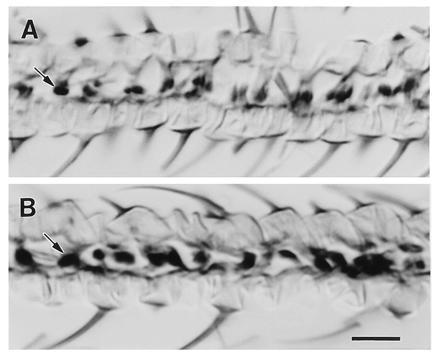
The distribution of β-integrin in cross-sections through pharate adult wings. To control for variations in staining, a pharate wing expressing DGsα*, thereby destined to form blisters (B), was mounted next to a pharate wing expressing DGsα, which will form a normal, unblistered wing (A). In each case, Gal4-30A was used to drive expression. The pairs were then sectioned and stained together for peroxidase activity. Sixteen such pairs were processed and the images presented represent the typical degree of variation observed both within and between samples. (Bar = 10 μm.)
DGsα*-Induced Blistering Occurs in the Absence of PKA.
Phenotypes arising from activation of DGsα pathways should critically depend on activation of PKA, given the traditional scheme developed in cultured mammalian cells. Consistent with this prediction, many studies have demonstrated that response of cultured cells to activation of this pathway through expression of Gsα* depends on PKA (15, 46, 47). To test whether this pathway is acting in wing epithelial cells to generate blistering, we initially employed a dominant-negative form of the regulatory subunit of PKA (Rdn) to inhibit PKA activity in these cells (33, 56). A number of studies have shown that during early stages of wing formation, PKA (as encoded by the dco gene) functions to repress the expression of signaling molecules like the transforming growth factor β homolog decapentaplegic (dpp) that mediate subsequent growth and pattern formation in the developing wing (31–33). Thus, in the absence of PKA, inappropriate expression of dpp leads to anterior wing duplications. As shown in Fig. 4A, GAL4-30A-mediated expression of Rdn results, as expected, in small wing duplications (arrows) in the most proximal portion of the anterior compartment of the wing, consistent with the expression pattern dictated by GAL4-30A in wing discs during early larval periods (Table 1). These duplications are similar to duplications generated in clones of cells homozygous for dco null mutations (31–33) consistent with previous results which indicate that dco encodes the sole cAMP-dependent protein kinase activity in these cells (28, 29). By constructing flies that carry both DGsα* and Rdn GAL4-sensitive transgenes, activation of DGsα pathways and inhibition of PKA occur simultaneously within cells of the wing epithelium when these GAL4 lines are used to drive expression. Coexpression of DGsα* and Rdn in these cells results in a superimposition of the DGsα* phenotype on the Rdn phenotype—i.e., wing duplications that contain blisters (Fig. 4B). Thus, each phenotype occurs independently when these proteins are coexpressed, indicating that PKA activity is not required to generate the blistering observed on activation of the DGsα pathway in wing epithelial cells.
Figure 4.

Phenotypes produced when Gal4-30A is used to drive expression of Rdn (A) or Rdn and DGsα* (B). Rdn expression produces a small anterior wing duplication while expression of both Rdn and DGsα* produces both the anterior wing duplication and blisters within the duplication.
Although Rdn expression generates a wing phenotype equivalent to that found in dco-null clones, biochemical studies of a variety of mutations in the dco gene suggest that these phenotypes can also be generated in flies containing up to roughly 10% the normal PKA activity (28, 33). Thus, the blistering observed on activation of DGsα pathways could depend on residual activity not eliminated by Rdn expression. To address this possibility, we eliminated all dco-dependent PKA activity by generating clones of cells within the developing wing which are homozygous for null alleles of dco. Clones of cells in which dco-generated PKA activity has been eliminated were again observed to produce anterior duplications of the normal wing pattern (Fig. 5B) (31–33) while expression of DGsα* by GAL4-10 produces large blisters in wings containing no dco-null clones (Fig. 5A). As shown in Fig. 5C, GAL4-driven expression of DGsα* in dco-null clones again results in the formation of blisters within large clones (arrows) generated by elimination of PKA activity. This result demonstrates that the blistering observed on expression of DGsα* in pharate wings occurs even in the complete absence of dco-encoded PKA activity.
Figure 5.
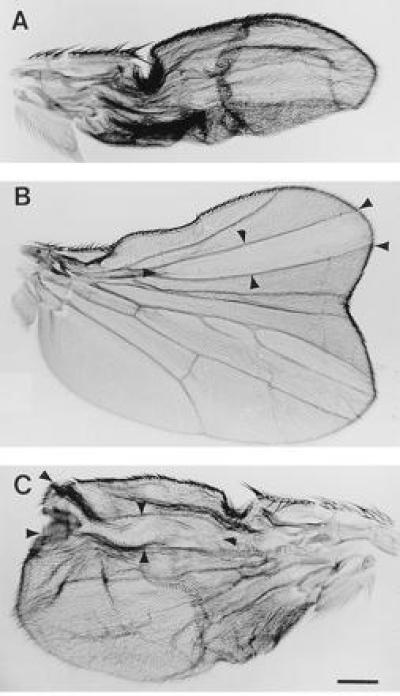
Wings from (A) HEM fly (Gal4-10, DGsα*) with severe wing blisters; (B) a fly with a dco-null wing clone (note large anterior duplication; and (C) a HEM fly carrying a dco-null wing clone. Position of clones are marked by arrow heads. Despite the absence of PKA in the region of the clone, the wing still blistered. (Bar = 200 μm.)
DISCUSSION
Previous studies have shown that each of the components of the traditional Gsα signaling pathway are present in Drosophila and specifically, that DGsα is the functional, as well as structural, homolog of its mammalian counterpart (24–26). Thus, we have been able to exploit a number of genetic techniques available in Drosophila to examine how activation of the ubiquitous Gsα signaling pathway in vivo might lead to the cell type-specific responses observed in many differentiated cells in culture. Here, we have used the GAL4/UAS system (35) to direct the expression of site-directed mutant forms of the DGsα that result in activation of this protein, and hence downstream components of the pathway defined by this signaling molecule. By use of GAL4 enhancer trap lines that drive expression in specific spatial and temporal patterns during development, we have been able to define a set of cell-specific phenotypes. By characterizing one of these phenotypes, wing blistering, in more detail, we have been able to further demonstrate that this phenotype represents a specific response of wing epithelial cells to activation of DGsα pathways, that the sensitivity of wing epithelial cells to activation of this pathway changes as they undergo their developmental program, and that blister formation does not result from misspecification of vein cells into intervein cells because blisters form in the complete absence of wing veins. Thus, blisters must result from some restricted alteration in the structure or function of intervein cells.
A number of observations suggest that the blistering phenotype does not result from inappropriate localization of exogenously expressed DGsα protein or in disruption of the normal architecture of intervein cells. In addition, blistering does not appear to be due to some indirect effect of DGsα* expression on the localization of integrin complexes that mediate the adhesion of dorsal and ventral wing surfaces. Blistering then is a very specific response of these cells to activation of the DGsα pathway. Because the overall organization of intervein cells is apparently unchanged by expression of DGsα* and the distribution of a number of critical proteins is not altered, these blisters must result from rupture of the basal adhesion complexes that hold the two epithelial layers together during wing expansion. Identical blistering phenotypes can be generated by mutations in genes encoding either the α or β subunit of the PS-integrins within the basal adhesion complex (42–45). Thus, these observations suggest that the function of the DGsα pathway within these specialized cells is to regulate the adhesive properties of the integrin molecules; a regulatory pathway that must play a role in the complex morphogenic changes that occur as the wing develops during pupal stages (38). This proposed molecular interaction between the Gsα pathway and integrins can be tested at the genetic level given existing mutations in each of the two α subunits and common β subunit which form these integrin complexes (42–45).
By two different methods, we have been able to demonstrate that the pathway by which activation of the DGsα pathway affects the properties of the basal adhesion complexes does not depend on PKA. Although expression of Rdn should inhibit all PKA activity within these cells, regardless of isoform, abundant genetic and biochemical evidence has demonstrated that the only functional PKA catalytic subunits in postembryonic flies are the product of the dco gene (28, 29). Consistent with this conclusion, expression of Rdn in wing discs results in the anterior wing duplications observed in clones of cells containing homozygous dco-null mutations. Since blistering occurs on co-expression of DGsα* with Rdn and on expression of DGsα* in dco-null clones, the traditional transduction pathway developed on the basis of biochemical studies in cultured mammalian cells; namely, Gsα activation of AC leading to stimulation of PKA does not lead to the generation of the blistering phenotype in these cells.
Although the molecular components of the DGsα pathway leading to blister formation remain to be identified, two general alternatives are possible. First, these studies may define a novel signal transduction pathway activated directly by DGsα. Recently, a number of studies in cultured cells have also proposed novel functions for Gsα which are independent of PKA (48–51). In each case, however, further study has shown that the effects observed are likely to be indirect and depend instead on the activation of PKA (46, 47). At this time then, the only effectors whose activity is clearly modulated directly by Gsα are AC isoforms. Alternatively, the primary mediator of the effects of cAMP in these cells may be a molecule other than PKA. There is, however, no method available for directly determining in vivo whether intracellular concentrations of cAMP are elevated on expression of DGsα* in wing epithelial cells. In any event, the genetic studies presented here show that novel Gsα pathways do exist, indicating that the in vivo response of cells to activation of this ubiquitous signaling pathway cannot be explained simply by activation of PKA but may represent the interaction of both PKA-dependent and novel PKA-independent pathways. Inputs from each of these pathway are likely to be coordinated with signals generated through other pathways to produce the complex, cell-specific responses observed following activation of Gsα pathways in different cell types in culture and in symptoms present in individuals with McCune–Albright syndrome generated by somatic mutations resulting in activated Gsα proteins (52, 53).
Many recent studies have pointed out the importance of genetic analysis in delineating and organizing the components of complex transmembrane signal transduction pathways. Only recently have these techniques been applied to pathways activated by heterotrimeric G proteins in metazoan organisms (54, 55). We anticipate that further application of the genetic tools available in Drosophila will not only delineate the components of the novel pathway activated by DGsα in wing epithelial cells but the components of pathways leading to specific responses of other cell types to activation of this and other Gα pathways.
Acknowledgments
We thank Hemlata Mistry for constructing the HEM recombinant chromosome, Dr. Dan Kalderon for dco and Rdn strains, Dr. Henry Bourne for HA-tagged rat Gsα constructs, and the Vollum Confocal Facility. This work was supported by Science and Engineering Research Council Grant GR/F94989 to C.O., by Human Frontiers Science Program Grant RG67-93 to C.O. and M.F., and grants from the National Institutes of Health to M.F.
Footnotes
Abbreviations: AC, adenylyl cyclase; PKA, protein kinase A; HA, hemagglutinin; β-gal, β-galactosidase.
References
- 1.Bourne, H. & Nicoll, R. (1993) Cell 72/Neuron 10, 65–75. [DOI] [PubMed]
- 2.Neer E. Protein Sci. 1994;3:3–14. doi: 10.1002/pro.5560030102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neer E. Cell. 1995;60:249–257. doi: 10.1016/0092-8674(95)90407-7. [DOI] [PubMed] [Google Scholar]
- 4.Tang W, Gilman A. Cell. 1992;70:869–872. doi: 10.1016/0092-8674(92)90236-6. [DOI] [PubMed] [Google Scholar]
- 5.Cooper D, Mons N, Karpen J. Nature (London) 1995;374:421–424. doi: 10.1038/374421a0. [DOI] [PubMed] [Google Scholar]
- 6.Bourne H, Sanders D, McCormick F. Nature (London) 1991;349:117–127. doi: 10.1038/349117a0. [DOI] [PubMed] [Google Scholar]
- 7.Insel P, Bourne H, Coffino P, Tomkins G. Science. 1975;190:896–898. doi: 10.1126/science.171770. [DOI] [PubMed] [Google Scholar]
- 8.Coffino P, Bourne H, Freidrich U, Hochmamm P, Insel I, Lemaire K, Melmon G, Tomkins G. Recent Prog Horm Res. 1976;32:669–684. doi: 10.1016/b978-0-12-571132-6.50037-3. [DOI] [PubMed] [Google Scholar]
- 9.Gottesman M. Cell. 1980;22:329–330. doi: 10.1016/0092-8674(80)90342-6. [DOI] [PubMed] [Google Scholar]
- 10.Gottesman M, Le Cam A, Bukowski M, Pastan I. Somatic Cell Genet. 1980;6:45–61. doi: 10.1007/BF01538695. [DOI] [PubMed] [Google Scholar]
- 11.Gottesman M. In: Molecular Cell Genetics. Gottesman M, editor. New York: Wiley; 1985. pp. 711–743. [Google Scholar]
- 12.Francis S, Corbin J. Annu Rev Physiol. 1994;56:237–272. doi: 10.1146/annurev.ph.56.030194.001321. [DOI] [PubMed] [Google Scholar]
- 13.Rosengurt E, Legg A, Strang G, Courtenay-Luck N. Proc Natl Acad Sci USA. 1981;78:4392–4396. doi: 10.1073/pnas.78.7.4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kabir A, Kozasa T, Kaziro Y, Nakamura S. Cell Signalling. 1993;5:443–452. doi: 10.1016/0898-6568(93)90084-y. [DOI] [PubMed] [Google Scholar]
- 15.Chen J, Iyengar R. Science. 1994;263:1278–1281. doi: 10.1126/science.8122111. [DOI] [PubMed] [Google Scholar]
- 16.Faure M, Bourne H. Mol Biol Cell. 1995;6:1025–1035. doi: 10.1091/mbc.6.8.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scott J, McCartney S. Mol Endocrinol. 1994;8:5–11. doi: 10.1210/mend.8.1.8152430. [DOI] [PubMed] [Google Scholar]
- 18.Zimmerman A. Curr Opin Neurobiol. 1995;5:296–303. doi: 10.1016/0959-4388(95)80041-7. [DOI] [PubMed] [Google Scholar]
- 19.Biel M, Zong X, Distler M, Bosse E, Klugbauer N, Murakami M, Flockerzi V, Hofmann F. Proc Natl Acad Sci USA. 1994;91:3505–3509. doi: 10.1073/pnas.91.9.3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Witz P, Amlaikey N, Plassat J, Maroteaux L, Borrelli E, Hen R. Proc Natl Acad Sci USA. 1990;87:8940–8944. doi: 10.1073/pnas.87.22.8940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nighorn A, Healy M J, Davis R L. Neuron. 1991;6:455–476. doi: 10.1016/0896-6273(91)90253-v. [DOI] [PubMed] [Google Scholar]
- 22.Levin L R, Han P L, Hwang P M, Feinstein P G, Davis R L, Reed R R. Cell. 1992;68:479–489. doi: 10.1016/0092-8674(92)90185-f. [DOI] [PubMed] [Google Scholar]
- 23.Smolik S, Rose R, Goodman R. Mol Cell Biol. 1992;12:4123–4132. doi: 10.1128/mcb.12.9.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quan F, Wolfgang W, Forte M. Proc Natl Acad Sci USA. 1989;86:4321–4325. doi: 10.1073/pnas.86.11.4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quan F, Forte M. Mol Cell Biol. 1990;10:910–917. doi: 10.1128/mcb.10.3.910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quan F, Thomas L, Forte M. Proc Natl Acad Sci USA. 1991;88:1898–1902. doi: 10.1073/pnas.88.5.1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kalderon D, Rubin G. Genes Dev. 1988;2:1539–1556. doi: 10.1101/gad.2.12a.1539. [DOI] [PubMed] [Google Scholar]
- 28.Lane M, Kalderon D. Genes Dev. 1993;7:1229–1243. doi: 10.1101/gad.7.7a.1229. [DOI] [PubMed] [Google Scholar]
- 29.Melendez A, Li W, Kalderon D. Genetics. 1995;141:1507–1520. doi: 10.1093/genetics/141.4.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lane M, Kalderon D. Genes Dev. 1994;8:2986–2995. doi: 10.1101/gad.8.24.2986. [DOI] [PubMed] [Google Scholar]
- 31.Jiang J, Struhl G. Cell. 1995;80:563–572. doi: 10.1016/0092-8674(95)90510-3. [DOI] [PubMed] [Google Scholar]
- 32.Lepage T, Cohen S, Diaz-Benjumea F, Parkhurst S. Nature (London) 1995;373:711–715. doi: 10.1038/373711a0. [DOI] [PubMed] [Google Scholar]
- 33.Li W, Ohlenmeyer M, Lane M, Kalderon D. Cell. 1995;80:553–562. doi: 10.1016/0092-8674(95)90509-x. [DOI] [PubMed] [Google Scholar]
- 34.Levis M, Bourne H. J Cell Biol. 1992;119:1297–1307. doi: 10.1083/jcb.119.5.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brand A, Perrimon N. Development (Cambridge, UK) 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 36.Wolfgang W, Spiegel A, Forte M. J Neurosci. 1990;10:1014–1024. doi: 10.1523/JNEUROSCI.10-03-01014.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Landis C, Masters S, Spada A, Pace A, Bourne H, Vallar L. Nature (London) 1989;340:692–696. doi: 10.1038/340692a0. [DOI] [PubMed] [Google Scholar]
- 38.Fristrom D, Wilcox M, Fristrom J. Development (Cambridge, UK) 1993;117:509–523. doi: 10.1242/dev.117.2.509. [DOI] [PubMed] [Google Scholar]
- 39.Fristrom D, Gotwals P, Eaton S, Kornberg T, Sturtevant M, Bier E, Fristrom J. Development (Cambridge, UK) 1994;120:2661–2671. doi: 10.1242/dev.120.9.2661. [DOI] [PubMed] [Google Scholar]
- 40.Sturtevant M, Bier E. Development (Cambridge, UK) 1995;121:785–801. doi: 10.1242/dev.121.3.785. [DOI] [PubMed] [Google Scholar]
- 41.Wedegaertner P, Bourne H. Cell. 1994;77:1063–1070. doi: 10.1016/0092-8674(94)90445-6. [DOI] [PubMed] [Google Scholar]
- 42.Brabant M, Brower D. Dev Biol. 1993;157:49–59. doi: 10.1006/dbio.1993.1111. [DOI] [PubMed] [Google Scholar]
- 43.Brower D, Jaffe S. Nature (London) 1989;342:285–287. doi: 10.1038/342285a0. [DOI] [PubMed] [Google Scholar]
- 44.Brower D, Bunch T, Mukai L, Adamson T, Wehrli M, Lam S, Friedlander E, Roote C, Zusman S. Development (Cambridge, UK) 1995;121:1311–1320. doi: 10.1242/dev.121.5.1311. [DOI] [PubMed] [Google Scholar]
- 45.Brown N. Development (Cambridge, UK) 1994;120:1221–1231. doi: 10.1242/dev.120.5.1221. [DOI] [PubMed] [Google Scholar]
- 46.Hanson S, Casanova J. J Cell Biol. 1994;126:677–687. doi: 10.1083/jcb.126.3.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hartzell H C, Mery P-F, Fischmeister R, Szabo G. Nature (London) 1991;351:573–576. doi: 10.1038/351573a0. [DOI] [PubMed] [Google Scholar]
- 48.Mattera R, Graziano M, Yatani Y, Zhou Z, Graf R, Codina J, Birnbaumer L, Gliman A, Brown A. Science. 1989;243:804–807. doi: 10.1126/science.2536957. [DOI] [PubMed] [Google Scholar]
- 49.Yatani A, Imoto Y, Codina J, Hamilton S, Brown A, Birnbaumer L. J Biol Chem. 1988;263:9887–9895. [PubMed] [Google Scholar]
- 50.Bomsel M, Mostov K. J Biol Chem. 1993;268:25824–25835. [PubMed] [Google Scholar]
- 51.Pimplikar S, Simons K. Nature (London) 1993;362:456–458. doi: 10.1038/362456a0. [DOI] [PubMed] [Google Scholar]
- 52.Schwindinger W, Francoman C, Levine M. Proc Natl Acad Sci USA. 1992;89:5152–5156. doi: 10.1073/pnas.89.11.5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weinstein L, Shenker A, Gejman P, Merion M, Friedman E, Spiegel A. N Engl J Med. 1991;325:1688–1740. doi: 10.1056/NEJM199112123252403. [DOI] [PubMed] [Google Scholar]
- 54.Mendel J, Korswagen H, Liu K, Hajdu-Cronin Y, Simon M, Plasterk R, Sternberg P. Science. 1995;267:1652–1655. doi: 10.1126/science.7886455. [DOI] [PubMed] [Google Scholar]
- 55.Segalat L, Elkes D, Kaplan J. Science. 1995;267:1648–1651. doi: 10.1126/science.7886454. [DOI] [PubMed] [Google Scholar]
- 56.Clegg C, Correll L, Cadd G, McKnight G. J Biol Chem. 1987;262:13111–13119. [PubMed] [Google Scholar]